
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Effect of vanadium doping on α-KxMnO2 as a positive electrode active material for rechargeable magnesium batteries†
Isaac
Oda-Bayliss
a,
Shunsuke
Yagi
 *a,
Masao
Kamiko
a,
Kai
Shimada
a,
Hiroaki
Kobayashi
b and
Tetsu
Ichitsubo
c
*a,
Masao
Kamiko
a,
Kai
Shimada
a,
Hiroaki
Kobayashi
b and
Tetsu
Ichitsubo
c
aInstitute of Industrial Science, The University of Tokyo, 4-6-1 Komaba, Meguro-ku, Tokyo 153-8505, Japan. E-mail: syagi@iis.u-tokyo.ac.jp
bDepartment of Chemistry, Faculty of Science, Hokkaido University, Kita 10, Nishi 8, Kita-ku, Sapporo, Hokkaido 060-0810, Japan
cInstitute for Materials Research, Tohoku University, 2-1-1 Katahira, Aoba-ku, Sendai 980-8577, Japan
First published on 7th June 2024
Abstract
α-MnO2 has recently attracted attention as a promising candidate for positive electrode active materials for rechargeable magnesium batteries (RMBs) due to its ability to accommodate Mg2+ ions without phase changes at limited concentrations. However, the stability of this phase and the kinetic barriers to Mg2+ insertion remain significant challenges. This study purposes doping of potassium stabilized hollandite MnO2 (α-KxMnO2) with vanadium ions as a method to improve the stability and cyclability of the α-KxMnO2 phase. RMBs with V-doped α-KxMnO2 positive electrode active materials exhibit discharge potentials over 0.2 V greater than that of undoped α-KxMnO2, and improved magnesiation and demagnesiation upon discharge and charge, respectively. V-doped α-KxMnO2 also exhibits greater preference for reduction via Mg2+ insertion, compared to conversion to disparate phase oxides, enhancing the stability of the α-KxMnO2 phase, and increasing capacity retention upon battery cycling.
1 Introduction
Currently lithium-ion batteries (LIBs) are employed across a wide array of practical applications, due to their high energy density and rate capability. However, as the scale of applications increases, the need for further increases in energy density has become apparent. One major factor limiting the energy density of LIBs is the low theoretical specific capacity of the carbonaceous negative electrode active materials (372 mA h g−1). While the use of a lithium metal negative electrode would result in a much greater capacity, the dendritic deposition of lithium metal upon charging and the associated short circuit risk make this substitution difficult.1 As a result, in recent years increased attention has turned to alternative batteries using metallic negative electrodes such as Mg, Ca, and Al.Among these candidates, one of the most promising is Mg; Mg metal has a high specific capacity (ca. 2200 mA h g−1) and exhibits non-dendritic plating,2–6 allowing for use as a safe battery negative electrode. Aurbach et al. pioneered the field of rechargeable magnesium batteries (RMBs) when they demonstrated Chevrel phase Mo6S8 as a positive electrode active material enabling reversible Mg insertion/extraction.7 However, this Chevrel phase Mo6S8 exhibited both low discharge potential (1.1–1.2 V vs. Mg/Mg2+) and capacity (∼75 mA h g−1). Therefore, subsequent research in the field of RMBs has focused on finding positive electrode active materials that improve these two key characteristics.
Due to the typically higher redox potentials of oxides compared to sulfides, significant efforts have been made to develop metal oxide positive electrode active materials with relatively high discharge potentials and capacities, such as olivine MgCoSiO4 (∼1.5 V vs. Mg/Mg2+, 250 mA h g−1),8 spinel MgMn2O4 (∼2.9 V vs. Mg/Mg2+, 180 mA h g−1),9 MgCo2O4 (∼2.9 V vs. Mg/Mg2+, 200 mA h g−1),10 and MgFe2O4 (∼2.5 V vs. Mg/Mg2+, 110 mA h g−1).11,12
While these spinel type oxides in particular possess high discharge potentials and initial capacities, they exhibit significant capacity degradation upon battery cycling, partly due to their conversion from the spinel to the rock-salt phase upon magnesiation.10 The high electrostatic interaction between Mg2+ ions and the host framework, combined with the high stability of the rock-salt phase greatly limit Mg2+ extraction upon charge, decreasing cyclability.
To improve battery cyclability at higher potentials, we turned to hollandite manganese oxide (α-MnO2) as a positive electrode active material for RMBs. α-MnO2 is a polymorph of manganese oxide formed by conjoined MnO6 octahedra and characterized by long 1D tunnels with side lengths of two octahedra. These 2 × 2 tunnels are often stabilized by a large cation located at the tunnel centre as shown in Fig. 1. One of the most common forms of hollandite α-MnO2 is cryptomelane (α-KxMnO2), in which K+ ions occupy the tunnel centre sites.
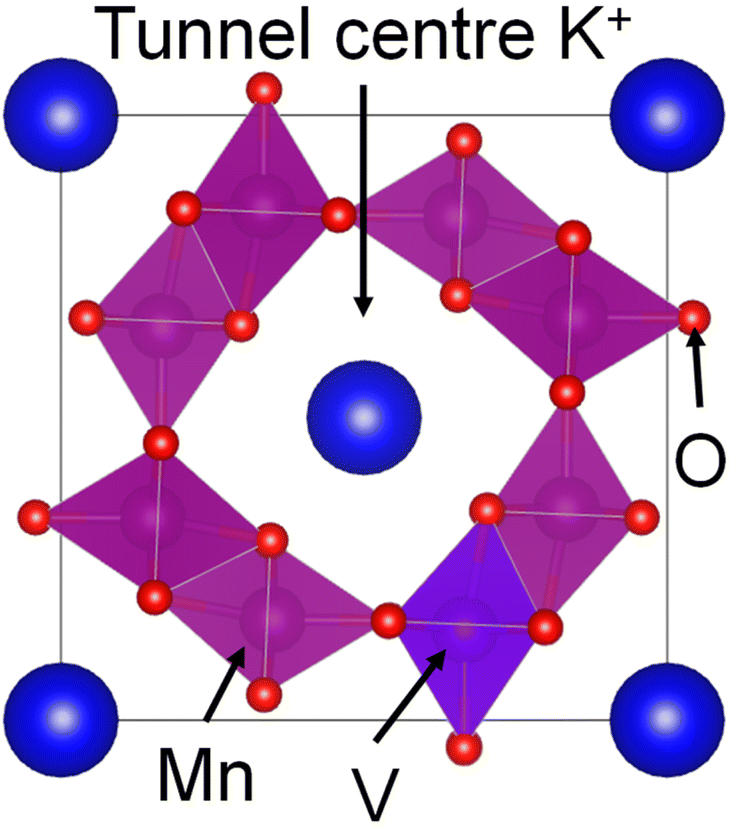 | ||
| Fig. 1 Schematic diagram of potassium stabilized hollandite manganese oxide (α-KxMnO2) with K, Mn, O and substituted V positions labelled. VESTA 3 is used for three-dimensional visualization of the crystal. | ||
α-MnO2 has been extensively studied as a positive electrode active material in LIBs and was an early metal oxide candidate considered for use in RMBs.13 However, Ling et al.14 and Arthur et al.15 showed that the primary mode of magnesiation of α-MnO2 was not by Mg2+ insertion, but by surface Mg enrichment and conversion to Mg–O and Mn–O compounds due to the high stability of MgO.
