
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
A redox-reversible A/B-site co-doped BaFeO3 electrode for direct hydrocarbon solid oxide fuel cells†
Haixia
Li
ac,
Wanhua
Wang
ac,
Kai
Zhao
a,
Ka-Young
Park
 a,
Taehee
Lee
a,
Ramin Babazadeh
Dizaj
a,
Andreas
Heyden
b,
Dong
Ding
*c and
Fanglin
Chen
*a
a,
Taehee
Lee
a,
Ramin Babazadeh
Dizaj
a,
Andreas
Heyden
b,
Dong
Ding
*c and
Fanglin
Chen
*a
aDepartment of Mechanical Engineering, University of South Carolina, Columbia, SC 29208, USA. E-mail: chenfa@cec.sc.edu
bDepartment of Chemical Engineering, University of South Carolina, Columbia, SC 29201, USA
cEnergy & Environmental Science and Technology, Idaho National Laboratory, Idaho Falls, ID 83401, USA. E-mail: dong.ding@inl.gov
First published on 3rd May 2024
Abstract
Solid oxide fuel cells (SOFCs) can directly convert the chemical energy in fuel to electrical energy with fuel flexibility; however, the conventional nickel-based anodes face great challenges due to coking upon direct oxidation of hydrocarbon fuels and redox instability. Thus, developing new anode materials which can provide high coking resistance as well as redox stability is crucial. In this work, Ba0.6La0.4Fe0.8Mo0.1Ni0.1O3−δ (BLFMN) has been synthesized in air using a sol–gel combustion method, resulting in a dual phase consisting of a cubic BLFMN main phase and scheelite BaMoO4 (BMO4) secondary phase. By heat-treating the BLFMN dual phase in H2 at 800 °C for 5 h, a metallic nanoparticle-decorated BLFMN triple phase compound comprising cubic BLFMN, cubic BaMoO3 (BMO3) and in situ exsolved FeNi3 alloy was obtained. BLFMN was subsequently investigated as an electrode material for La0.8Sr0.2Ga0.83Mg0.17O3−δ (LSGM) electrolyte (300 μm) supported SOFCs. Symmetrical cells using BLFMN as electrodes with the cell configuration of BLFMN//LSGM//BLFMN showed excellent redox reversibility and a peak power density (PPD) of 1.32 W cm−2 at 850 °C when using H2 as fuel. Single cell with the cell configuration of BLFMN//LSGM//LSCF (La0.6Sr0.4Co0.2Fe0.8O3−δ) reached PPD of 1.61 and 0.41 W cm−2 at 850 °C when operating with H2 and CH4 fuel, respectively. Moreover, the single cell exhibit excellent stability (over 300 h) upon direct oxidation of hydrocarbon fuels of CH4 and C3H8. This study indicates that BLFMN is a promising redox reversible and coking resistant anode for SOFCs.
1. Introduction
Solid oxide fuel cells (SOFCs) have emerged as one of the most promising technologies for sustainable and efficient energy conversion. These high-temperature electrochemical devices offer the unique advantage of directly converting the chemical energy stored in fuels into electricity with remarkable efficiency. The versatility of SOFCs allows them to utilize a wide range of fuels, including natural gas, coal gas, and gasified carbonaceous solids, making them attractive candidates for a variety of applications from distributed power generation to mobile and stationary power systems.1,2 Despite their remarkable potential, the widespread commercialization of SOFCs has been hindered by the challenge of direct oxidation of carbon-containing fuels. For example, the typical extensively studied Ni-based cermet anode for SOFCs suffers from severe carbon deposition or coking when carbon-containing fuels are utilized, even though Ni has been considered as an effective catalyst for direct oxidation of hydrocarbon fuels with the advantages of high catalytic activity, high electrical conductivity, and low cost. These carbon deposits can obstruct the electrochemical reactions, impeding the efficient flow of ions and electrons, and ultimately decreasing the overall performance and durability of the SOFC.3,4Addressing the coking issue of electrocatalysts is crucial for advancing SOFC technology and unlocking its full potential. To achieve this, extensive research endeavors are underway with the objective of identifying novel anode materials possessing not only exceptional electrocatalytic activity but also superior resistance to coking. In recent years, perovskite-based oxide anodes, exemplified by chromite (La0.75Sr0.25Cr0.5Mn0.5O3−δ),5 titanate (La0.2Sr0.8TiO3),6 and double perovskite oxides such as Sr2Fe1.5Mo0.5O6−δ (SFM),7–9 have garnered substantial attention from researchers. This heightened interest can be attributed to their commendable mixed ionic–electronic conductivity (MIEC), redox stability, excellent sulfur tolerance, and resistance to coking.10 However, these alternative anodes still show insufficient catalytic activity for fuel oxidation when compared with the state-of-the-art Ni based anode, resulting in significantly lower cell performance.11
Therefore, further advancements and refinements are imperative to enhance the catalytic efficacy of perovskite-based anodes and shorten the cell performance gap of fuel oxidation in comparison with their Ni-based counterparts. Numerous strategies have been proposed to optimize perovskite-based anodes with the aim of enhancing the performance of SOFCs. One such approach involves the fabrication of composite anodes by incorporating perovskite with oxygen ion conductive oxides such as Ce0.8Sm0.2O2−δ (SDC) or Ce0.9Gd0.1O2 (GDC), a technique designed to bolster the catalytic activity of the anode by increasing the oxygen ion conductivity.12 Additionally, the infiltration of active catalysts onto the surface of the perovskite backbone has proven to be an effective method for enhancing catalytic activity towards fuel oxidation, thereby improving overall cell performance.13,14 Furthermore, A-site or B-site doping has been demonstrated as an effective means to tune the properties of perovskite materials, including thermal expansion coefficient, electrical conductivity, and structural stability. Lee et al., for instance, have simultaneously doped Sr to the A site and Ge to the B site in PrBaFe2O5+δ, resulting in the composition of PrBa0.5Sr0.5Fe1.9Ge0.1O5+δ. This modification yielded a notable enhancement in cell performance, as evidenced by a critical improvement in the maximum power density (MPD, 838.4 mW cm−2 at 800 °C) relative to the undoped PrBaFe2O5+δ (497.6 mW cm−2).15 Another method of novel SOFC anode exploration involves the addition of a transition metal to the B site, which can in situ exsolve from the perovskite structure as nanosized metal particles functioning as catalysts.16 For example, Ni/Co doping into the B site of a ferrite perovskite, leading to the exsolution of NiFe/CoFe alloy nanoparticles, has demonstrated catalytic efficacy in improving cell performance and coking resistance in SOFCs.17,18
Apart from the above extensively explored perovskite anodes, the class of BaFeO3-based perovskites has garnered considerable attention among researchers due to their noteworthy attributes, including low cost, high oxygen-ionic conductivity, and excellent catalytic activity for the oxygen evolution reaction.19,20 However, obtaining a pure phase BaFeO3 is challenging owing to the ion radius mismatch between Ba2+ and Fe3+. This challenge has been effectively addressed through A-site or B-site doping, or A/B site co-doping, resulting not only in the stabilization of a single cubic structure but also in the optimization of the properties of the doped BaFeO3 to meet the requirements to serve as an anode of SOFCs.21,22 For example, judiciously doping different amounts of La into the A site of BaFeO3 led to the attainment of a cubic perovskite structure when 10–20% La was introduced to the A site, while 40% La doping yielded the composition with the highest electrical conductivity in air within the temperature range of 100–800 °C. Remarkably, a certain amount of La doping was proven to be instrumental in reducing the area-specific resistance (ASR) of SOFCs using an La-doped BaFeO3 cathode, demonstrating the tunability of the material properties of BaFeO3 through doping.20
However, when functioning as an anode, structural and chemical stability becomes an essential property, particularly considering the reducing atmosphere that anode materials typically encounter. Doping with high-valence elements is regarded as an effective approach to stabilize the structure of perovskites.23–25 For instance, Li et al. proposed a co-doped composition, Ba0.95La0.05Fe0.9Nb0.1O3−δ, for use as the electrodes of symmetric SOFCs, exhibiting remarkable structural stability against wet H2-reduction, which is attributed to the considerably high valence state of Nb.24 Furthermore, the redox couple Mo6+/Mo5+ is commonly employed as a dopant to achieve structural stability, owing to its variable valence in different atmospheres. Thus, Mo was introduced into the B site of LaFeO3 to enhance its structural stability. Using La0.5Ba0.5Fe0.95Mo0.05O3−δ as electrodes, a symmetrical cell supported by a 300 μm LSGM electrolyte achieved a peak power density of 0.96 W cm−2 at 800 °C, exhibiting 25 hour short-term stability and 20 hour redox stability.25 Moreover, in situ exsolution has been demonstrated to be an effective strategy to increase the material's catalytic activity, electrical conductivity and coking resistance.17 Thus, a B-site Mo/Ni co-doped composition, (PrBa)0.95Fe1.9−xNixMo0.1O6−δ (x = 0, 0.1, 0.2, 0.3, 0.4), has been explored. Ni-doping has been found to be beneficial in increasing the catalytic activity of the anode material. Notably, when x = 0.3, the cell exhibited a very small polarization resistance of 0.028 Ω cm2 at 750 °C and maintained stability for 50 hours in wet propane, demonstrating coking resistance with FeNi alloy nanoparticles as a catalyst.26 In addition, an A-site-deficient (Ba0.9La0.1)0.95Co0.7Fe0.2Nb0.1O3−δ (BLCFN) perovskite oxide with in situ exsolved Co–Fe nanoparticles has been developed and the cells with BLCFN as an anode exhibit improved electrocatalytic activity and coking resistance when using CH4 as fuel, resulting in a stable voltage output over 200 h with an applied constant current of 0.2 A cm−2.27
In this work, we introduce a novel composition derived from BaFeO3, featuring A-site La doping and B-site Mo/Ni co-doping. Specifically, the synthesized electrode material, Ba0.6La0.4Fe0.8Mo0.1Ni0.1O3, underwent a systematic evaluation on an LSGM electrolyte supported SOFC, encompassing redox stability, catalytic activity, electrochemical performance, and coking resistance. Our comprehensive investigation of the Ba0.6La0.4Fe0.8Mo0.1Ni0.1O3 material in this study reveals not only remarkable redox reversibility but also excellent electrocatalytic activity towards a variety of fuels and outstanding resistance to coking with carbon-containing fuels. This work highlights the potential of BLFMN as a promising anode candidate of SOFCs.
