
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Phenyl-incorporated polyorganosilica membranes with enhanced hydrothermal stability for H2/CO2 separation†
Vinh T.
Bui
 a,
Varun R.
Satti
a,
Elizabeth
Haddad
a,
Ameya Manoj
Tandel
a,
Narjes
Esmaeili
a,
Sai Srikar
Chundury
a,
Fathy
Attia
a,
Lingxiang
Zhu
bc and
Haiqing
Lin
*a
a,
Varun R.
Satti
a,
Elizabeth
Haddad
a,
Ameya Manoj
Tandel
a,
Narjes
Esmaeili
a,
Sai Srikar
Chundury
a,
Fathy
Attia
a,
Lingxiang
Zhu
bc and
Haiqing
Lin
*a
aDepartment of Chemical and Biological Engineering, University at Buffalo, The State University of New York, Buffalo, NY 14260, USA. E-mail: haiqingl@buffalo.edu
bU.S. Department of Energy, National Energy Technology Laboratory, Pittsburgh, PA 15236, USA
cNETL Support Contractor, 626 Cochrans Mill Road, P. O. Box 10940, Pittsburgh, PA 15236, USA
First published on 17th June 2024
Abstract
Ultrathin silica membranes can be prepared by oxygen plasma treatment of polysiloxane-based membranes and exhibit excellent H2/CO2 separation properties. However, silica often faces hydrothermal instability, reducing gas selectivity. Here, we incorporate hydrophobic phenyl groups into polysiloxane precursors by copolymerizing with vinyl-terminated polyphenylmethylsiloxane (vPPMS) and thoroughly investigate the chemical and structural properties and H2/CO2 separation stability of the resulting polyorganosilica membranes. Significantly, adding phenyl groups enhances the hydrothermal stability of the polyorganosilica membranes, as corroborated by their relatively stable surface atomic compositions. For instance, the hydrothermal challenge decreases the H2/CO2 selectivity by 74% for a phenyl-free membrane and the O/Si molar ratio from 1.97 to 1.72; by contrast, it reduces the selectivity by only 17% for a membrane derived from a polysiloxane containing 60 mass% PPMS, which is accompanied by a smaller change of the O/Si molar ratio from 2.0 to 1.85. This study unravels the mechanism of hydrothermal instability of silica membranes and demonstrates that enhancing hydrophobicity can effectively improve their potential applicability involving water vapor.
Introduction
Silica membranes with unique porous structures that permeate H2 and reject larger gas molecules such as CO2, CH4, and C2H6 are attractive for hydrogen purification and recovery, an essential component of a hydrogen economy.1–4 These membranes are often fabricated using a sol–gel or chemical vapor deposition (CVD) method, and the selective layer thickness can range from 0.2 to 10 μm, rendering good separation properties.5,6 However, inorganic substrates are often used in these membranes, making large-scale production a great challenge.3,7–9Recently, we demonstrated that rapid oxygen plasma treatment of polysiloxanes induces a silica-like layer (polyorganosilica or POSi) as thin as 10 nm, achieving superior H2/CO2 separation properties.10,11 Oxygen plasma generates ions and radicals, which cleave organic groups and form siloxane linkages (Si–O–Si), creating a tight silica network with strong molecular sieving ability. Importantly, polysiloxanes can be fabricated into thin-film composite (TFC) membranes with polymeric substrates, which have been commercially produced on a large scale. The integration of thin inorganic layers and polymeric substrates makes them attractive for practical applications.12 For instance, POSi membranes derived from 120 second plasma treatment of poly(dimethylsiloxane-co-methylhydroxidesiloxane) (poly(DMS-co-MHOS)) exhibited a mixed-gas H2 permeance of 370 GPU (1 GPU = 10−6 cm3(STP) cm−2 s−1 cmHg−1) and H2/CO2 selectivity of 68 at 150 °C. However, exposure to 7.0 mol% water vapor decreased the H2 permeance by 51% to 180 GPU and the H2/CO2 selectivity by 59% to 25, and upon shifting back to dry conditions, the separation performance did not recover, indicating irreversible chemical and structural changes.11 Specifically, the siloxane linkages underwent hydrolysis and produced silanol (Si–OH) groups, which condensed to rearrange the networks and decrease H2/CO2 separation properties.6,13–16
The hydrothermal stability of silica membranes can be improved by doping with metal ions to form stable metal–silica linkages17–19 or enhancing hydrophobicity,5,6,20–23 as shown in Table S1.† Particularly incorporating nonhydrolyzable organic groups (such as fluorine,5 ethyl,22,24 and phenyl25 groups) has emerged as a versatile approach due to the enormous choices of precursors and the flexibility of chemical functionalization. For example, tetraethyl orthosilicate (TEOS)-derived silica membranes exhibited a dramatic decrease of H2/CO2 selectivity from 1500 to 520 after exposure to water vapor at 16.5 kPa at 600 °C for 130 h,18 while the phenyl-containing membranes derived from dimethoxydiphenylsilane (DMDPS) exhibited stable H2 permeance at 2100 GPU and an H2/N2 selectivity of ∼1000 after exposure to water vapor at 3.4 kPa at 300 °C for 226 h.25 Nevertheless, these approaches were only demonstrated for silica membranes prepared using the sol–gel or CVD method. To the best of our knowledge, they have not been examined for plasma-derived POSi membranes with silica layers as thin as ∼10 nm.