| MnO2 + yMg2+ + 2ye− → yMgO + MnO2−y, |
This irreversible reaction mechanism was thought to limit the utility of α-MnO2 as a positive electrode active material for RMBs.
However, Hatakeyama et al. recently showed at elevated temperatures (150 °C) that α-MnO2 could exhibit insertion and extraction of Mg2+ up to 220 mA h g−1 with an initial discharge potential of 2.6 V vs. Mg/Mg2+ without phase transformation.16
| MnO2 + yMg2+ + 2ye− ↔ MgyMnO2, |
While these findings elucidated the role of reaction kinetics in the magnesiation pathway, there were still several limitations to the application of α-MnO2 as a positive electrode active material for RMBs. Namely, the high electrostatic interaction of Mg2+ ions and the α-MnO2 host framework resulted in magnesium buildup upon cycling and capacity degradation, indicating the need for further decrease in kinetic barriers.
To minimize the kinetic barriers to magnesiation, we looked to two potential methods by which α-MnO2 can be modified that have been shown to improve battery performance in LIBs.
These methods were partial substitution of framework Mn with disparate transition metal ions,17–21 and modification of the concentration and species of the tunnel centre cations.22–24
Of particular interest for the motivation of this work were two previous studies. First, Poyraz et al. examined the effect of tunnel centre K+ concentration in the α-KxMnO2 electrode on LIB performance and found that decreased tunnel centre K+ concentration improved discharge capacity and rate capability.22 Second, Yoo et al. determined that transition metal ion doping of α-KxMnO2 with V and with Fe both lead to improvements in cyclability in LIBs, which were attributed to improved stability of the α-KxMnO2 phase due to the doped transition metal ions.
In this study we aimed to minimize the kinetic barriers to magnesiation and improve the stability of the α-KxMnO2 phase by framework doping of α-KxMnO2 with vanadium. V was chosen as the dopant due to its similar ionic radius to Mn and its higher valence (+5) compared to Mn (4+). Namely, we attempted the framework substitution of the higher valence V to stabilize the α-KxMnO2 phase, while simultaneously decreasing tunnel centre potassium occupancy due to the need to maintain charge neutrality.
2 Experimental
2.1 Preparation of vanadium doped α-KxMnO2
α-KxMnO2 with variable concentrations of doped vanadium was synthesized using a hydrothermal method reported by Polverejan et al.25 14.3 mmol MnSO4·5H2O (Nacalai Tesque, INC., Japan), 21.45 mmol K2SO4 (Nacalai Tesque, INC., Japan), and 21.45 mmol K2S2O8 (FUJIFILM Wako Pure Chemical Co., Japan) were dissolved in 60 mL of deionized water. Vanadium was introduced by addition of Na3VO4 (MP Biomedicals, Inc., USA), and precursor concentration was controlled by using the V/(Mn + V) mole ratio. Four samples with ratios of 0, 5, 10, and 20 mol% were prepared. The solutions were each transferred to 100 mL PTFE (polytetrafluoroethylene)-lined stainless steel autoclaves and heated at 200 °C for 48 h. The obtained suspensions were filtered, washed three times with deionized water, and dried at 80 °C for 24 h to remove excess water. The obtained powders were ground and passed through a 150 μm mesh to minimize the impact of particle size on the electrochemical measurements. The expected synthesis reaction is:| MnSO4 + K2S2O8 + 2H2O → MnO2 + K2SO4 + 2H2SO4, |
Excess K2SO4 is used to provide K+ ions used to stabilize the tunnel structure. The resulting solution is acidic due to the H2SO4 formed as a by-product of the synthesis reaction.
2.2 Materials characterization
Crystallographic characterization of the 4 obtained samples was performed by synchrotron X-ray diffraction (SXRD) at the BL02B2 beamline in SPring-8, using a 0.59999 Å incident beam, calibrated with a CeO2 standard. X-ray photoelectron spectroscopy (XPS) measurements were carried out using an ULVAC-PHI Quantera Instrument to analyse the valences of framework Mn and V. The morphologies and elemental distributions within the obtained powders were observed using a JEOL JSM-6010LA analytical scanning electron microscope with energy dispersive X-ray (EDX) analysis. Focused ion beam scanning electron microscopy (FIB-SEM) was performed using a Thermo fisher Scios 2 DualBeam microscope. Finally, bulk elemental compositions were analysed using an SPS 3500 series inductively coupled plasma (ICP) spectrometer.2.3 Electrochemical measurements
All electrochemical tests were performed using a three-electrode beaker cell (Fig. S1†). The working electrode (WE) was composed of a slurry of the α-KxMnO2 active material, activated carbon conductive agent (Super P, Hohsen Co., Japan), and PVDF binder (KF polymer, Hohsen Co., Japan) at an 8![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1:1 mass ratio, suspended in 600 μL of N-methyl-2-pyrrolidone (NMP, Nacalai Tesque, INC., Japan). The slurry was mixed using a rotation-revolution mixer (ARE-310, Thinky, Inc., Japan) at 2000 rpm for 20 min with an additional defoaming step at 2200 rpm for 2 min, before coating on a platinum current collector, and dried at 80 °C under vacuum for 24 h. The counter electrode (CE) was a 99.9% Mg ribbon, polished using #150 sandpaper under an Ar atmosphere. Finally, an Ag/Ag+ reference electrode (RE) composed of an Ag wire immersed in 450 μL solution composed of 0.01 M AgNO3, 0.1 M Mg[TFSA]2, and (TFSA: bis(trifluoromethanesulfonyl)-amide) in triethylene glycol dimethyl ether (G3) in a glass tube with a porous Vycor® glass tip was employed. The electrolyte was prepared using a 10 mL volumetric flask by dissolving 0.5 mmol of magnesium bis(trifluoromethanesulfonamide) (Mg[TFSA]2) (FUJIFILM Wako Pure Chemical Co., Japan) and 0.5 mmol tetraethyleneglycol dimethyl ether (G4) (FUJIFILM Wako Pure Chemical Co., Japan) in N-methyl-N-propylpyrrolidinium TFSA ([Pyr1,3][TFSA]) (FUJIFILM Wako Pure Chemical Co., Japan). This electrolyte will henceforth be denoted as 0.5 M [Mg(G4)][TFSA]2/[Pyr1,3][TFSA].26 The conversion from the Ag/Ag+ reference potential to the Mg/Mg2+ potential was determined experimentally using the Mg deposition/dissolution potential for a Pt electrode at 100 °C as shown in Fig. S2.† The equation 0 V vs. Mg/Mg2+ = −2.6 V vs. Ag/Ag+ was used to convert all recorded potentials. All electrochemical measurements were performed using a potentiostat (VSP-300, BioLogic, France).