2. Experimental section
2.1 Materials synthesis
The Ba0.6La0.4Fe0.8Mo0.1Ni0.1O3−δ (BLFMN) powder was synthesized by a sol–gel method. A stoichiometric quantity of the precursor chemicals of Ba(NO3)2, La(NO3)3·6H2O, Fe(NO3)3·9H2O, (NH4)6Mo7O24·4H2O and Ni(NO3)2·6H2O was dissolved in distilled water. Subsequently, glycine and citric acid were added into the solution as chelating agents, with a molar ratio of metal cations to citric acid to glycine of 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1.5:2. The obtained transparent solution was then heated at 80 °C with continuous agitation until gelation occurred. The resulting gel was further heated on a hot plate, reaching a temperature of 300 to 400 °C until self-ignition occurred. The precursor powder was pulverized, and subsequently calcined at 1100 °C for 5 hours in air, yielding the BLFMN powder. A similar procedure was employed for the synthesis of BMO4 powder. Additionally, the LSCF air electrode powder was purchased from Fuel Cell Materials.
:1.5:2. The obtained transparent solution was then heated at 80 °C with continuous agitation until gelation occurred. The resulting gel was further heated on a hot plate, reaching a temperature of 300 to 400 °C until self-ignition occurred. The precursor powder was pulverized, and subsequently calcined at 1100 °C for 5 hours in air, yielding the BLFMN powder. A similar procedure was employed for the synthesis of BMO4 powder. Additionally, the LSCF air electrode powder was purchased from Fuel Cell Materials.
2.2 Characterization
The crystalline phases of the synthesized BLFMN and BMO4 powders, as well as those subjected to various heat-treatments in different atmospheres, were characterized by powder X-ray diffraction (XRD) analysis utilizing a Rigaku MiniFlex II instrument. CuKα radiation with a wavelength (λ) of 0.15415 nm was employed in the XRD measurement. The morphology of the BLFMN powder and the microstructure of electrolyte-supported single cells were characterized using a field emission scanning electron microscope (FE-SEM, Zeiss Ultra Plus FESEM) together with a high-angle annular dark-field transmission electron microscope (HAADF-TEM, TEI Tecnai F30) equipped with an energy-dispersive X-ray (EDX) analyser. Furthermore, thermogravimetric analysis (TGA) was carried out using a thermal analyzer (STA 449 F3, NETZSCH, Germany) to assess the weight variations of the BLFMN and BMO4 powders under distinct gas atmospheres. The temperature range extended from 30 to 900 °C, with a heating rate of 5 °C min−1. Air and 4% H2/N2 were employed for redox reversibility of BLFMN powder investigation with a gas flow rate of 50 mL min−1. Hydrogen temperature-programmed reduction (H2-TPR) (Auto Chem II 2920, Micromeritics Instrument Corporation, USA) was performed on BLFMN and BMO4 powders by heating the powder (100 mg) in 5% H2–N2 (50 mL min−1) at 5 °C min−1 from 50 to 900 °C after drying the powder in Ar at 200 °C for 1 h. H2 consumption results were recorded using a thermal conductivity detector (TCD) for the subsequent analysis. BLFMN bar samples with dimensions of approximately 34 × 6.9 × 0.7 mm3 were fabricated by uniaxially pressing the corresponding powders mixed with 4% polyvinyl butyral (PVB) under 7 MPa pressure for 1 min. The resulting bar samples were then sintered in air at 1350 °C for 5 hours to obtain dense samples, and the electrical conductivity of the dense samples was then determined using the DC four-probe method at temperatures ranging from 300 to 850 °C.2.3 Cell fabrication and electrochemical performance testing
Dense La0.8Sr0.2Ga0.83Mg0.17O3−δ (LSGM) electrolyte with a thickness of approximately 300 μm was prepared via the same procedure as that in previous work.28,29 To prepare the electrode ink, BLFMN or LSCF powder was mixed with a V006 binder using an agate mortar and pestle in a weight ratio of 1:1.5. The resulting anode or cathode ink was then brushed to both sides of the LSGM pellets, with an active area of 0.33 cm2 to fabricate single cells with the cell configuration of BLFMN/LSGM/LSCF. These single cells were subjected to calcination at 1000 °C for 2 hours in an air environment to achieve a good bonding between the electrode and the electrolyte. Symmetrical cells were prepared in the same way but the same BLFMN ink was applied on both sides of the LSGM electrolyte pellets, resulting in the cell configuration of BLFMN//LSGM//BLFMN. For the polarization resistance (Rp) evaluation, silver mesh affixed with gold paste was utilized as a current collector on the electrode surface of symmetrical cells with an active electrode area of 0.33 cm2.
For cell electrochemical performance testing, gold paste was applied to the top of both the electrodes as current collectors. Subsequently, the dried gold paste was calcined at 800 °C for 1 h to eliminate the organic substances. Lastly, the assembled single cells were hermetically sealed onto an alumina tube using a ceramic sealant (Ceramabond 552, Aremco, USA). The current density–voltage–power density (I–V–P) curves of the SOFCs were obtained within the temperature range of 650–850 °C, utilizing a Versa STAT 3-400 electrochemical workstation. In addition, electrochemical impedance spectra (EIS) were acquired at the open circuit voltage (OCV) for both single and symmetrical cells with a frequency range of 106 to 0.01 Hz and an AC amplitude of 10 mV.