Here, we demonstrate that incorporating phenyl groups into plasma-derived POSi membranes can enhance hydrothermal stability and retain their attractive molecular sieving ability for H2/CO2 separation, as shown in Fig. 1. First, copolymers of poly(methylphenylsiloxane-co-dimethylsiloxane-co-methylhydrogensiloxane) (poly(PMS-co-DMS-co-MHS)) were synthesized via a hydrosilylation reaction, and the unreacted hydrosilane (–SiH) groups in MHS were hydrolyzed to –SiOH by the water vapor or oxygen in the atmosphere, resulting in poly(PMS-co-DMS-co-MHOS). The phenyl content can be systematically varied by changing the PMS content between 0 and 80 wt%. The obtained polysiloxanes were thoroughly characterized for thermal transitions and gas sorption and permeation properties. Second, these polysiloxanes were fabricated into TFC membranes, and their morphology and gas permeance were determined. Third, the membranes were treated with oxygen plasma under optimized conditions identified in our prior study11 to render phenyl-containing POSi membranes with strong size-sieving ability. The effect of the PPMS loading on H2/CO2 separation properties was systematically investigated. Finally, the membranes with various amounts of phenyl groups were exposed to saturated water vapor at 100 °C for 24 h, and its effect on surface atomic compositions and separation properties was elucidated. Introducing phenyl groups enhances surface chemistry stability and increases the hydrothermal stability of the POSi membranes. This study sheds light on the hydrolysis mechanisms of thin organosilica layers and provides an effective route to improve their stability for applications in humid environments.
 | ||
| Fig. 1 Our synthesis approach to POSi membranes (POSixx) derived from poly(PMS-co-DMS-co-MHOS). (a) Synthesis of poly(PMS-co-DMS-co-MHS) by hydrosilylation reaction followed by hydrolysis and oxidation of the unreacted Si–H groups. (b) Preparation of POSi membranes, including fabrication of polysiloxane TFC membranes (PMxx) and oxygen plasma treatment. Photos of (c) a freestanding polysiloxane film and (d) a POSi membrane. | ||
Experimental
Materials
A commercial polymeric porous support (ultrafiltration membrane with pore sizes of 10–50 nm) was kindly provided by Membrane Technology and Research, Inc. (MTR, Newark, CA). Wacker Dehesive 944 (vinyl-terminated PDMS, vPDMS), cross-linker V24 (polymethylhydrosiloxane, PMHS), and catalyst OL (1 mass% platinum in PDMS) were procured from Wacker Chemical Corporation (Ann Arbor, MI). PMV-9925 (vinyl-terminated polyphenylmethylsiloxane, vPPMS, 2000–3000 g mol−1) was purchased from Gelest (Morrisville, PA). Toluene (99.85%) and dimethylformamide (DMF, 99.8%) were procured from Fisher Scientific (Waltham, MA). Gas cylinders of H2, CO2, N2, and CH4 with purity >99.9% were obtained from Airgas Inc. (Buffalo, NY).Preparation of polysiloxanes and TFC membranes
Polysiloxanes were synthesized from two monomers (vPPMS and vPDMS) and a crosslinker (PMHS) via a hydrosilylation reaction between vinyl and –SiH groups. Table S2† summarizes the compositions of the monomers and crosslinker. Briefly, a ∼5 mass% prepolymer solution was prepared by dissolving the monomers and crosslinker at a desired ratio in toluene, and then catalyst OL (60 mg) was added. The reaction was carried out at ≈23 °C for 10 min. The solution was poured into a Teflon Petri dish and slowly dried to obtain a freestanding film with a thickness of ∼200 μm (Fig. 1c). The films are denoted as Pxx, where xx is the vPPMS mass percentage in the monomers and cross-linker.Polysiloxane TFC membranes were prepared by coating 2 mass% of P0, P17, or P38 solution onto the porous support using a dip coater (MTI Corporation, CA).11 A 5 mass% solution was used to prepare the P60 membrane to avoid microdefect formation, leading to a selective layer thicker than other membranes. P60 membranes prepared from <5% solutions were defective because of the lower molecular weight of vPPMS than vPDMS and a higher tendency to pore penetration than P0, P17, or P38. After drying at 60 °C overnight, the membrane was exposed to oxygen plasma to prepare POSi membranes using Phantom III (Trion Technology, Inc., Clearwater, FL) at an oxygen pressure of 100 mTorr with a flow rate of 20 sccm for 120 s.11 The plasma was generated at an inductively coupled plasma (ICP) power of 200 W and reactive ion etching (RIE) of 20 W. Polysiloxane membranes and plasma-treated ones (Fig. 1d) are denoted as PMxx and POSixx, respectively. The membranes were kept under room conditions before use.