:1:1 mass ratio, suspended in 600 μL of N-methyl-2-pyrrolidone (NMP, Nacalai Tesque, INC., Japan). The slurry was mixed using a rotation-revolution mixer (ARE-310, Thinky, Inc., Japan) at 2000 rpm for 20 min with an additional defoaming step at 2200 rpm for 2 min, before coating on a platinum current collector, and dried at 80 °C under vacuum for 24 h. The counter electrode (CE) was a 99.9% Mg ribbon, polished using #150 sandpaper under an Ar atmosphere. Finally, an Ag/Ag+ reference electrode (RE) composed of an Ag wire immersed in 450 μL solution composed of 0.01 M AgNO3, 0.1 M Mg[TFSA]2, and (TFSA: bis(trifluoromethanesulfonyl)-amide) in triethylene glycol dimethyl ether (G3) in a glass tube with a porous Vycor® glass tip was employed. The electrolyte was prepared using a 10 mL volumetric flask by dissolving 0.5 mmol of magnesium bis(trifluoromethanesulfonamide) (Mg[TFSA]2) (FUJIFILM Wako Pure Chemical Co., Japan) and 0.5 mmol tetraethyleneglycol dimethyl ether (G4) (FUJIFILM Wako Pure Chemical Co., Japan) in N-methyl-N-propylpyrrolidinium TFSA ([Pyr1,3][TFSA]) (FUJIFILM Wako Pure Chemical Co., Japan). This electrolyte will henceforth be denoted as 0.5 M [Mg(G4)][TFSA]2/[Pyr1,3][TFSA].26 The conversion from the Ag/Ag+ reference potential to the Mg/Mg2+ potential was determined experimentally using the Mg deposition/dissolution potential for a Pt electrode at 100 °C as shown in Fig. S2.† The equation 0 V vs. Mg/Mg2+ = −2.6 V vs. Ag/Ag+ was used to convert all recorded potentials. All electrochemical measurements were performed using a potentiostat (VSP-300, BioLogic, France).
3 Results and discussion
3.1 Chemical and structural characterization
The primary modifier examined in this study was the effect of V-doping of the α-KxMnO2 positive electrode active material on RMB performance. We analyzed the V and K contents for each of the obtained samples using ICP spectroscopy as tabulated in Table 1. The concentrations of doped V in the obtained powders were measured to be 4.3, 7.2, and 12.0 mol% for the 5, 10, and 20 mol% precursor mixtures, respectively.| Precursor V/(Mn + V) mol% | Obtained V/(Mn + V) mol% (ICP) | Obtained K/(Mn + V) mol% (ICP) |
|---|---|---|
| 0 | 0 | 9.8 |
| 5 | 4.3 | 12.8 |
| 10 | 7.2 | 10.1 |
| 20 | 12.0 | 10.1 |
Surprisingly, the concentration of potassium for each of the above four samples, measured by using K/(Mn + V) mole ratios, did not exhibit significant changes with values of 9.8, 12.8, 10.1, and 10.1 mol% respectively.
Fig. 2a–d show the morphology and elemental mapping of V, Mn, and K measured for the 7.2% V-doped sample. We observe a homogeneous distribution of V, Mn, and K across the sample. Fig. 2e shows the SXRD peak profiles of the doped samples. Excluding slight contamination of the 4.3% V-doped sample with what appears to be Mn2V2O7, the profiles exhibit characteristic peaks of α-KxMnO2. The presence of these characteristic peaks and the homogeneous distribution of V across the sample show that the added V doped the host α-KxMnO2, as opposed to forming separate V compounds. Additionally, we observed no significant shifts in peak position, and a slight broadening of peaks as the concentration of the dopant increases. At higher V concentrations crystallinity is decreased, as evidenced by the broadening of the peaks visible in the 12.0% V-doped sample. The samples with V concentrations greater than 12.0% were not considered for battery testing, due to the formation of separate V compounds during synthesis.
 | ||
| Fig. 2 (a) SEM image of 7.2% V-doped α-KxMnO2 and EDS elemental mapping of the corresponding area for (b) potassium, (c) manganese, and (d) vanadium. (e) SXRD profiles of undoped, 4.3% V-doped, 7.2% V-doped, and 12.0% V-doped α-KxMnO2. | ||
The location within the crystal lattice occupied by the doped V was analyzed by conducting four separate Rietveld refinements of the single SXRD profile for 7.2% V-doped α-KxMnO2 with the V position constrained to different Wyckoff positions within the α-KxMnO2 lattice. The 7.2% V-doped sample was chosen for this refinement due to the combination of high crystallinity and lack of visible contaminants as compared with the SXRD profiles for the other V-doped samples.
As described in detail in Fig. S3 and S4,† the Rietveld refinement with the V position constrained to the 8 h site illustrated improved fit to the SXRD peaks compared to the cases with other constrained positions. Despite there being no constraint on the location of the doped V beyond the Wyckoff position, the location of the doped V was only slightly offset from the location of the framework Mn. It is therefore likely that the doped-V mainly substituted the framework Mn. This result is reinforced by Raman spectroscopy measurements reported by Polverejan et al.25 illustrating that only a single additional Raman peak, corresponding to the V–O single bond was present in the V-doped α-KxMnO2 samples.