3. Results and discussion
3.1 Crystal structure
Fig. 1 depicts the XRD patterns of both pristine and heat-treated BLFMN powders. The XRD pattern of pristine BLFMN powder (Fig. 1(a)) can be indexed to a cubic phase (PDF#97-018-5273, Ba0.5La0.5FeO3 (BLF)) along with a scheelite BaMoO4 (BMO4) (PDF#97-005-6109) second phase. This observation aligns with previous reports indicating that the BMO4 impurity phase forms when the Mo doping level at the Fe site exceeds the stoichiometry of 0.1.25 Upon heat-treating the pristine BLFMN powder in H2 at 800 °C for 5 h (Fig. 1(b)), a triple phase is observed, comprising the BLF main cubic phase, BaMoO3 (BMO3) (PDF#97-019-2726), and FeNi3 alloy (PDF#97-018-8242). Remarkably, upon re-calcination of the reduced powder in air at 800 °C for 5 hours (Fig. 1(c)), the triple phase of the reduced powder reverts to the pristine dual phase. This exceptional redox reversibility of BLFMN can be attributed to the stabilized BLF main cubic structure through high-valence Mo doping and the fully redox reversible nature of the BMO4 secondary phase. As illustrated in Fig. S1,† the prepared pristine BMO4 powder exhibits a tetragonal structure, well indexed to PDF#97-025-0487 (Fig. S1(a)†). After reducing the powder in H2 for 5 hours, the BMO4 powder with a tetragonal phase transform into the cubic phase BMO3 (PDF#97-019-2726) (Fig. S1(b)†). Notably, BMO3 can be easily reverted to BMO4 by heat treatment in air at 800 °C for 5 hours (Fig. S1(c)†). These distinctive behaviors underscore the dynamic redox characteristics of the BLFMN material, providing insights into the reversible structural transformations induced by a redox environment. In addition, BLFMN also shows good chemical compatibility with LSGM electrolyte upon calcining the well mixed BLFMN and LSGM powder with a weight ratio of 1:1, as displayed in Fig. S2.†
 | ||
| Fig. 1 X-ray diffraction patterns of (a) the as-prepared pristine BLFMN, (b) BLFMN reduced in H2 at 800 °C for 5 h, and (c) reduced BLFMN being re-oxidized at 800 °C for 5 h in air. | ||
3.2 SEM analysis
SEM was employed to characterize the surface morphology and single-cell microstructure. As described in the XRD analysis, BLFMN samples underwent phase transformation upon heat treatment in H2. Consequently, the morphologies of the pristine and reduced BLFMN have been observed, as shown in Fig. 2(a and b), respectively, revealing a porous microstructure with interconnected grains. After reduction in H2, spherical nanosized particles are uniformly dispersed on the smooth surface across the reduced BLFMN samples. The high-angle annular dark-field scanning transmission electron microscopy (HAADF-STEM) image and the corresponding EDX elemental mapping are shown in Fig. 2(c). As can be seen, there are nanoparticles with size of around 40 nm and Ba, La, Ni, Fe, Mo and O elements are uniformly distributed in the BLFMN substrate, while Ni and Fe are rich in the exsolved nanoparticles. Moreover, the HAADF-STEM image and the element distribution along a line scan of a typical BLFMN surface with exsolved nanoparticles are shown in Fig. 2(d). Both the EDX mapping and the line scanning confirm that the nanoparticles are composed of Fe and Ni, which is consistent with the XRD analysis above. | ||
| Fig. 2 Surface morphology of the (a) pristine BLFMN powder and (b) BLFMN powder treated in H2 at 800 °C for 5 h, (c) HAADF-STEM image and the corresponding EDX element mapping images of an NiFe alloy nanoparticle, and (d) HAADF-STEM image and corresponding EDX line scan of the nanoparticles. | ||
Fig. S3(a)† presents the cross-section of an electrolyte-supported single cell, featuring LSCF as the cathode and BLFMN as the anode. The anode measures approximately 22 μm while the cathode is about 14 μm, and the porous electrodes exhibit good adherence to the dense LSGM electrolyte (300 μm). Fig. S3(b)† is a magnified view of the LSCF cathode/LSGM electrolyte interface with a gold current collector layer on top of the LSCF cathode.
3.3 Thermogravimetric and temperature programmed reduction analysis
Hydrogen temperature-programmed reduction (H2-TPR) measurements and thermogravimetric analysis (TGA) were conducted to assess the thermal stability of BLFMN and BMO4 in different atmospheres. The TPR diagram in Fig. 3(a) for BLFMN reveals two distinct peaks at 275 °C and 357 °C, corresponding to the reduction of Fe4+ to Fe3+, and Mo6+ to Mo5+, respectively.25 As the temperature increases, the TPR curve ascends from around 650 °C, signifying the further reduction of Fe3+ to Fe2+ or Fe0, Ni2+ to Ni0, and Mo5+ to Mo4+.26 In contrast, the TPR signal of BMO4 emerges at around 650 °C, which is primarily attributable to the reduction of the Mo element.30 | ||
| Fig. 3 (a) TPR plot for BLFMN and BMO4 powders from 50 to 900 °C, (b) TGA in air and 4% H2/N2 for BLFMN and BMO4 powders from 50 to 900 °C, and (c) weight change of BLFMN and BMO4 powders by switching the gas between air and 4% H2/N2; N2 was purged to the chamber for 20 min to swap the remaining air or 4% H2/N2 for each gas switching. | ||
The TGA results, illustrated in Fig. 3(b), depict the weight losses of the samples upon heating to 900 °C in either air or a 4% H2–N2 atmosphere. For BLFMN, a gradual weight loss is observed with an increase in temperature up to 900 °C, amounting to a total weight loss of 0.85%. In contrast, BMO4 exhibits no observable weight loss when exposed to an air atmosphere, indicating its exceptional thermal stability. On the other hand, in a 4% H2–N2 atmosphere, BLFMN displays two distinct stages of weight loss. The first stage, occurring at 200–350 °C, is attributed to the loss of surface oxygen or adsorbed moisture, while the second stage, observed at 350–900 °C, corresponds to the large amount of lattice oxygen loss with increasing temperature in an H2-contained atmosphere. These findings agree well with the above TPR analysis and the observations of the other reported perovskite materials.22,31 However, BMO4 exhibits much less weight loss (0.18%) than BLFMN (2.73%), demonstrating its good thermal stability even under a reducing atmosphere at elevated temperatures. To further validate the redox reversibility characteristic observed by the XRD analysis, TGA was employed with an atmospheric switch during testing to monitor weight changes in different environments.32 The sample was initially ramped up to 900 °C in air. After the temperature reached 900 °C and held for 30 min, the weight of the sample was recorded for 2 h to monitor the stabilization of sample weight in an air atmosphere. As shown in Fig. 3(c), the weight loss of BMO4 and BLFMN was stabilized at 99.85% and 98.85%, respectively. Subsequently, the gas in the TGA testing chamber was switched from air to N2 to flush the chamber system for 20 min, and then switched to 4% H2/N2, and the sample was exposed to this H2 containing atmosphere for 1.5 hours, resulting in a decrease in weight due to the loss of lattice oxygen in a lower oxygen partial pressure environment. Upon reverting the chamber gas back to air, the weight of the sample returned to its initial value. Specifically, after reabsorbing oxygen from air, the weights of both BMO4 and BLFMN were restored to 99.83% and 98.84%, respectively. This sequence of weight changes confirms the excellent redox reversibility of both BLFMN and BMO4 materials.