Characterization of polymers and membranes
Freestanding films of Pxx were thoroughly characterized for chemical and physical properties. Fourier transform infrared (FTIR) spectra were obtained using a Vertex 70 Burker spectrometer (Billerica, MA). Gel content was determined from the sample mass difference before and after immersion in toluene at 35 °C for 24 h. Polymer density was determined by Archimedes' principle using a Mettler Toledo XS 64 analytical balance with a density kit. Decane with a density of 0.73 g cm−3 was used as an auxiliary solvent.26 X-ray diffraction (XRD) patterns were obtained using a Rigaku Ultima IV X-ray diffractometer (Rigaku Analytical Devices, MA) with a CuKα source (1.54 Å−1). Glass transition temperature (Tg) was determined using a Differential Scanning Calorimeter (DSC, Q2000, TA instruments, New Castle, DE). Thermal stability was evaluated using a thermogravimetric meter (TGA, SDT Q600, TA Instruments, DE) with a temperature ramping from 100 to 1000 °C at 10 °C min−1 in N2.The pure-gas permeability of Pxx films was determined using a constant-volume and variable-pressure apparatus at 60 psig and 35 °C.10,27 Sorption isotherms of CO2 and C2H6 were determined using a pressure decay method with a dual-volume and dual-transducer apparatus at 35 °C.28 Gas solubility (SA, cm3(STP) cm−3 atm−1) at an equilibrium pressure of pA (atm) is calculated using eqn (1):
| SA = CA/pA | (1) |
To elucidate the effect of plasma treatment on surface chemistry, polysiloxane films were coated on Si wafers before the plasma treatment. X-ray photoelectron spectroscopy (XPS) was performed using a PHI5000 VersaProbe III scanning probe from Physical Electronics Inc. (Chanhassen, MN) with a spot size of 100 μm, and CasaXPS was used to derive the atomic concentration.
The surface and cross-section of the TFC membranes were imaged using a focused ion beam scanning electron microscope (FIB-SEM, Carl Zeiss Auriga CrossBeam, Germany). To determine the selective layer thickness, a sample was immersed in DMF at ≈23 °C for 24 h to dissolve the porous support, and the selective layer was collected using a Si wafer and then dried at 150 °C for 2 days before measurement using an F-20 (Filmetrics, San Diego, CA).
The pure-gas permeance of the membranes was determined using a constant-pressure and variable-volume apparatus with a feed pressure of 60 psig at 35 °C for PMxx and at 100 °C for POSixx.11 The hydrothermal stability of POSi membranes was investigated in three steps. First, pure-gas H2/CO2 separation properties were determined at 100 °C under dry feed conditions. Second, the membrane was taken out of the permeation cell and exposed to the air saturated by water vapor at 100 °C for 12 h using a custom-built apparatus, and then it was dried overnight at ≈23 °C. Finally, the sample was re-tested for H2/CO2 separation properties. The membranes after hydrothermal treatment are denoted as HT-POSixx.
Results and discussion
Characterization of polysiloxane freestanding films
Fig. 2a presents the FTIR spectra of Pxx freestanding films containing 0–60 mass% PPMS. All samples exhibit two peaks at 1086 and 1007 cm−1 corresponding to the siloxane backbone (Si–O–Si),29–32 but no Si–H characteristic peak at 2165 cm−1 (Fig. S1†). Increasing the PPMS content increases the intensity of the peaks at 695 and 730 cm−1 (aromatic C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C vibration) and 1430 cm−1 (Si–Ph vibration),30,31 and decreases the peak intensity for 907 cm−1 (Si–O stretching in Si–OH induced from the hydrolysis and oxidation of Si–H),29,32,33 consistent with the high gel content in the polymers (>90%, Fig. S2a†). The peak of 907 cm−1 almost disappears for P60, indicating the almost complete reaction of PMHS. On the other hand, the gel content decreases with increasing PPMS content. P80 shows a gel content of only 83%, and it fails to be made into defect-free TFC membranes.
C vibration) and 1430 cm−1 (Si–Ph vibration),30,31 and decreases the peak intensity for 907 cm−1 (Si–O stretching in Si–OH induced from the hydrolysis and oxidation of Si–H),29,32,33 consistent with the high gel content in the polymers (>90%, Fig. S2a†). The peak of 907 cm−1 almost disappears for P60, indicating the almost complete reaction of PMHS. On the other hand, the gel content decreases with increasing PPMS content. P80 shows a gel content of only 83%, and it fails to be made into defect-free TFC membranes.