XPS was employed to determine the averaged valence states of V and Mn. Fig. 3a compares the XPS profiles for the V 2p3/2 peaks of 7.2% V-doped α-KxMnO2 compared to a V2O5 reference. A charge reference of 284.8 eV was used for the measured adventitious C 1s peaks. The V 2p1/2 peak was not considered for this fitting due to the partial overlap with the O 1s peak. It was concluded that the valence of the doped V was most likely +5. A slight difference in the binding energies of the V5+ peaks of V2O5 and V-doped α-KxMnO2 is generally expected due to the differences in the bonding environments of the two samples.27 The satellite (sat.) peaks are thought to be related to surface adsorption.
 | ||
| Fig. 3 X-ray photoelectron spectra: (a) V 2p3/2 peak of V2O5 and 7.2% V-doped α-KxMnO2 and (b) Mn 2p peaks of undoped α-KxMnO2 and 7.2% V-doped α-KxMnO2. | ||
The average valence of Mn was analyzed by fitting of Mn 2p peak profiles measured by XPS for undoped and 7.2% V-doped α-KxMnO2, respectively, as shown in Fig. 3b. The average Mn valence for each sample, calculated using the ratio of Mn3+ and Mn4+ peak area, was roughly +3.6. Detailed area ratios are provided in Table S2.† This result is corroborated by the good fit of the O 1s lattice oxygen peaks, provided in Fig. S5 and Table S3,† when constrained by the area ratios in accordance with the Mn3+ and Mn4+ ratios calculated in the above Mn 2p profiles. The addition of V5+ without change in the average Mn valence necessitates a change in either oxygen vacancy concentration, or tunnel cation concentration to maintain charge neutrality of the synthesized sample. These mechanisms and the effects on RMB performance will be discussed in greater detail in Section 3.5.
3.2 Constant current discharge–charge analysis
We evaluated the discharge performance of the RMBs with undoped and V-doped α-KxMnO2 positive electrode active materials to determine the effect of V-doping on the discharge potential and initial discharge capacity. The four α-KxMnO2 variants were tested at 100 °C using a constant discharge current density of 10 mA g−1, with a capacity cutoff of 200 mA h g−1. The capacity cutoff of 200 mA h g−1 was implemented as α-MnO2 had been shown to exhibit irreversible conversion to rock-salt phase MgO–MnO at capacities exceeding 220 mA h g−1.16The discharge profiles for the four α-KxMnO2 samples are shown in Fig. 4a. The first point of interest is the 0.2 V – 0.3 V increase in initial discharge potential seen in every V-doped α-KxMnO2 sample compared to the undoped α-KxMnO2. This increased discharge potential is ascribed to a decrease in overpotential for the Mn4+/Mn3+ reduction reaction as opposed to a separate V5+/V4+ redox reaction, as the theoretical discharge capacity for the V5+/V4+ redox couple is limited to 12.7, 21.3, and 35.6 mA h g−1 for the 4.3%, 7.2% and 12.0% V-doped α-KxMnO2, respectively.
 | ||
| Fig. 4 (a) Constant current discharge curves of undoped and V-doped α-KxMnO2, (b) SXRD profiles of undoped and V-doped α-KxMnO2 following constant current discharge to 200 mA h g−1 shown in (a). (c) Constant current discharge–charge curves of undoped and 7.2% V-doped α-KxMnO2. | ||
Secondly, there is a retention of higher discharge potentials in the case of the 7.2% V-doped sample, as the discharge potential exceeds 2 V vs. Mg/Mg2+ at the 200 mA h g−1 cutoff. In contrast, all other samples experienced significant declines in discharge potential before the capacity cutoff. The SXRD profiles of the four samples following discharge are provided in Fig. 4b. In the undoped and 4.3% V-doped samples, we observe an additional Mn2O3 peak that is not present in the 7.3% and 12.0% V-doped samples. The presence of Mn2O3 peaks in addition to the α-KxMnO2 peaks shows that some portion of reduction during discharge progressed by conversion of the framework α-KxMnO2 as opposed to reduction accompanied by Mg2+ insertion. The lack of visible Mn2O3 peaks in the 7.2% and 12.0% V-doped samples suggest that the presence of the V within the α-KxMnO2 lattice limits the favorability of the conversion mechanism.
Given the improved performance of the 7.2% V-doped sample, we further investigated 7.2% V-doped α-KxMnO2 in detail.
Fig. 4c compares the discharge–charge profiles of 7.2% V-doped and undoped α-KxMnO2. The 7.2% V-doped sample exhibits a charge plateau at about 2.8 V vs. Mg/Mg2+ that is not observed in the undoped sample. Elemental composition of post discharge and post discharge–charge samples for both undoped α-KxMnO2 and 7.2% V-doped α-KxMnO2 was analyzed using ICP spectroscopy. The analyzed magnesium content for each sample is provided in Table 2. V-doping increases the Mg concentration after discharge and enables greater Mg extraction after subsequent charging. This greater magnitude of insertion and extraction indicates that V-doping improves Mg2+ mobility within the host framework both for discharge and charge processes. Additionally, there is no decrease in V concentration in the 7.2% V-doped α-KxMnO2 across the charge cycle. Therefore, the oxidative reaction observed at 2.8 V vs. Mg/Mg2+ is likely caused by the extraction of magnesium from the host framework, and not by extraction or dissolution of the doped V. Another point to consider is the lower total concentration of magnesium compared to theoretical values. This implies the presence of background electrolyte decomposition, the contribution of other cations such as protons, or potential dissolution of magnesium-based conversion products, all leading to lower-than-expected values of detected magnesium.28
| Sample description | Experimental condition | Theoretical Mg/(Mn + V) calc. from electrochemical capacity (mol%) | Experimental Mg/(Mn + V) estimated by using ICP (mol%) | Experimental V/(Mn + V) in powder samples estimated by using ICP (mol%) | Experimental V/(Mn + V) estimated by using ICP (mol%) |
|---|---|---|---|---|---|
| Undoped α-KxMnO2 | Discharge to 200 mA h g−1 | 33.9 | 1.23 (3.6%) | 0 | 0 |
| Charge to 200 mA h g−1 following discharge | 0 | 0 | 0 | 0 | |
| 7.2% V-doped α-KxMnO2 | Discharge to 200 mA h g−1 | 33.9 | 5.66 (16.7%) | 7.2 | 7.2 |
| Charge to 200 mA h g−1 following discharge | 0 | 0.65 | 7.2 | 8.1 |
3.3 Effect of V-doping on redox pathways
To better determine the effect of V-doping of α-KxMnO2 on the reduction and oxidation mechanisms, we performed cyclic voltammetry (CV) starting at the open circuit potential with 1 V vs. Mg/Mg2+ and 3.6 V vs. Mg/Mg2+ as the lower and upper bounds, respectively. The voltammograms of undoped and 7.2% V-doped α-KxMnO2 are presented in Fig. 5. All voltammetry measurements were performed at 100 °C to match the conditions of the discharge–charge tests. | ||
| Fig. 5 (a) Cyclic voltammogram of undoped α-KxMnO2 measured at a scan rate of 25 μV s−1 at 100 °C. (b) Cyclic voltammogram of 7.2% V-doped α-KxMnO2 measured at a scan rate of 25 μV s−1 at 100 °C. | ||
The two separate cathodic peaks at 2.1 V vs. Mg/Mg2+ and 1.7 V vs. Mg/Mg2+ seen in each scan illustrate the presence of two separate reduction reactions. The V-doped sample exhibits higher current densities for the 2.1 V vs. Mg/Mg2+ peak compared to the undoped sample, and significantly lower current densities for the 1.7 V vs. Mg/Mg2+ redox peak, which agrees well with the discharge curves shown in Fig. 4a. V-doping improves the selectivity of the redox reaction with the higher potential, while suppressing the reaction at 1.7 V vs. Mg/Mg2+.