3.4 Electrical conductivity
The electrical conductivity of the anode plays a pivotal role in facilitating efficient electron transport and current collection for practical SOFC applications. Fig. 4 depicts the temperature-dependent electrical conductivity of BLFMN within the temperature range of 300–850 °C. The conductivity trend demonstrates variation with temperature under different atmospheres. In an air environment, the conductivity initially increases with rising temperature, reaching a peak at 500 °C (208 S cm−1). This behavior indicates a semi-conductive nature, elucidated by a thermal-activated small polaron hopping mechanism illustrated in eqn (1).20 Subsequently, the conductivity decreases as the temperature continues to rise, which is attributed to the loss of lattice oxygen and the reduction in charge carriers (electron holes) at elevated temperatures, indicated by the reverse reaction of eqn (1).27w | (1) |
 represents an oxygen vacancy,
represents an oxygen vacancy,  a lattice oxygen and h˙ an electron hole.
a lattice oxygen and h˙ an electron hole.
 | ||
| Fig. 4 Conductivity variation of BLFMN with temperature in air and 5% H2/N2. | ||
Upon switching the atmosphere to 5% H2/N2, the sample underwent stabilization for 12 hours, resulting in a conductivity reduction to 0.8 S cm−1 at 850 °C. This decline in conductivity within a hydrogen-containing atmosphere can be attributed to the loss of lattice oxygen, owing to the reduction of Fe, Mo, and Ni cations, as described by the above TPR and TGA analyses in Fig. 3(a and b). Consequently, the decrease in electron holes as charge carriers leads to a significant reduction in electrical conductivity.26,31
3.5 Electrocatalytic activity
Electrocatalytic activity, typically reflected by polarization resistance (Rp), is an important factor that dictates the electrochemical performance of SOFCs. Therefore, the Rp values of the BLFMN electrode in an air or H2 atmosphere are determined from electrochemical impedance spectra (EIS) on a symmetrical cell with the cell configuration of BLFMN//LSGM//BLFMN. With the high-frequency induction tail and the ohmic resistance subtracted, the distance between the two points of the impedance intersecting the real axis of the Nyquist plot is defined as Rp to evaluate the electrocatalytic activity. Shown in Fig. 5(a and b) are the EIS in the testing temperature range of 650–800 °C in air and an H2 atmosphere, respectively. The Rp values of the BLFMN anode decrease rapidly with an increase in temperature from 650 to 800 °C whether in air or hydrogen, indicating that the increase of the working temperature can effectively accelerate the electrode reaction process and lower the electrode polarization resistance.41 The Rp values were determined to be 0.47, 0.19, 0.08, and 0.04 Ω cm2 in air while being 0.99, 0.50, 0.23, and 0.13 Ω cm2 in hydrogen at 650, 700, 750 and 800 °C, respectively. Comparing the Rp values obtained in this study with those reported for electrode materials tested under similar conditions, as listed in Table 1, the lower Rp values observed both in air and in H2 in this work indicate the strong electrocatalytic activity of BLFMN for the oxygen reduction reaction (ORR) and hydrogen oxidation reaction (HOR). | ||
| Fig. 5 Electrochemical impedance spectra of BLFMN symmetric cells measured in air (a) and H2 (b) at 650, 700, 750, and 800 °C, respectively. Resulting DRT plots of the BLFMN symmetric cell under air (c) and H2 (d) atmospheres at 650, 700, 750, and 800 °C, respectively. | ||
| Electrode | R p in air (Ω cm2) | R p in H2 (Ω cm2) | Ref. |
|---|---|---|---|
| Sr2Co0.4Fe1.2Mo0.4O6−δ | 0.10 | 0.14 | 18 |
| La0.5Ba0.5Fe0.95Mo0.05O3−δ | 0.07 | 0.12 | 25 |
| La0.1Sr1.9Fe1.4Ni0.1Mo0.5O6−δ | 0.06 | 0.34 | 28 |
| Sr2Fe1.4Ni0.1Mo0.5O6−δ | 0.09 | 0.52 | 28 |
| Ca0.25Sr1.75Fe1.5Mo0.5O3−δ | 0.09 | 0.20 | 33 |
| Sr2Fe1.5Mo0.5O6−δ | 0.24 | 0.27 | 34 |
| La0.5Sr0.5Fe0.9W0.1O3−δ | 0.08 | 0.16 | 35 |
| Pr0.4Sr0.6Fe0.875Mo0.125O3−δ | 1.02 | 1.60 | 36 |
| La0.5Sr0.5Fe0.9Nb0.1O3−δ | 0.06 | 0.24 | 37 |
| Sr2Ti0.8Co0.2FeO6 | 0.12 | 0.38 | 38 |
| La0.8Sr1.2Fe0.9Co0.1O4−δ | 0.29 | 1.14 | 39 |
| Pr0.5Sr0.5Cr0.1Fe0.9−xNixO3−δ | 0.09 | 1.28 | 40 |
| BLFMN | 0.04 | 0.13 | This work |
At the same time, the Arrhenius plot of ASR as a function of the operating temperature is presented in Fig. S4,† and is linearly fitted to calculate the activation energy of the electrode reaction in air or in a hydrogen atmosphere. The values of Ea for the electrode reaction process at the temperature range of 650–800° are 1.40 and 1.17 eV in air and in hydrogen, respectively, which are close to those of some reported electrode materials, such as Ba0.95La0.05Fe0.9Nb0.1O3−δ (1.09 eV in H2),24 (Ba0.9La0.1)0.95Co0.7Fe0.2Nb0.1O3−δ (1.28 eV in 5%H2/Ar),27 Pr0.4Bi0.1Ba0.5MnO3−δ (1.12 eV in H2),42 (Sr0.3La0.7)0.8(Fe0.7Ti0.3)0.9Ni0.1O3−δ (1.31 eV in air),43 and (PrBa)0.95(Fe0.9Mo0.1)2O5+δ (1.31 eV in air),44 and lower Ea values in H2 than in air represent a smaller chemical barrier for fuel oxidation reaction at the electrode.43
The distribution of relaxation times (DRT) method has been proven effective in understanding the electrochemical reaction processes in SOFCs.45 To elucidate the kinetics of the ORR and HOR on the BLFMN electrode, the EIS plots obtained in an air or H2 atmosphere were deconstructed using the DRT method with a Gaussian function.46 By analysing different time constants directly from the EIS data, distinct processes can be highlighted through specific peaks in the deconvoluted DRT curves. The most prominent peak in the plot signifies the rate limiting step of the electrode reaction.
Fig. 5(c and d) present the results of the DRT analysis derived from the EIS data of Fig. 5(a and b), respectively. In the air atmosphere (Fig. 5(c)), the progression from low to high frequency involves oxygen diffusion in the cathode (∼0.1 Hz), surface oxygen adsorption and dissociation (∼100 to 200 Hz), and the charge transfer process and O2− transfer across the electrode/electrolyte interface (∼1000 to 2000 Hz).35,37 The DRT plots in the air atmosphere at different temperatures exhibit three distinct peaks marked as P1, P2, and P3, indicating the electrode reaction can be delineated into three sub-steps. The primary peak of DRT plots is temperature-sensitive, suggesting that the dominant rate limiting step of the cathodic reaction varies with temperature. A substantial shift from 30 Hz to ∼1033 Hz is observed as the temperature rises from 650 °C to 800 °C, signifying that at lower temperatures, the limiting step is surface oxygen adsorption and dissociation processes, while at higher temperatures, the charge transfer and ionic transport process predominantly take effect. In the H2 atmosphere (Fig. 5(d)), four characteristic peaks represented as P1, P2, P3, and P4 are observed in the frequency range from 100 to 105 Hz, indicating that the electrode reactions may be governed by four sub-steps. Unlike the DRT curve in the air atmosphere, the main peak of DRT data in the anodic atmosphere exhibits a smaller temperature dependence, suggesting that the limiting step for the HOR remains consistent at different temperatures. However, with increasing temperature, the intensity of P2, P3, and P4 decreases or even disappears (P3, P4), which is likely associated with the electro-oxidation of H2, indicating an increase in the catalytic activity of the electrode for H2 oxidation at elevated temperatures.37,40