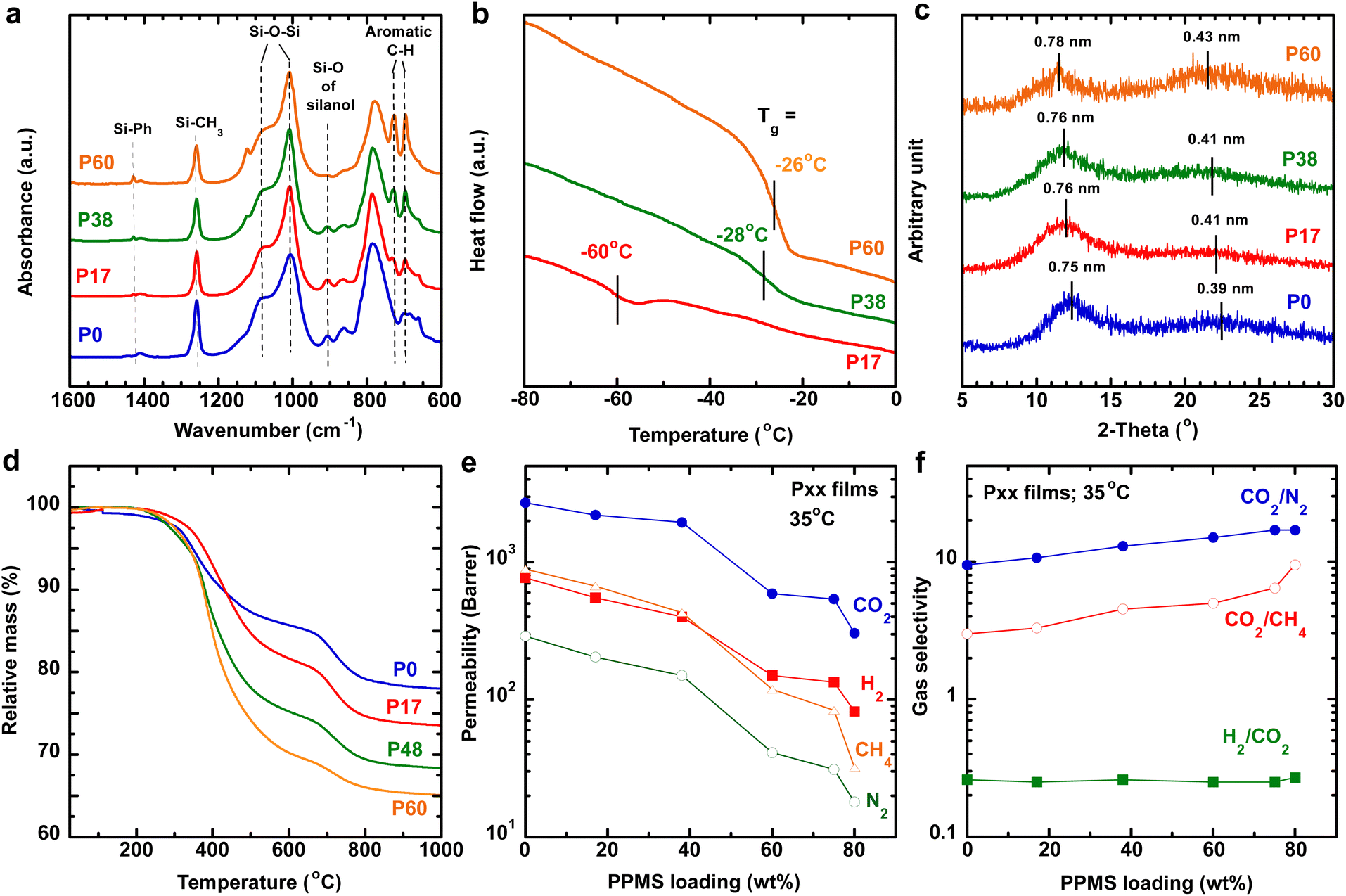 | ||
| Fig. 2 Characterization of Pxx freestanding films as a function of PPMS loadings. (a) FTIR spectra, (b) DSC thermograms, (c) XRD patterns, (d) TGA curves, (e) pure-gas permeability at 35 °C, and (f) CO2/gas selectivity at 35 °C. | ||
Fig. 2b and Table S2† compare the thermograms and Tg of Pxx films. P0 exhibits Tg below our instrument's detection limit (−90 °C). Adding 17 and 60 mass% PPMS increases Tg to −60 and −26 °C, respectively. Similar behavior has been reported for other phenyl-containing polysiloxanes.31
Fig. 2c presents the XRD patterns of the polysiloxanes. P0 exhibits characteristic peaks at 2θ of 12.4° and 22.4°, corresponding to the d-spacings of 0.75 and 0.39 nm, respectively. These two peaks are attributed to the folding of siloxane chains34 and short-range order reflecting the distance between Si atoms,35 respectively. P60 exhibits d-spacing values of 0.78 and 0.43 nm, higher than those of P0, indicating that incorporating bulky pendant phenyl groups loosens siloxane chain packing.31 Increasing the PPMS content also increases the density from 0.997 g cm−1 for P0 to 1.159 g cm−1 for P60 (Fig. S2b†). Additionally, polysiloxanes show thermal stability up to 250 °C, presenting their potential for use in H2/CO2 separation at 100–250 °C (Fig. 2d).
Fig. 2e and f exhibit the gas transport properties of Pxx films at 60 psig and 35 °C. Incorporating 60 mass% PPMS decreases the CO2 permeability (PA) from 2700 to 540 barrer (1 barrer = 10−10 cm3(STP) cm cm−2 s−1 cmHg−1) and increases the CO2/N2 selectivity from 9.5 to 15 and CO2/CH4 selectivity from 3.0 to 5.2 (Table S2†). On the other hand, the H2/CO2 selectivity of all polymers is roughly 0.25, as expected for rubbery polysiloxanes.10 Fig. S3† presents the gas sorption isotherms of Pxx films.
Increasing the PPMS content slightly decreases the CO2 solubility and drastically decreases the CO2 diffusivity because of the decreased polymer chain flexibility, as reflected by the increased Tg.