In the anodic scan, the voltammogram of 7.2% V-doped α-KxMnO2 shows a significant anodic peak at 3.1 V vs. Mg/Mg2+ which is not present in the undoped α-KxMnO2 profile, indicating a separate oxidation reaction. In both voltammograms there is an increase in anodic current starting above 3 V vs. Mg/Mg2+ up to the upper potential limit of 3.6 V vs. Mg/Mg2+. We believe this anodic current to be caused by oxidative electrolyte decomposition as the magnitude and onset potentials align with previously reported values for oxidative electrolyte decomposition of the [Mg(G4)][TFSA]2/[Pyr1,3][TFSA] electrolyte.12,29
To investigate the reduction mechanisms for the two separate cathodic peaks at 2.1 V vs. Mg/Mg2+ and 1.7 V vs. Mg/Mg2+, we performed two additional linear sweep voltammetry (LSV) experiments from OCP to 2 V vs. Mg/Mg2+ and OCP to 1 V vs. Mg/Mg2+ for undoped α-KxMnO2. Fig. 6a compares the two LSV profiles, and Fig. 6b compares the two SXRD profiles corresponding to the samples following each experiment. The SXRD profile following the potential sweep from OCP to 2.1 V vs. Mg/Mg2+ shows characteristic α-KxMnO2 peaks without obvious impurity peaks. In contrast, we see the presence of many new peaks in addition to the α-KxMnO2 peaks following a cathodic potential sweep to 1 V vs. Mg/Mg2+. Notably there are several peaks corresponding to Mn3O4, but no Mn2O3 peaks as previously observed in the discharged samples, possibly due to further conversion of Mn2O3 to Mn3O4. In addition, there were several peaks that could not be identified, likely caused by distortion of the α-KxMnO2 tunnels upon high levels of magnesiation. These SXRD profiles indicate that the redox reaction at 2.1 V vs. Mg/Mg2+ corresponds to reduction of α-KxMnO2via magnesiation, while the 1.7 V vs. Mg/Mg2+ redox reaction is a conversion from α-KxMnO2 to different Mg–Mn oxide phases. The significant increase in the 2.1 V vs. Mg/Mg2+ cathodic peak in the 7.2% V-doped cyclic voltammogram in Fig. 5b therefore correlates with improved magnesium mobility, while the significantly smaller peak at 1.7 V vs. Mg/Mg2+ shows that the addition of vanadium limited the progression of the conversion reaction by improving the stability of the α-KxMnO2 phase.
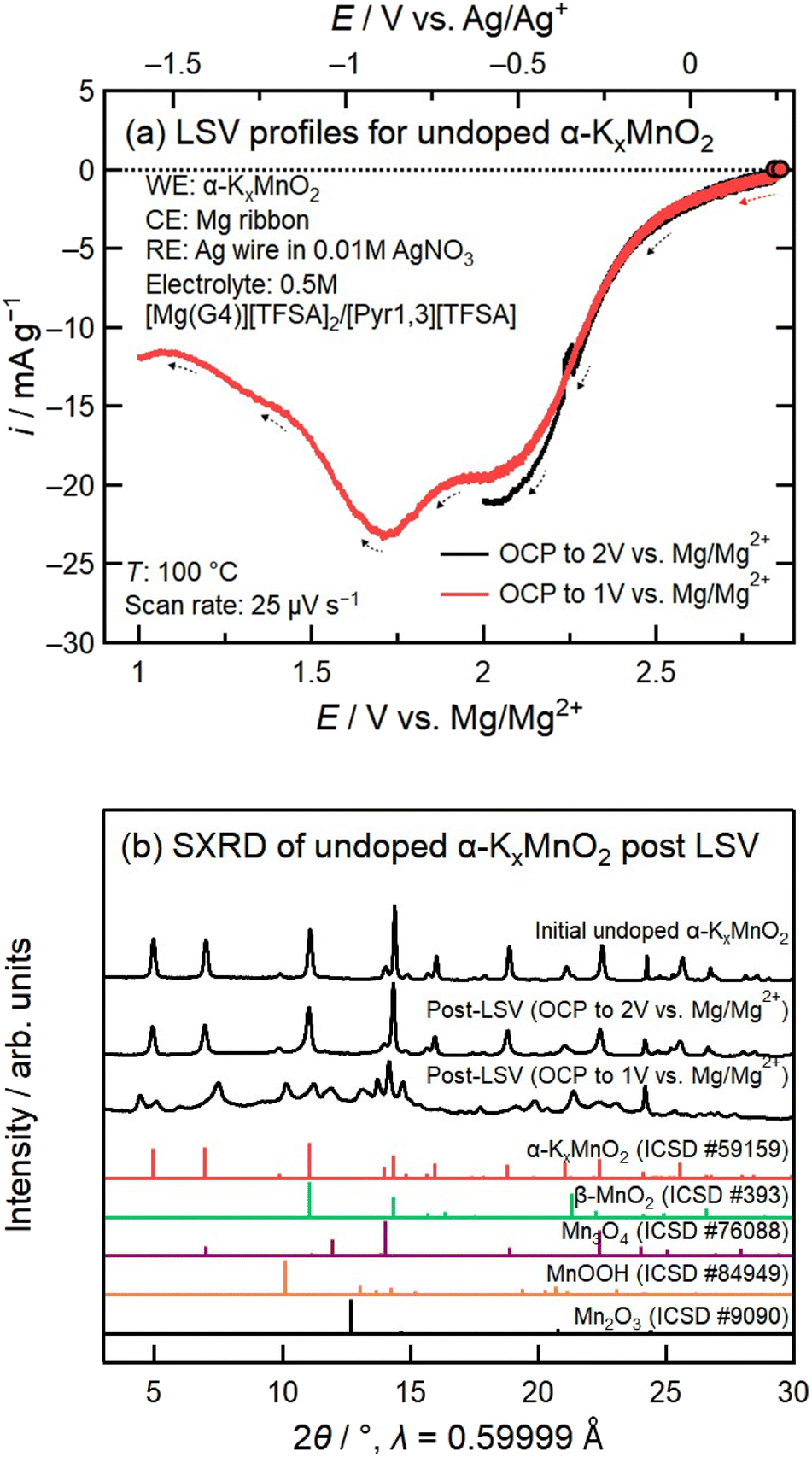 | ||
| Fig. 6 (a) Linear sweep voltammograms of undoped α-KxMnO2 from OCP to 2 V vs. Mg/Mg2+ and OCP to 1 V vs. Mg/Mg2+. (b) SXRD profiles of undoped α-KxMnO2 as-synthesized powder and composite electrodes containing undoped α-KxMnO2 following LSV from OCP to 2 V vs. Mg/Mg2+ and OCP to 1 V vs. Mg/Mg2+. | ||
3.4 Effect of V-doping on RMB cyclability
The effect of V-doping on the cyclability of α-KxMnO2 was examined using galvanostatic cycling with potential limitation (GCPL) across ten discharge–charge cycles. Undoped and 7.2% V-doped α-KxMnO2 samples were evaluated at a constant current density of 10 mA g−1 with potential cutoffs of 2 V and 3.6 V vs. Mg/Mg2+. The 2 V vs. Mg/Mg2+ cutoff was set for two reasons. Firstly, the cutoff at 2 V vs. Mg/Mg2+ prevents discharge by the conversion mechanism. As shown in Section 3.3, discharge progresses by Mg2+ insertion at potentials above 2 V vs. Mg/Mg2+, but switches to a conversion mechanism at lower potentials. Secondly, the cutoff prevents reductive electrolyte decomposition upon discharge, as the onset of reductive electrolyte decomposition is not expected above 2 V vs. Mg/Mg2+.Fig. 7a and b plot the discharge potentials for undoped α-KxMnO2 and 7.2% V-doped α-KxMnO2, respectively across the ten cycles. There is a clear decrease in capacity during cycling in both samples; however, the magnitude of this decrease is suppressed in the V-doped sample as shown in Fig. 7c. This improvement in cyclability caused by V-doping alone is promising, as there remain many factors that can be modified to further increase cyclability. Investigation into the effects of different cation species within the tunnel centre, co-insertion of Mg2+ as part of a chelated complex or other electrolyte additives and multiple possible framework dopants provide many options for further improvements.