3.6 Cell electrochemical performance using H2 as fuel
The electrochemical performance of single cells with the cell configuration of BLFMN//LSGM//LSCF was assessed by supplying H2 to the anode while exposing the cathode to the ambient air. As illustrated in the XRD, SEM and TEM analysis, after being reduced in an H2 atmosphere, the exsolved FeNi3 alloy nanoparticles from the parent cubic BLFMN phase were expected to possess high catalytic activity toward fuel oxidation reactions.7 Moreover, BMO3, derived from reduction-induced phase transformation from a scheelite-type BMO4 oxide, was reported to be a good electrocatalyst for hydrogen evolution because the BMO3 perovskite made of interconnected MoO6 octahedra is found to be more hydrogen evolution reaction active than the BMO4 consisting of isolated MoO4 tetrahedra, due to the stabilized Mo tetravalent state, the increased surface oxygen vacancy concentration, and the several orders improved electrical conductivity of BMO3.47–49 In addition, the presence of Fe2+/Fe3+/Fe4+ and Mo5+/Mo6+ redox couples in reduced BLFMN was supposed to induce electrical properties while losing lattice oxygen, facilitating catalytic oxidation of fuel and electron transport.50 Therefore, BLFMN is expected to be an excellent anode material with promising single cell (SC) performance. As shown in Fig. 6(a), the maximum cell power density (Pmax) was 1.61 W cm−2 at 850 °C. Correspondingly, the Rp values (Fig. 6(c)) of single cells were determined to be 0.16 Ω cm2 at 850 °C. The observed trend reveals an increase in Pmax and a decrease in Rp with a rise in the cell operating temperature, which is primarily attributed to the enhanced catalytic activity for H2 oxidation, as discussed in the DRT analysis. | ||
| Fig. 6 (a and c) I–V–P and EIS curves for the BLFMN//LSGM//LSCF single cell (SC) operating from 650 to 850 °C. (b and d) I–V–P and EIS curves for the BLFMN//LSGM//BLFMN symmetric cell (SMC) operating from 650 to 850 °C. (e) Redox cycling stability of the BLFMN electrode with cell configuration of BLFMN//LSGM//BLFMN at 800 °C between the mode of the symmetric cell and the single cell. | ||
As reported, A/B-site doped LaFeO3 materials can act as good cathode materials for SOFCs due to their good mixed ionic and electronic conductivity and excellent ORR activity.21,25,51 Therefore, symmetric cells (SMCs) with BLFMN as both the anode and cathode (BLFMN//LSGM//BLFMN) were fabricated and the electrochemical performance was evaluated. The I–V–P curves as well as the corresponding EIS are shown in Fig. 6(b and d), respectively. The RΩ, Rp and Pmax values of the SC and SMC are listed in Table S1.† The Rp values of the SMC are higher than those for single cells at 850 °C, resulting in a slightly lower Pmax. However, upon comparison with other electrode materials as listed in Table 2 and Fig. S5,† which display the Rp or Pmax of the listed electrode materials at the temperature range of 650–850 °C, BLFMN demonstrates comparable or even lower Rp and higher Pmax values, whether acting as an anode in a single cell or serving as both the anode and cathode in a symmetric cell at an intermediate temperature range, indicating its excellent catalytic activity for the HOR and ORR. However, the I–V curves for the symmetric cells exhibit non-linearity at 800 °C and 850 °C (as shown in the inset in Fig. 6(b)), indicating an increased activation polarization occurring at the cathode.52 The sluggish performance of the ORR is attributed to the limited charge transfer and ionic transport processes at elevated temperatures, as elucidated in the DRT analysis.
| Anode | Cell type | Cell configuration (anode/FL/electrolyte/cathode) | T (°C) | R p (Ω cm2) | P max (mW cm−2) | Ref. |
|---|---|---|---|---|---|---|
| a Single cell (SC); symmetrical cell (SMC); functional layer (FL); La0.8Sr0.2Ga0.87Mg0.13O3−δ or La0.8Sr0.2Ga0.8Mg0.2O3−δ (LSGM); La0.6Sr0.4Co0.2Fe0.8O3−δ (LSCF); Ce0.8Sm0.2O2−δ (SDC); La0.4Ce0.6O2 (LDC); Ce0.9Gd0.1O2 (GDC); PrBa0.5Sr0.5Co1.5Fe0.5O5+δ (PBSCF). | ||||||
| Sr2V0.4Fe0.9Mo0.7Ni0.4O6−δ (SVFNM) | SC | SVFNM/SDC/LSGM/LSCF | 850 | 0.18 | 828 | 7 |
| Sr2Co0.4Fe1.2Mo0.406−δ (SCFM) | SC | SCFM/LSGM (300 μm)/LSCF | 850 | 0.17 | 1139 | 18 |
| Sr2Co0.4Fe1.2Mo0.406−δ (SCFM) | SMC | SCFM/LSGM (300 μm)/SCFM | 850 | 0.12 | 993 | 18 |
| La0.1Sr1.9Fe1.4Ni0.1Mo0.5O6−δ (LSFMN) | SC | LSFMN/LSGM (300 μm)/LSCF | 850 | 0.18 | 1371 | 28 |
| Pr0.1Sr1.9Fe1.4Ni0.1Mo0.5O6−δ (PSFMN) | SC | PSFMN/LSGM (300 μm)/LSCF | 850 | 0.20 | 1127 | 28 |
| Sr2Fe1.4Ni0.1Mo0.5O6−δ (SFMN) | SC | SFMN/LSGM (300 μm)/LSCF | 850 | 0.22 | 1083 | 28 |
| Sr2Fe1.5Mo0.5O6−δ (SFM) | SMC | SFM/LSGM (265 μm)/SFM | 800 | 0.47 | 470 | 34 |
| La0.5Sr0.5Fe0.9W0.1O3−δ (LSFW) | SMC | LSFW/LSGM (250 μm)/LSFW | 800 | 0.41 | 617 | 35 |
| La0.5Sr0.5Fe0.9Nb0.1O3−δ (LSFNb) | SMC | LSFNb/LSGM (300 μm)/LSFNb | 800 | 0.19 | 820 | 37 |
| La0.8Sr1.2Fe0.9Co0.1O4−δ (LSFCo) | SMC | LSFCo/LSGM (1000 μm)/LSFCo | 800 | 0.38 | 237 | 39 |
| Sr2FeMo0.6Mg0.25Ga0.15O6−δ (SFMMG) | SC | SFMMG/LDC/LSGM (300 μm)/PBSCF | 850 | 0.16 | 818 | 53 |
| Sr1.9Fe1.4Ni0.1Mo0.5O6−δ (S1.9FNM) | SC | SMNF-SDC/LDC/LSGM/LSCF | 800 | 0.26 | 968 | 54 |
| La0.6Sr0.4Mn0.2Fe0.8O3−δ (LSMF) | SC | LSMF/LSGM (280 μm)/LSCF-GDC | 800 | 0.11 | 720 | 55 |
| La0.6Sr0.4Mn0.2Fe0.8O3−δ (LSMF) | SMC | LSMF-GDC/LSGM (280 μm)/LSMF-GDC | 800 | 0.51 | 640 | 55 |
| La0.6Sr0.4Fe0.95Pt0.05O3−δ (LSFPt) | SMC | LSFPt/LSGM (300 μm)/LSFPt | 850 | 0.58 | 719 | 56 |
| La0.95Fe0.80Ni0.05Ti0.15O3 (LFNT) | SC | LFNT/LSGM (350 μm)/LSC | 800 | 0.51 | 280 | 57 |
| La0.6Sr0.4Fe0.95Ru0.05O3−δ (LSFRu) | SMC | LSFRu/LSGM (300 μm)LSFRu | 850 | 0.31 | 602 | 58 |
| BLFMN | SC | BLFMN/LSGM (300 μm)/LSCF | 850 | 0.16 | 1610 | This work |
| BLFMN | SMC | BLFMN/LSGM (300 μm)/BLFMN | 850 | 0.19 | 1320 | This work |
Considering the outstanding redox reversibility of BLFMN, which was demonstrated by the XRD and TGA analyses, the cycling stability of the BLFMN electrode was further evaluated in a symmetric cell at 800 °C. Specifically, the cell was initially operated in a single cell mode, namely, the atmosphere of one side of the electrode was H2 while that of the other side was air. Single cells were operated under a constant current load of 0.3 A cm−2 for 24 h to obtain stable cell performance. Then, the current load was removed, and the first Rp and Pmax in the single cell mode were obtained from EIS and I–V curves, respectively. Subsequently, the atmosphere of the anode side of the single cell was flushed with N2 for 0.5 h, and then switched to air, so that both the anode and cathode were exposed to air, and the cell was in a symmetric cell mode in an air atmosphere. After exposing both electrodes in air for 2.5 h, the EIS plot was measured to obtain the Rp in the symmetric cell mode in air. The anode was then flushed with N2 for 0.5 h, and subsequently switched to H2 for 2.5 h, while the cathode was still exposed to air, and the EIS plot was recorded to obtain the Rp and the V–I curve was measured to obtain Pmax in the single cell mode. The cell operation mode was illustrated in the inset in Fig. 6(e). By switching the anode atmosphere between H2 and air for 12 cycles, the Rp as well as Pmax values of the symmetrical cell remained almost unchanged (Fig. 6(e)), indicating the excellent redox cycling reversibility of the BLFMN electrode.