Chemical and structural characterization of TFC membranes
Fig. 3a–d present the cross-sectional SEM images of PMxx membranes, where the polysiloxane layer thickness varies between 200 and 400 nm, except for PM60 (950 nm), which was prepared using a more concentrated solution (5 mass%, instead of 2 mass%). The thickness values are consistent with those measured using the F20 (Table S3†),11 which has a larger scanning window than the SEM. Therefore, the thickness (l, nm) from the F20 measurement is used for further analysis. | ||
| Fig. 3 Cross-sectional SEM images for the membranes of (a) PM0, (b) PM17, (c) PM38, (d) PM60, (e) POSi0, (f) POSi17, (g) POSi38, and (h) POSi60. Comparison of Si 2p and C 1s peaks (i) between P0 and POSi0 and (j) between P38 and POSi38 by XPS. | ||
Oxygen plasma treatment was conducted using optimized conditions for balanced H2/CO2 separation properties based on our previous study.11 Interestingly, all POSi membranes show wrinkles after the plasma treatment (Fig. 3e–h) because of the stiffness mismatch between the rigid silica skin layer and soft bulk during the expansion and contraction of the composites.36,37 The plasma can heat the polysiloxane to as high as 230 °C;38 upon cooling, the soft polysiloxane bulk contracts, but the rigid silica skin layer does not, resulting in the wrinkles.36 However, no direct correlation between the phenyl content and wrinkle structures can be observed (Table S3†). Notably, the wrinkled silica layers are still defect-free, and the wrinkles increase the surface area by 4–15%, which could potentially improve the gas permeance,39 considering the ultrathin nature of the silica layer (∼10 nm).10
To better understand chemical changes, XPS analysis was performed on the polymers and POSi samples coated on Si wafers. Polysiloxanes exhibit an O/Si ratio of ≈1.18 (Table S4†), consistent with their theoretical values.11 Adding 60 mass% PPMS increases the C/Si ratio from 2.18 to 2.71 due to the replacement of –CH3 with phenyl groups. The oxygen plasma treatment decreases the C content and increases the O content because of the scissoring of Si–C bonds and the formation of the silica structure.10,11 For instance, plasma treatment decreases the C content from 52 to 25 mol% and increases the O content from 25 to 48 mol%. Increasing the PPMS content increases the C/Si molar ratio while retaining the O/Si molar ratio in the POSi samples. The O/Si ratio of ∼2.0 in the POSi samples is consistent with the silica structure (Si–(O)4).
Fig. 3i and j present the deconvolution of Si 2p and C 1s peaks for P0 and P38 and their corresponding POSi samples (i.e., POSi0 and POSi38). The Si 2p peak is deconvoluted into Si–(O)2 at 102.1 eV, Si–(O)3 at 102.8 eV, and Si–(O)4 at 103.4 eV, while the C 1s is deconvoluted into C–H/C–C at 284.6 eV and C–O at 286.0 eV.10,40 P0 and P38 exhibit two characteristic peaks for Si–(O)2 and Si–(O)3, corresponding to the Si–O–Si backbones and Si–OH groups, respectively.11,29 As expected, both polysiloxanes show only C–H and C–C without C–O in the C 1s region (Table S5†). By contrast, the plasma treatment leads to Si–(O)4 and C–O peaks, further confirming the oxidation and formation of silica structures.10,11
Gas transport properties of POSi membranes
Fig. 4a presents the pure-gas permeance (QA in GPU) of PMxx membranes as a function of penetrant kinetic diameter at 100 °C and 60 psig. Gas permeance follows an order of N2 < CH4 < H2 < CO2, similar to that for Pxx films (Fig. 2d). Increasing the PPMS content decreases the gas permeance partially because of the decreased permeability of the selective layer. All membranes show an H2/CO2 selectivity of 0.65–0.72, higher than those at 35 °C (Fig. S5 and Table S6†). Table S6† also presents the ratio of the estimated permeability (PA,est, given by PA,est = QA × l) to the measured permeability for the corresponding freestanding films. All membranes exhibit a permeability ratio close to 1 (suggesting their defect-free nature) except for PM60 because of the microdefects. | ||
| Fig. 4 Pure-gas transport properties of PM and POSi membranes at 100 °C. Comparison of gas permeance in (a) PM and (b) POSi membranes. (c) H2/gas selectivity of POSi membranes. (d) Schematic of a series-parallel flow model in POSi membranes. | ||
Table S7† shows that pure-gas permeance in POSi membranes is independent of feed pressures (20, 40, and 60 psig) at 100 °C. Fig. 4b and c present H2/gas separation properties at 60 psig as a function of penetrant kinetic diameter. Increasing the penetrant kinetic diameter from H2 (2.89 Å) to CO2 (3.3 Å) rapidly decreases the gas permeance, leading to a very high H2/CO2 selectivity. For instance, POSi0 exhibits an H2 permeance of 140 GPU with an H2/CO2 and H2/N2 selectivity of 57 and 490, respectively, much higher than those of the PMxx membranes, highlighting the effectiveness of oxygen plasma in creating silica structures for molecular separation. Adding PPMS increases the CO2, N2, and CH4 permeance in POSi membranes and slightly decreases the H2 permeance, reducing the H2/gas selectivity. For example, incorporating 60 mass% PPMS increases the CO2 permeance from 2.3 to 5.8 GPU and decreases the H2 permeance from 140 to 120 GPU, decreasing the H2/CO2 selectivity from 57 to 21 and H2/N2 selectivity from 490 to 90. Interestingly, the CH4 (3.8 Å) permeance is higher than the N2 (3.64 Å) permeance in all POSi membranes despite its larger kinetic diameter than N2.