 | ||
| Fig. 7 (a) Galvanostatic cycling with potential limitation (GCPL) profile for undoped α-KxMnO2 measured at a current density of 10 mA g−1 and 2 V and 3.6 V vs. Mg/Mg2+ potential cutoffs. (b) GCPL profile for 7.2% V-doped α-KxMnO2 measured at a current density of 10 mA g−1 and 2 V and 3.6 V vs. Mg/Mg2+ potential cutoffs. (c) Discharge capacity plotted against cycle number for undoped and 7.2% V-doped α-KxMnO2. | ||
3.5 Screening of mechanisms for improved RMB performance
We have thus far shown how V-doping of α-KxMnO2 leads to greater Mg2+ mobility, as evidenced by the greater current densities corresponding to Mg2+ insertion and extraction, and greater phase stability. Here we posit that the presence of V5+ can explain the improved stability, while a decrease in H+ occupying the α-KxMnO2 tunnel in the V-doped sample is responsible for the increase in Mg2+ mobility.Several factors were considered as explanations for the increased mobility and stability. We considered a decrease in oxygen vacancy concentration, effects of V–O bonding, a decrease in grain size, an increase in tunnel diameter, and a decrease in cation concentrations within the tunnel structure in the V-doped sample as possible explanations. Each factor above was considered as they can offer explanations for the observed improvement in the Mg2+ mobility or α-KxMnO2 phase stability.
An increase in oxygen vacancy concentration has been shown across multiple studies to decrease the stability of transition metal oxide active materials in LIBs, causing cracking and dissolution of the transition metal ions.30,31 The concentrations of oxygen vacancies in both samples were calculated using the ratio of atomic concentrations of Mn, V and O obtained via XPS. There was no appreciable difference in atomic concentration of O across the two samples, with atomic percentages of 63.7% and 63.0% for the undoped and 7.2% V-doped samples, respectively (Table S4†). Combined with the minimal difference in the ratio of lattice and absorbed oxygen peak areas of 67.7% for undoped α-KxMnO2 and 68.8% for 7.2% V-doped α-KxMnO2 detailed in Table S3,† it can be concluded that there is minimal change in oxygen vacancy concentration caused by V-doping, and that changes in oxygen vacancy concentration are unlikely to be the explanation for the improved phase stability. Consequently, we considered differences between the framework Mn and doped V that could explain the improved stability. Differences in the valence, ionic size, and M–O bond distance of the doped V from those of the framework Mn may offer explanations. Density functional theory (DFT) calculations of transition metal dopants in LIBs by Brady et al. showed that transition metal dopants such as Nb5+ may be able to suppress tunnel breakage at high degrees of lithiation.21 In particular, they reported that these dopants improve the stability of the α-MnO2 phase during lithiation by preventing the breakage of bonds within the framework MO6 octahedra. While we believe a similar comprehensive review of transition metal dopants of α-MnO2 for RMBs is required to fully elucidate the mechanism, the greater difference in charge of the V5+–O2− bond compared to the Mn3.6+–O2− bond could be responsible for a similar suppression of tunnel breakage.