3.7 Cell electrochemical performance using hydrocarbon fuel
Coking resistance for direct oxidation of hydrocarbon fuels of the BLFMN electrode was investigated in single cells with the cell configuration of BLFMN//LSGM//LSCF. As shown in Fig. 7(a), when CH4 was supplied to the BLFMN anode, the Pmax values of the single cells were 0.415, 0.203 and 0.076 W cm−2 at 850, 800 and 750 °C, respectively. The electrochemical performance of the single cells using CH4 as fuel is lower than that of the single cells using H2 as fuel, indicating the BLFMN anode shows lower catalytic activity for the CH4 oxidation reaction mainly accounting for the extremely stable CH4 molecules,18,59 which is evident from the larger polarization resistance of the single cells using CH4 as fuel than using H2 as fuel as shown in Fig. 7(c) and 6(c). On the other hand, when C3H8 was used as the fuel, the Pmax of the single cell was 0.412 W cm−2 at 750 °C, as presented in Fig. 7(b). Due to the lower activation energy strength of the C–H bond in C3H8 (∼415 kJ mol−1) than that in CH4 (∼435 kJ mol−1), the single cell showed higher OCV (∼1 V) at 750 °C when C3H8 was used as fuel compared with that using CH4 as fuel (∼0.8 V) at 750 °C. Such an OCV difference with CH4 and C3H8 is consistent with the theoretical OCV calculated from the Nernst equation.3,60 Meanwhile, the single cell shows lower polarization using C3H8 as fuel (1.10 Ω cm2) than that using CH4 as fuel (1.48 Ω cm2) at 750 °C, as shown in Fig. 7(c and d), respectively. | ||
| Fig. 7 (a) I–V–P and (c) EIS curves for the single cell operating from 750 to 850 °C with CH4 as fuel. (b) I–V–P and (d) EIS curves for the single cell operating at 750 °C with C3H8 as fuel. | ||
3.8 Cell performance stability
The stability of cell performance for direct CH4 or C3H8 oxidation with BLFMN as the anode was assessed by subjecting single cells to constant current loads over time. Fig. 8(a) illustrates the good stability observed when H2 was used as fuel. Fig. 8(b and c) depict the absence of a significant voltage drop over 300 h under constant current loads of 0.1 A cm−2 at 800 °C for CH4 and 0.3 A cm−2 at 750 °C for C3H8, respectively. SEM analysis was employed to evaluate the microstructure of the cell before and after ∼325 h of stability testing using CH4 as fuel. Fig. 9(a and b) present cross-sections of the fresh cell and the cell after stability testing with CH4, respectively. After ∼325 h of operation, no discernible delamination between the electrode and electrolyte was observed. Furthermore, image analysis using ImageJ software was conducted to estimate the average grain size of the electrodes. Fig. 9(e) shows that the fresh BLFMN electrode had an average grain size of 382 nm, which increased to 425 nm after ∼325 h of operation in dry CH4. The growth rate of the grain size (an increase of 11%) was notably lower compared to that of traditional Ni-YSZ electrodes (which typically show increases ranging from 20% to over 100%).61,62 The excellent cell stability upon direct oxidation of hydrocarbon fuel indicates that the BLFMN electrode is superior to the conventional Ni-based anode, which has been shown to be easily deactivated when directly operating with dry hydrocarbon fuels.63–65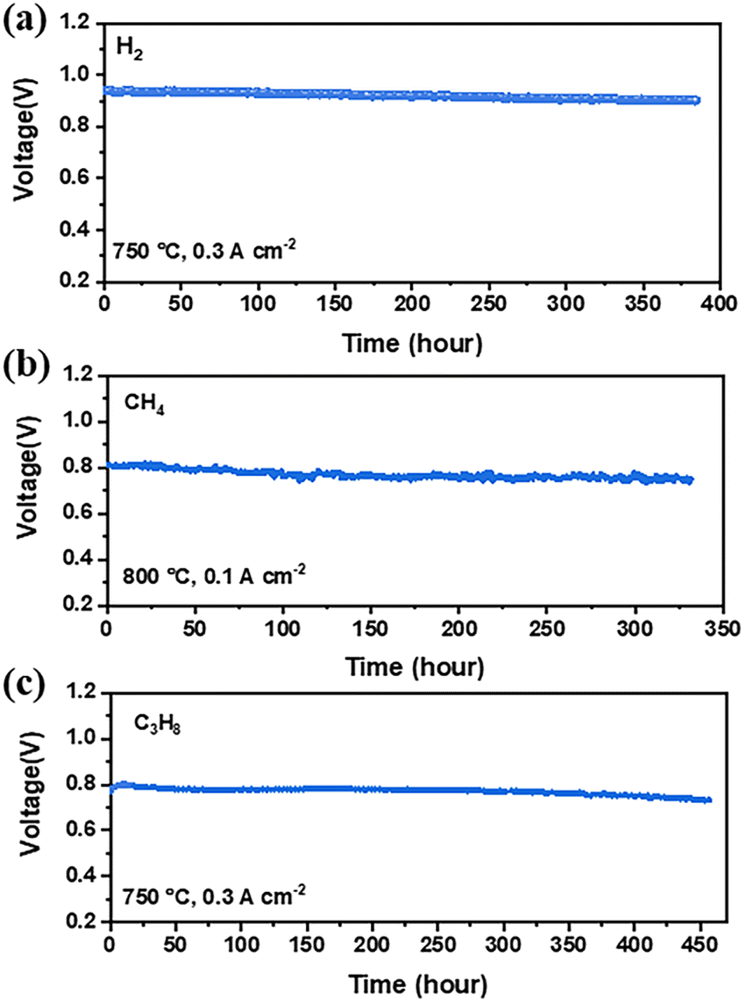 | ||
| Fig. 8 Performance stability test for the single cells with the BLFMN|LSGM|LSCF configuration under a constant current load of (a) 0.3 A cm−2 at 750 °C in H2, (b) 0.1 A cm−2 at 800 °C in CH4, and (c) 0.3 A cm−2 at 750 °C in C3H8. | ||
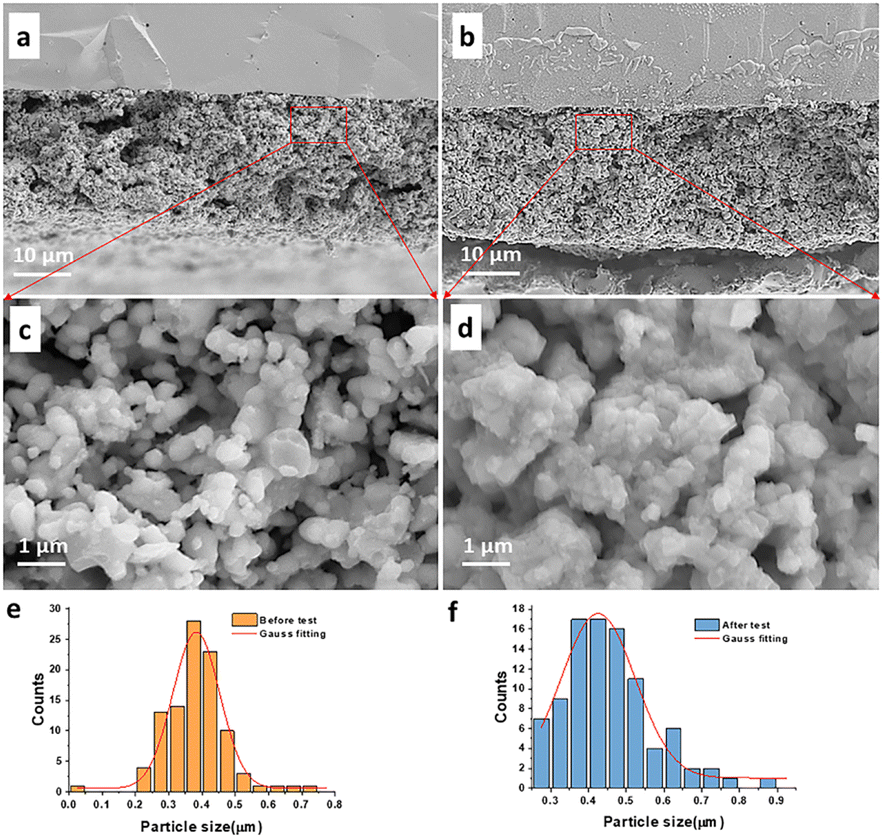 | ||
| Fig. 9 (a and b) SEM image of the cross section of the fresh cell and the cell after tested with CH4; (c and d) magnified fractured cross-sectional SEM image of the BLFMN anode for the area in the red rectangle in (a) and (b), respectively; (e and f) the particle size distribution analysis of the fresh BLFMN anode and BLFMN anode after being tested in dry CH4 for ∼325 h, respectively. | ||
The improved coking resistance can be explained by several aspects.18,26,54,60 Firstly, the cell will generate water vapor under operating conditions, and the deposited carbon will be gradually removed through a wet reforming process.18,66 Secondly, a bi-metallic alloy, such as FeNi3, embedded on the perovskite backbone can preferentially oxidize carbon atoms, removing them from the surface of the electrocatalyst.26,54 Most importantly, according to the working principle of oxygen ion conducting SOFCs, when cells are operated under a constant current load, oxygen ions generated in the cathode will be transported to the anode through the oxygen ion conducting LSGM electrolyte, and then the deposited carbon in the anode will be gradually oxidized.60 Above all, the good performance and stability from single cells using BLFMN as the anode indicate that BLFMN is a promising anode material with good coking resistance.