We hypothesize that the higher permeance of CH4 than N2 can be ascribed to the microdefects in the silica layer with Knudsen diffusion (Fig. 4d).41 Within this framework, gas permeation is described using a series-parallel flow model, as presented in eqn (2):41
 | (2) |
 | (3) |
To simplify the approach, we assume that the dense silica portion can completely reject CH4, i.e., the amorphous silica layer has pores smaller than 3.8 Å. Therefore, CH4 permeance is completely derived from the microdefects through Knudsen diffusion, and its transport resistance in the PM membranes is negligible. Additionally, as POSi membranes achieve H2/CH4 selectivity (>100) much higher than their Knudsen selectivity (2.8), ε is expected to be much lower than 1. Consequently, eqn (2) and (3) can be combined and reduced to:
 | (4) |
Table S8† summarizes the estimated H2/CO2 separation properties of the dense organosilica phase in POSi membranes. Increasing the PPMS content increases CO2QA,dSi values, consistent with the QA values of POSi membranes, presumably because of the larger pores caused by the bulky phenyl groups. In contrast, H2QA,dSi appears to increase with increasing PPMS content, opposite to the trend observed for the QA values (Fig. 4b). For example, introducing 60 mass% PPMS decreases H2QA from 140 to 120 GPU but increases QA,dSi from 150 to 190 GPU. This can be attributed to the increasingly significant resistance to H2 transport of the PM bulk layer with increasing PPMS content because of the decreased permeability and increased thickness. Transport resistance of the polysiloxane layer in POSi60 is 38% for H2 but less than 2% for CO2 and CH4 (Table S8†). Increasing the PPMS content increases all gas permeance and decreases the H2/CO2 selectivity of the dense silica layer. For example, the estimated H2/CO2 selectivity of the dense silica decreases from 73 for POSi0 to 38 for POSi60. This can be attributed to the addition of bulky phenyl groups enlarging the silica nanopores, increasing the H2 and CO2 gas permeance but decreasing the size-sieving ability.42,43 These self-consistent results validate our assumption of the dense silica with pores smaller than 3.8, though they cannot be directly validated using the techniques available.
Hydrothermal stability of POSi membranes for H2/CO2 separation
To evaluate the hydrothermal stability, all POSi membranes were exposed to saturated water vapor at 100 °C for 12 h, a typical condition used for accelerated tests in the literature.19Fig. 5a presents the relative values of H2 permeance and H2/CO2 selectivity after the exposure, and the absolute values are recorded in Table S9.† POSi0 exhibits a 12% increase in H2 permeance with a 74% decrease in H2/CO2 selectivity, reflecting the complicated nature of the structure rearrangement of the silica networks. On the other hand, POSi38 exhibits a 32% reduction in H2 permeance but only a 54% reduction in H2/CO2 selectivity, and POSi60 shows only a 17% decrease in H2/CO2 selectivity, suggesting improved hydrothermal stability with the PPMS addition. The enhanced stability is also consistent for organically decorated silica networks.3,17,25,44 Notably, HT-POSi38 exhibits the highest H2/CO2 selectivity (19) compared to HT-POSi0 (16) and HT-POSi60 (11) (Table S9†), though the values would likely vary under different hydrothermal treatment conditions. | ||
| Fig. 5 Hydrothermal stability of POSi membranes after exposure to saturated water vapor at 100 °C for 12 h. (a) Relative H2 permeance and H2/CO2 selectivity. (b) C/Si molar ratio and (c) O/Si molar ratio. Si 2p peak deconvolution of (d) POSi0 and (e) POSi60. | ||
Surface XPS was performed to better understand the structural changes of POSi membranes after hydrothermal treatment (Fig. 5b–e, Tables S4 and S5†). Water vapor exposure decreases the O/Si molar ratio because of the condensation of silanol groups, forming water molecules and losing oxygen atoms.3,19,45,46 Intriguingly, it increases the C/Si molar ratio, presumably because the polysiloxane chains with higher C/Si molar ratios migrate from the bulk to the surface.47,48 Phenyl-containing POSi membranes show smaller changes in the atomic ratios than POSi0 (Fig. 5b and c). For example, hydrothermal treatment decreases the O/Si ratio from 1.97 to 1.72 and increases the C/Si ratio from 0.82 to 1.07 for POSi0, while it slightly decreases the O/Si ratio from 1.86 to 1.79 and increases the C/Si ratio from 1.21 to 1.27 for POSi38. The stabler composition in POSi38 than in POSi0 is consistent with the more stable H2/CO2 separation properties.
Fig. 5d, e and Table S5† present the Si 2p peak deconvolution of POSi0 and POSi60 before and after hydrothermal treatment. Hydrothermal treatment of POSi0 decreases the Si–(O)4 peak contribution from 73% to 60% and increases the Si–(O)3 peak contribution from 15% to 25%. The conversion toward Si–(O)3 indicates that the silica networks become loosened, consistent with the reduced molecular sieving ability. In comparison, both Si–(O)4 and Si–(O)3 peak contributions of POSi60 barely change after hydrothermal treatment, suggesting unchanged silica networks and validating the almost unchanged H2/CO2 separation properties. The lower degree of Si 2p redistribution in phenyl-POSi membranes can be attributed to the enhanced hydrophobicity protecting the “core” network structure and bulky phenyl groups making it difficult for the silica structure to reorganize.