A smaller average grain size caused by V-doping was investigated as a possible explanation for the improved Mg2+ mobility. A smaller grain size has been shown to correlate with improved battery performance in RMBs.32,33 As the size of the grain decreases, the ratio of surface area to volume increases. In their study on α-MnO2 active materials in RMBs, Ling et al. showed that Mg2+ insertion upon discharge was often limited to the exterior surface of the α-MnO2 particle due to the sluggish diffusion of Mg2+.14 As such we considered that a smaller grain size could explain the observed increase in current density. The grain size of the undoped and 7.2% V-doped samples was compared using high-resolution SEM as shown in Fig. 8a and b. Both samples exhibit similar morphologies, composed of numerous capsule-shaped grains, with dimensions of approximately 50 nm × 100 nm. The cross-section images of the two samples (Fig. S6†) also show similar clustering of these small grains. As such, we concluded that there is no appreciable change in grain size due to V-doping, and that a decrease in grain size due to V-doping is not likely to be the mechanism by which RMB performance is improved.
 | ||
| Fig. 8 High-resolution SEM images of (a) undoped α-KxMnO2 and (b) 7.2% V-doped α-KxMnO2. | ||
Second, we posited an increase in tunnel diameter due to V-doping as a mechanism for improved Mg2+ mobility. It is expected that a greater tunnel diameter would result in lower electrostatic interaction. Similar methods have been employed to great success in layered oxides with larger interlayer spacing leading to improved Mg2+ kinetics.34–37 The horizontal lattice constant a was considered as it is proportional to the tunnel diameter. The lattice constants for both samples, calculated via Rietveld refinement are equivalent, with calculated values of 9.839 Å and 9.842 Å for undoped and 7.2% V-doped α-KxMnO2, respectively. We concluded that the tunnel size is not affected by V-doping, and that modified tunnel size is not the mechanism by which RMB performance was improved.
Lastly, a change in the concentration of cations within the α-MnO2 tunnel structure is considered. A decrease in tunnel centre cation concentration may decrease the electrostatic repulsion with the inserted Mg2+ ions. Proyaz et al. previously showed that a smaller K+ concentration in α-KxMnO2 improved performance in LIBs.22 As previously discussed in Section 3.1, ICP analysis of K/(Mn + V) molar ratios shows equivalent concentrations of tunnel centre K+ across all samples. A change in K+ concentration therefore cannot explain the improved performance. However, the α-KxMnO2 tunnel may contain molecules other than K+, as both H2O and H+ may be introduced during the hydrothermal synthesis. Both H2O and H+ are capable of occupying the tunnel complex of α-MnO2, as shown by Dai et al.38 and Johnson et al.39 A decrease in H+ concentration from the undoped to the 7.2% V-doped sample provides an explanation for the mechanism by which charge neutrality is maintained in the V-doped sample, with a decrease in H+ concentration accompanying the substitution of Mn3.6+ with V5+. The decrease in H+ concentration would consequently decrease the electrostatic repulsion experienced by the Mg2+ during insertion and extraction, as there would be fewer cationic species within the tunnel to obstruct the Mg2+ diffusion. Further investigation is needed to isolate the impact of tunnel centre H+ in α-MnO2 active materials on RMBs.
4 Conclusions
In conclusion, doping of α-KxMnO2 with V ions was carried out to observe the effects of the introduction of higher valence transition metal ions on RMB performance. We successfully doped α-KxMnO2 with three different concentrations of vanadium via a hydrothermal process. Successful doping was characterized by the even distribution of V within the host α-KxMnO2 and Rietveld refinement of the SXRD profiles. We showed that V-doping took place via substitution of V for framework Mn. The substituted V had a valence of +5, while the Mn valence did not change due to V-doping with an average valence of +3.6 in both the undoped and V-doped samples.Furthermore, the effect of this V-doping on α-KxMnO2 positive electrode performance in RMBs was evaluated. We determined that V-doping leads to an increase in initial RMB discharge potential by 0.2 V to 0.3 V compared to that of undoped α-KxMnO2. Additionally, we determined that at limited concentrations such as 7.2% V, the addition of V stabilized the α-KxMnO2 framework, leading to a greater degree of reduction with Mg2+ insertion, and a significant decrease in reduction accompanied by conversion of α-KxMnO2 to disparate Mn oxide phases.
We also observed that V-doping improved the mobility of Mg2+ ions during oxidation, resulting in a significant increase in Mg2+ extraction observed in both cyclic voltammograms and constant current charge profiles. This improved Mg2+ mobility allowed for an improvement in RMB cyclability.
We investigated possible mechanisms to explain the greater Mg2+ mobility and α-KxMnO2 stability in the V-doped sample. We posited that changes in the concentration of H+ occupying the α-KxMnO2 tunnel could be responsible for the improved Mg2+ insertion kinetics, while a greater difference in charge of the V–O bond compared to the Mn–O bond could be responsible for the improved stability. We also ruled out the possibility of changes in grain size, tunnel dimensions, and oxygen vacancy concentrations as explanations for the observed improvements.
The significant improvement in the RMB performance of V-doped α-KxMnO2 paves the way for further improvements to achieve RMB practical applications by way of transition metal doping.
Author contributions
Isaac Oda-Bayliss: investigation, writing – original draft, writing – review & editing. Shunsuke Yagi: supervision, writing – review & editing, funding acquisition. Masao Kamiko: resources, validation. Kai Shimada: investigation. Hiroaki Kobayashi: discussion. Tetsu Ichitsubo: supervision.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This study was supported by the GteX Program Japan Grant Number JPMJGX23S1 and the Japan Society for the Promotion of Science (JSPS: 23H05452). SXRD measurements were conducted at the BL02B2 beamline (Proposal No. 2023A1728 and 2023B1635) at SPring-8. The data shown in Fig. 8a and b were observed at Komaba Analysis Core, Institute of Industrial Science, The University of Tokyo, and data analysis was supported by Dr Atsushi Fukuda. The authors acknowledge their support.References
- J. M. Tarascon and M. Armand, Nature, 2001, 414, 359–367 CrossRef CAS.