4. Conclusions
In this work, dual phase BLFMN consisting of a cubic perovskite and scheelite BMO4 was synthesized using a sol–gel combustion method. By heat-treating the dual phase BLFMN in a reducing atmosphere, a composite comprising the cubic BLF main phase, cubic BMO3 secondary phase and in situ-exsolved nanoparticles of the FeNi3 bimetallic alloy phase was achieved. By heat-treating the reduced powders in an oxidizing atmosphere, the composite can transform back to the initial dual phase, showing excellent redox reversibility of the BLFMN samples. The catalytic activity of BLFMN was evaluated using a symmetric cell with the cell configuration of BLFMN//LSGM//BLFMN, exhibiting a low Rp in air (0.04 Ω cm2) or in H2 (0.13 Ω cm2) at 800 °C. Furthermore, the symmetrical cells exhibited excellent redox reversibility by switching the atmosphere in the anode between air and H2 with 12 cycles. Single cells using BLFMN as the anode with the cell configuration of BLFMN//LSGM//LSCF have demonstrated Pmax values of 1.61 W cm−2 at 850 °C, using H2 as the fuel. Moreover, BLFMN has also demonstrated good catalytic activity and performance stability (over 300 h) for direct oxidation of CH4 and C3H8, indicating that the newly designed BLFMN is a promising electrode material for direct hydrocarbon SOFCs.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors would like to acknowledge the support of the National Science Foundation (DMR-1832809) and the Range Extenders for Electric Aviation with Low Carbon and High Efficiency (REEACH) program in Advanced Research Projects Agency-Energy (ARPA-E) under the DoE Idaho Operations Office under contract no. DE-AC07-05ID14517.References
- Q. Xu, Z. Guo, L. Xia, Q. He, Z. Li, I. Temitope Bello, K. Zheng and M. Ni, Energy Convers. Manage., 2022, 253, 115175 CrossRef CAS.
- S. Zarabi Golkhatmi, M. I. Asghar and P. D. Lund, Renewable Sustainable Energy Rev., 2022, 161, 112339 CrossRef CAS.
- W. Zhang, J. Wei, F. Yin and C. Sun, Mater. Chem. Front., 2023, 7, 1943–1991 RSC.
- S. He, Y. Zou, K. Chen and S. P. Jiang, Interdiscip. Mater., 2023, 2, 111–136 CrossRef.
- M. van den Bossche, R. Matthews, A. Lichtenberger and S. McIntosh, J. Electrochem. Soc., 2010, 157, B392 CrossRef CAS.
- K. Bin Yoo, B. H. Park and G. M. Choi, Solid State Ionics, 2012, 225, 104–107 CrossRef.
- L. Song, D. Chen, J. Pan, X. Hu, X. Shen, Y. Huan and T. Wei, ACS Appl. Mater. Interfaces, 2023, 15, 48296–48303 CrossRef CAS PubMed.
- Q. Liu, D. E. Bugaris, G. Xiao, M. Chmara, S. Ma, H. C. Zur Loye, M. D. Amiridis and F. Chen, J. Power Sources, 2011, 196, 9148–9153 CrossRef CAS.
- Z. Han, H. Dong, Y. Wang, Y. Yang, H. Yu and Z. Yang, J. Mater. Chem. A, 2023, 11, 18820–18831 RSC.
- X. M. Ge, S. H. Chan, Q. L. Liu and Q. Sun, Adv. Energy Mater., 2012, 2, 1156–1181 CrossRef CAS.
- A. N. Zainon, M. R. Somalu, A. M. Kamarul Bahrain, A. Muchtar, N. A. Baharuddin, M. A. Muhammed, N. Osman, A. Abdul Samat, A. K. Azad and N. P. Brandon, Int. J. Hydrogen Energy, 2023, 48, 20441–20464 CrossRef CAS.
- W. Zhang, F. Yin, Z. Cheng, S. Peng and C. Sun, Fuel, 2023, 354, 129356 CrossRef CAS.
- D. Ding, X. Li, S. Y. Lai, K. Gerdes and M. Liu, Energy Environ. Sci., 2014, 7, 552–575 RSC.
- H. Rostaghi Chalaki, A. Babaei, A. Ataie and S. V. Seyed-Vakili, Ionics, 2020, 26, 1011–1018 CrossRef CAS.
- D. Lee, D. Kim, S. J. Son, Y. il Kwon, Y. Lee, J. H. Ahn and J. H. Joo, J. Power Sources, 2019, 434, 226743 CrossRef CAS.
- Z. Sun, C. Hao, S. Toan, R. Zhang, H. Li, Y. Wu, H. Liu and Z. Sun, J. Mater. Chem. A, 2023, 11, 17961–17976 RSC.
- C. Yang, Z. Yang, C. Jin, G. Xiao, F. Chen and M. Han, Adv. Mater., 2012, 24, 1439–1443 CrossRef CAS PubMed.
- P. Qiu, X. Yang, W. Wang, T. Wei, Y. Lu, J. Lin, Z. Yuan, L. Jia, J. Li and F. Chen, ACS Appl. Mater. Interfaces, 2020, 12, 13988–13995 CrossRef CAS PubMed.
- C. Wang, H. Miao, X. Zhang, J. Huang and J. Yuan, J. Power Sources, 2024, 596, 234112 CrossRef CAS.
- X. Ding, X. Gao, W. Zhu, J. Wang and J. Jiang, Int. J. Hydrogen Energy, 2014, 39, 12092–12100 CrossRef CAS.
- G. Nandikes, P. Pathak, M. Karthikeyan, A. A. M. Abahussain and L. Singh, Int. J. Hydrogen Energy, 2024, 52, 627–641 CrossRef CAS.
- J. Wang, M. Saccoccio, D. Chen, Y. Gao, C. Chen and F. Ciucci, J. Power Sources, 2015, 297, 511–518 CrossRef CAS.
- Y. Zhang, X. Zhang, J. Ni and C. Ni, ACS Appl. Mater. Interfaces, 2023, 15, 3974–3984 CrossRef CAS PubMed.
- Z. Li, J. Wang, J. Zhu, Y. Sun, Z. Wang, Y. Zhao, Z. Wei, Z. Zhu and Q. Zheng, Sustainable Energy Fuels, 2021, 5, 5975–5984 RSC.
- H. Cai, J. Xu, M. Wu, W. Long, L. Zhang, Z. Song and L. Zhang, J. Eur. Ceram. Soc., 2020, 40, 4361–4365 CrossRef CAS.