Conclusion
Incorporating phenyl groups into polysiloxane precursors improves the hydrophobicity and hydrothermal stability of the resulting POSi membranes, achieving superior H2/CO2 separation properties. Polysiloxanes containing 0–60% PPMS were synthesized and fabricated into TFC membranes with <1 μm selective layers. Rapid oxygen plasma treatment (120 s) was used to generate composites comprising an ultrathin, wrinkled silica layer (∼10 nm) on top of the rubbery polymer network, which is thoroughly validated by the O/Si and C/Si molar ratios determined using XPS. POSi38 exhibits a balanced H2 permeance of 140 GPU and H2/CO2 selectivity of 41 at 100 °C, superior to those of polymeric materials. Importantly, after exposure to water vapor at 100 °C for 12 h, POSi38 and POSi60 lost H2/CO2 selectivity by 54% and 17%, respectively, lower than that of POSi0 lacking phenyl groups (74% reduction). The improved hydrothermal stability of POSi38 is further validated by stable values of the O/Si and C/Si molar ratios compared to POSi0.This study illustrates the mechanism of hydrothermal reactions in ultrathin silica layers derived from oxygen plasma treatment of polysiloxanes, facilitating the design of robust silica membranes for practical applications. Further optimization of POSi membranes includes increasing phenyl groups or other hydrophobic groups and evaluating performance with real gas streams containing various water contents. Notably, the feed gas is often dehydrated before entering membrane systems to avoid water condensation in the membrane modules, and therefore, there can be a balanced requirement of water removal and hydrothermal stability of the POSi membranes.
Author contributions
All authors contributed to the scientific discussion and manuscript preparation. H. L. and V. B. conceived the approach and conducted experimental designs. V. B., V. R. S., and E. H. fabricated and characterized materials. A. T., S. S. C., N. E., L. Z., and F. A. contributed to materials characterization. V. B. wrote the first draft of the manuscript, and all authors contributed to manuscript editing. H. L. supervised the project.Conflicts of interest
The authors declare no competing financial interest.Acknowledgements
We gratefully acknowledge the financial support from the U. S. National Science Foundation (2044623).References
- U. Anggarini, H. Nagasawa, M. Kanezashi and T. Tsuru, J. Membr. Sci., 2022, 642, 119962 CrossRef CAS.
- R. M. De Vos and H. Verweij, Science, 1998, 279, 1710–1711 CrossRef CAS PubMed.
- V. Bui, A. M. Tandel, V. R. Satti, E. Haddad and H. Lin, Adv. Membr., 2023, 3, 100064 CrossRef.
- A. Ali, R. Pothu, S. H. Siyal, S. Phulpoto, M. Sajjad and K. H. Thebo, Mater. Sci. Energy Technol., 2019, 2, 83–88 Search PubMed.
- P. H. T. Ngamou, J. P. Overbeek, R. Kreiter, H. M. van Veen, J. F. Vente, I. M. Wienk, P. F. Cuperus and M. Creatore, J. Mater. Chem. A, 2013, 1, 5567–5576 RSC.
- X. Yu, H. Nagasawa, M. Kanezashi and T. Tsuru, J. Mater. Chem. A, 2018, 6, 23378–23387 RSC.
- W.-W. Yan, K. Wakimoto, N. Moriyama, H. Nagasawa, M. Kanezashi and T. Tsuru, J. Membr. Sci., 2024, 702, 122798 CrossRef CAS.
- W.-W. Yan, K. Wakimoto, N. Moriyama, H. Nagasawa, M. Kanezashi and T. Tsuru, J. Membr. Sci., 2024, 122535 CrossRef CAS.
- H. Wang, S.-T. B. Lundin, K. Takanabe and S. T. Oyama, J. Mater. Chem. A, 2022, 10, 12869–12881 RSC.
- L. Zhu, L. Huang, S. R. Venna, A. K. Blevins, Y. Ding, D. P. Hopkinson, M. T. Swihart and H. Lin, ACS Nano, 2021, 15, 12119–12128 CrossRef CAS PubMed.
- V. Bui, V. R. Satti, E. Haddad, L. Hu, E. Deng, L. Zhu, W.-I. Lee, Y. Yin, K. Kisslinger, Y. Zhang, T. T. Bui, M. B. Rajapakse, L. Velarde, C.-Y. Nam and H. Lin, J. Membr. Sci., 2023, 688, 122099 CrossRef CAS.
- L. Hu, V. T. Bui, N. Esmaeili and H. Lin, CCST, 2024, 10, 100150 CAS.
- M. Kanezashi, T. Matsutani, H. Nagasawa and T. Tsuru, J. Membr. Sci., 2018, 549, 111–119 CrossRef CAS.
- G. Gong, H. Nagasawa, M. Kanezashi and T. Tsuru, ACS Appl. Mater. Interfaces, 2016, 8, 11060–11069 CrossRef CAS PubMed.
- M. Kanezashi, T. Shioda, T. Gunji and T. Tsuru, AIChE J., 2012, 58, 1733–1743 CrossRef CAS.
- N. W. Ockwig and T. M. Nenoff, Chem. Rev., 2007, 107, 4078–4110 CrossRef CAS PubMed.
- T. Tsuru, J. Chem. Eng. Jpn., 2018, 51, 713–725 CrossRef CAS.
- Y. Gu, P. Hacarlioglu and S. T. Oyama, J. Membr. Sci., 2008, 310, 28–37 CrossRef CAS.
- M. ten Hove, M. W. J. Luiten-Olieman, C. Huiskes, A. Nijmeijer and L. Winnubst, Sep. Purif. Technol., 2017, 189, 48–53 CrossRef CAS.
- A. P. Dral, K. Tempelman, E. J. Kappert, L. Winnubst, N. E. Benes and J. E. ten Elshof, J. Mater. Chem. A, 2017, 5, 1268–1281 RSC.
- H. Song, Y. Wei and H. Qi, J. Mater. Chem. A, 2017, 5, 24657–24666 RSC.
- N. Moriyama, A. Takeyama, T. Yamatoko, K.-i. Sawamura, K. Gonoi, H. Nagasawa, M. Kanezashi and T. Tsuru, Nat. Commun., 2023, 14, 7641 CrossRef CAS PubMed.
- S. Aoyama, H. Nagasawa, M. Kanezashi and T. Tsuru, ACS Nano, 2022, 16, 10302–10313 CrossRef CAS PubMed.
- H. L. Castricum, A. Sah, R. Kreiter, D. H. A. Blank, J. F. Vente and J. E. ten Elshof, J. Mater. Chem., 2008, 18, 2150–2158 RSC.
- Y. Ohta, K. Akamatsu, T. Sugawara, A. Nakao, A. Miyoshi and S.-I. Nakao, J. Membr. Sci., 2008, 315, 93–99 CrossRef CAS.
- L. Hu, V. T. Bui, S. Fan, W. Guo, S. Pal, Y. Ding and H. Lin, J. Mater. Chem. A, 2022, 10, 10872–10879 RSC.
- K. M. Rodriguez, W. Wu, T. Alebrahim, Y. Cao, B. D. Freeman, D. Harrigan, M. Jhalaria, A. Kratochvil, S. Kumar, W. Lee, Y. Lee, H. Lin, J. M. Richardson, Q. Song, B. Sundell, R. Thur, I. Vankelecom, A. Wang, L. Wang, C. Wiscount and Z. P. Smith, J. Membr. Sci., 2022, 659, 120746 CrossRef.
- W. Guo, T. N. Tran, H. Mondal, S. Schaefer, L. Huang and H. Lin, J. Membr. Sci., 2022, 648, 120352 CrossRef CAS.
- A. Esteves, J. Brokken-Zijp, J. Laven, H. Huinink, N. Reuvers, M. Van and G. De With, Polymer, 2009, 50, 3955–3966 CrossRef CAS.
- P. Kaali, D. Momcilovic, A. Markström, R. Aune, G. Czel and S. Karlsson, J. Appl. Polym. Sci., 2010, 115, 802–810 CrossRef CAS.
- Y. Pan, Y. Guo, J. Liu, H. Zhu, G. Chen, Q. Liu, G. Liu and W. Jin, Angew. Chem., Int. Ed., 2022, 61, e202111810 CrossRef CAS PubMed.
- N. Steinbrück and G. Kickelbick, J. Polym. Sci., Part B: Polym. Phys., 2019, 57, 1062–1073 CrossRef.
- K. Efimenko, W. E. Wallace and J. Genzer, J. Colloid Interface Sci., 2002, 254, 306–315 CrossRef CAS PubMed.
- F. Schilling, M. Gomez and A. Tonelli, Macromolecules, 1991, 24, 6552–6553 CrossRef CAS.
- J. Lancastre, N. Fernandes, F. Margaça, I. M. Salvado, L. Ferreira, A. Falcão and M. Casimiro, Radiat. Phys. Chem., 2012, 81, 1336–1340 CrossRef CAS.
- N. Bowden, S. Brittain, A. G. Evans, J. W. Hutchinson and G. M. Whitesides, Nature, 1998, 393, 146–149 CrossRef CAS.
- M. Nania, O. K. Matar and J. T. Cabral, Soft Matter, 2015, 11, 3067–3075 RSC.
- M. W. Kiehlbauch and D. B. Graves, J. Vac. Sci. Technol., 2003, 21, 660–670 CrossRef CAS.
- S. Karan, Z. Jiang and A. G. Livingston, Science, 2015, 348, 1347–1351 CrossRef CAS PubMed.
- H. Hillborg, J. Ankner, U. W. Gedde, G. Smith, H. Yasuda and K. Wikström, Polymer, 2000, 41, 6851–6863 CrossRef CAS.
- K. A. Lundy and I. Cabasso, Ind. Eng. Chem. Res., 1989, 28, 742–756 CrossRef CAS.
- M. Guo, J. Qian, R. Xu, X. Ren, J. Zhong and M. Kanezashi, J. Membr. Sci., 2022, 643, 120018 CrossRef CAS.
- G. Li, M. Kanezashi and T. Tsuru, J. Membr. Sci., 2011, 379, 287–295 CrossRef CAS.
- M. Kanezashi, K. Yada, T. Yoshioka and T. Tsuru, J. Am. Chem. Soc., 2009, 131, 414–415 CrossRef CAS PubMed.
- M. C. Duke, J. D. Da Costa, D. D. Do, P. G. Gray and G. Q. Lu, Adv. Funct. Mater., 2006, 16, 1215–1220 CrossRef CAS.
- C. J. Brinker, G. W. Scherer and E. Roth, J. Non-Cryst. Solids, 1985, 72, 345–368 CrossRef CAS.
- M. Morra, E. Occhiello, R. Marola, F. Garbassi, P. Humphrey and D. Johnson, J. Colloid Interface Sci., 1990, 137, 11–24 CrossRef CAS.
- J. Kim, M. K. Chaudhury and M. J. Owen, J. Colloid Interface Sci., 2000, 226, 231–236 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4ta02713b |
| This journal is © The Royal Society of Chemistry 2024 |