- S. Xue, N. Shi, Y. Wan, Z. Xu, D. Huan, S. Zhang, C. Xia, R. Peng and Y. Lu, J. Mater. Chem. A, 2019, 7, 21783–21793 RSC.
- H. Hu, M. Li, H. Min, X. Zhou, J. Li, X. Wang, Y. Lu and X. Ding, ACS Catal., 2022, 12, 828–836 CrossRef CAS.
- H. Li, W. Wang, J. Lin, K.-Y. Park, T. Lee, A. Heyden, D. Ding and F. Chen, Clean Energy, 2023, 7, 70–83 CrossRef.
- H. Li, W. Wang, L. Wang, M. Wang, K.-Y. Park, T. Lee, A. Heyden, D. Ding and F. Chen, ACS Appl. Mater. Interfaces, 2023, 15, 43732–43744 CrossRef CAS PubMed.
- L. D. S. Alencar, A. Mesquita, C. A. C. Feitosa, R. Balzer, F. D. Probst, D. C. Batalha, M. G. Rosmaninho, H. V Fajardo and M. I. B. Bernardi, Ceram. Int., 2017, 43, 4462–4469 CrossRef CAS.
- F. Dong, D. Chen, Y. Chen, Q. Zhao and Z. Shao, J. Mater. Chem., 2012, 22, 15071–15079 RSC.
- S. Liu, Q. Liu and J. Luo, ACS Catal., 2016, 6, 6219–6228 CrossRef CAS.
- Z. Xu, X. Hu, Y. Wan, S. Xue, S. Zhang, L. Zhang, B. Zhang and C. Xia, Electrochim. Acta, 2020, 341, 136067 CrossRef CAS.
- Q. Liu, X. Dong, G. Xiao, F. Zhao and F. Chen, Adv. Mater., 2010, 22, 5478–5482 CrossRef CAS PubMed.
- S. Wang, B. Wei and Z. Lü, Int. J. Hydrogen Energy, 2021, 46, 30101–30111 CrossRef CAS.
- D. Zhang, K. Zhang, T. He, N. Yu, Y. Zhao, Y. Wang and T. Liu, Int. J. Hydrogen Energy, 2020, 45, 21825–21835 CrossRef CAS.
- L. Bian, C. Duan, L. Wang, L. Zhu, R. O'Hayre and K. C. Chou, J. Power Sources, 2018, 399, 398–405 CrossRef CAS.
- B. Niu, C. Lu, W. Yi, S. Luo, X. Li, X. Zhong, X. Zhao and B. Xu, Appl. Catal., B, 2020, 270, 118842 CrossRef CAS.
- J. Zhou, T. H. Shin, C. Ni, G. Chen, K. Wu, Y. Cheng and J. T. S. Irvine, Chem. Mater., 2016, 28, 2981–2993 CrossRef CAS.
- J. Yan, H. Chen, Y. W. Li, S. D. Li and Z. Shao, J. Mater. Chem. A, 2023, 11, 21839–21845 RSC.
- W. Bian, W. Wu, B. Wang, W. Tang, M. Zhou, C. Jin, H. Ding, W. Fan, Y. Dong, J. Li and D. Ding, Nature, 2022, 604, 479–485 CrossRef CAS PubMed.
- Y. Yang, Z. Chen, M. Li, M. Rao, F. Jin, Y. Ling, P. Feng and S. Wang, J. Mater. Chem. A, 2023, 11, 7995–8002 RSC.
- M. B. Hanif, J. T. Gao, K. Shaheen, Y. P. Wang, M. Yasir, C. J. Li and C. X. Li, Int. J. Hydrogen Energy, 2021, 46, 8778–8791 CrossRef CAS.
- H. Ding, Z. Tao, S. Liu and J. Zhang, Sci. Rep., 2015, 5, 18129 CrossRef CAS PubMed.
- M. Ghamarinia, A. Babaei, C. Zamani and H. Aslannejad, Chem. Eng. J. Adv., 2023, 15, 100503 CrossRef CAS.
- T. H. Wan, M. Saccoccio, C. Chen and F. Ciucci, Electrochim. Acta, 2015, 184, 483–499 CrossRef CAS.
- B. K. Maji, H. Jena, R. Asuvathraman and K. V. G. Kutty, J. Alloys Compd., 2015, 640, 475–479 CrossRef CAS.
- X. Xu, Y. Pan, Y. Zhong, L. Ge, S. P. Jiang and Z. Shao, Composites, Part B, 2020, 198, 108214 CrossRef CAS.
- Somia, S. Mehmood, Z. Ali, I. Khan, F. Khan and I. Ahmad, J. Electron. Mater., 2019, 48, 1730–1739 CrossRef CAS.
- L. Shu, J. Sunarso, S. S. Hashim, J. Mao, W. Zhou and F. Liang, Int. J. Hydrogen Energy, 2019, 44, 31275–31304 CrossRef CAS.
- Z. Ma, C. Sun, C. Ma, H. Wu, Z. Zhan and L. Chen, Cuihua Xuebao, 2016, 37, 1347–1353 CAS.
- Z. Lyu, H. Li, Y. Wang and M. Han, J. Power Sources, 2021, 485, 229237 CrossRef CAS.
- M. Zhang, Y. Li, Z. Du, Y. Zhang and H. Zhao, J. Power Sources, 2024, 594, 234031 CrossRef CAS.
- G. Xiao, S. Wang, Y. Lin, Z. Yang, M. Han and F. Chen, J. Electrochem. Soc., 2014, 161, F305–F310 CrossRef CAS.
- Y. S. Chung, T. Kim, T. H. Shin, H. Yoon, S. Park, N. M. Sammes, W. B. Kim and J. S. Chung, J. Mater. Chem. A, 2017, 5, 6437–6446 RSC.
- A. P. Panunzi, L. Duranti, I. Luisetto, N. Lisi, M. Marelli and E. Di Bartolomeo, Chem. Eng. J., 2023, 471, 144448 CrossRef CAS.
- C. Ni, Q. Zeng, D. He, L. Peng, D. Xie, J. T. S. Irvine, S. Duan and J. Ni, J. Mater. Chem. A, 2019, 7, 26944–26953 RSC.
- M. Marasi, L. Duranti, I. Luisetto, E. Fabbri, S. Licoccia and E. Di Bartolomeo, J. Power Sources, 2023, 555, 232399 CrossRef CAS.
- W. Wu, L. C. Wang, H. Hu, W. Bian, J. Y. Gomez, C. J. Orme, H. Ding, Y. Dong, T. He, J. Li and D. Ding, ACS Catal., 2021, 11, 12194–12202 CrossRef CAS.
- S. Vecino-Mantilla, E. Quintero, C. Fonseca, G. H. Gauthier and P. Gauthier-Maradei, ChemCatChem, 2020, 12, 1453–1466 CrossRef CAS.
- D. Ding, Z. Liu, L. Li and C. Xia, Electrochem. Commun., 2008, 10, 1295–1298 CrossRef CAS.
- P. Tanasini, M. Cannarozzo, P. Costamagna, A. Faes, J. Van Herle, A. Hessler-Wyser and C. Comninellis, Fuel Cells, 2009, 9, 740–752 CrossRef CAS.
- K. Wei, X. Wang, R. A. Budiman, J. Kang, B. Lin, F. Zhou and Y. Ling, J. Mater. Sci., 2018, 53, 8747–8765 CrossRef CAS.
- Q. Yang, F. Chai, C. Ma, C. Sun, S. Shi and L. Chen, J. Mater. Chem. A, 2016, 4, 18031–18036 RSC.
- A. Mishra, R. Dudek, A. Gaffney, D. Ding and F. Li, Catal. Today, 2023, 424, 112462 CrossRef CAS.
- X. Li, M. Liu, S. Y. Lai, D. Ding, M. Gong, J. P. Lee, K. S. Blinn, Y. Bu, Z. Wang, L. A. Bottomley, F. M. Alamgir and M. Liu, Chem. Mater., 2015, 27, 822–828 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4ta01957a |
| This journal is © The Royal Society of Chemistry 2024 |