- S. Yagi, A. Tanaka, T. Ichitsubo and E. Matsubara, ECS Electrochem. Lett., 2012, 1, D11–D14 CrossRef CAS.
- M. Matsui, J. Power Sources, 2011, 196, 7048–7055 CrossRef CAS.
- M. Morita, N. Yoshimoto, S. Yakushiji and M. Ishikawa, Electrochem. Solid-State Lett., 2001, 4, A177–A179 CrossRef CAS.
- S. Yagi, A. Tanaka, Y. Ichikawa, T. Ichitsubo and E. Matsubara, J. Electrochem. Soc., 2013, 160, C83–C88 CrossRef CAS.
- S. Yagi, A. Tanaka, Y. Ichikawa, T. Ichitsubo and E. Matsubara, Res. Chem. Intermed., 2014, 40, 3–9 CrossRef CAS.
- D. Aurbach, Z. Lu, A. Schechter, Y. Gofer, H. Gizbar, R. Turgeman, Y. Cohen, M. Moshkovich and E. Levi, Nature, 2000, 407, 724–727 CrossRef CAS.
- Y. Nuli, Y. Zheng, Y. Wang, J. Yang and J. Wang, J. Mater. Chem., 2011, 21, 12437–12443 RSC.
- T. Hatakeyama, N. L. Okamoto, K. Shimokawa, H. Li, A. Nakao, Y. Uchimoto, H. Tanimura, T. Kawaguchi and T. Ichitsubo, Phys. Chem. Chem. Phys., 2019, 21, 23749–23757 RSC.
- S. Okamoto, T. Ichitsubo, T. Kawaguchi, Y. Kumagai, F. Oba, S. Yagi, K. Shimokawa, N. Goto, T. Doi and E. Matsubara, Adv. Sci., 2015, 2, 1500072 CrossRef.
- J. Han, S. Yagi and T. Ichitsubo, J. Power Sources, 2019, 435, 226822 CrossRef CAS.
- J. Han, S. Yagi, H. Takeuchi, M. Nakayama and T. Ichitsubo, J. Mater. Chem. A, 2021, 9, 26401–26409 RSC.
- R. Zhang, X. Yu, K. W. Nam, C. Ling, T. S. Arthur, W. Song, A. M. Knapp, S. N. Ehrlich, X. Q. Yang and M. Matsui, Electrochem. Commun., 2012, 23, 110–113 CrossRef CAS.
- C. Ling, R. Zhang, T. S. Arthur and F. Mizuno, Chem. Mater., 2015, 27, 5799–5807 CrossRef CAS.
- T. S. Arthur, R. Zhang, C. Ling, P. A. Glans, X. Fan, J. Guo and F. Mizuno, ACS Appl. Mater. Interfaces, 2014, 6, 7004–7008 CrossRef CAS.
- T. Hatakeyama, H. Li, N. L. Okamoto, K. Shimokawa, T. Kawaguchi, H. Tanimura, S. Imashuku, M. Fichtner and T. Ichitsubo, Chem. Mater., 2021, 33, 6983–6996 CrossRef CAS.
- H. N. Yoo, D. H. Park and S. J. Hwang, J. Power Sources, 2008, 185, 1374–1379 CrossRef CAS.
- A. M. Hashem, H. M. Abuzeid, D. Mikhailova, H. Ehrenberg, A. Mauger and C. M. Julien, J. Mater. Sci., 2012, 47, 2479–2485 CrossRef CAS.
- J. G. Radich, Y.-S. Chen and P. V. Kamat, ECS J. Solid State Sci. Technol., 2013, 2, M3178–M3181 CrossRef CAS.
- T. Sasaki, S. Komaba, N. Kumagai and I. Nakai, Electrochem. Solid-State Lett., 2005, 8, A471 CrossRef CAS.
- A. B. Brady, K. R. Tallman, E. S. Takeuchi, A. C. Marschilok, K. J. Takeuchi and P. Liu, J. Phys. Chem. C, 2019, 123, 25042–25051 CrossRef CAS.
- A. S. Poyraz, J. Huang, C. J. Pelliccione, X. Tong, S. Cheng, L. Wu, Y. Zhu, A. C. Marschilok, K. J. Takeuchi and E. S. Takeuchi, J. Mater. Chem. A, 2017, 5, 16914–16928 RSC.
- L. M. Housel, L. Wang, A. Abraham, J. Huang, G. D. Renderos, C. D. Quilty, A. B. Brady, A. C. Marschilok, K. J. Takeuchi and E. S. Takeuchi, Acc. Chem. Res., 2018, 51, 575–582 CrossRef CAS.
- M. M. Thackeray, Prog. Solid State Chem., 1997, 25, 1–71 CrossRef CAS.
- M. Polverejan, J. C. Villegas and S. L. Suib, J. Am. Chem. Soc., 2004, 126, 7774–7775 CrossRef CAS.
- T. Mandai, K. Tatesaka, K. Soh, H. Masu, A. Choudhary, Y. Tateyama, R. Ise, H. Imai, T. Takeguchi and K. Kanamura, Phys. Chem. Chem. Phys., 2019, 21, 12100–12111 RSC.
- J. F. Watts and J. Wolstenholme, The Electron Spectrum: Qualitative and Quantitative Interpretation, Wiley, 2003 Search PubMed.
- X. Ye, H. Li, T. Hatakeyama, H. Kobayashi, T. Mandai, N. L. Okamoto and T. Ichitsubo, ACS Appl. Mater. Interfaces, 2022, 14, 56685–56696 CrossRef CAS.
- J. Han, S. Yagi, H. Takeuchi, M. Nakayama and T. Ichitsubo, J. Phys. Chem. C, 2022, 126, 19074–19083 CrossRef CAS.
- Q. Li, D. Ning, D. Wong, K. An, Y. Tang, D. Zhou, G. Schuck, Z. Chen, N. Zhang and X. Liu, Nat. Commun., 2022, 13, 1123 CrossRef CAS.
- Z. K. Tang, Y. F. Xue, G. Teobaldi and L. M. Liu, Nanoscale Horiz., 2020, 5, 1453–1466 RSC.
- W. Chen, X. Zhan, B. Luo, Z. Ou, P. C. Shih, L. Yao, S. Pidaparthy, A. Patra, H. An, P. V. Braun, R. M. Stephens, H. Yang, J. M. Zuo and Q. Chen, Nano Lett., 2019, 19, 4712–4720 CrossRef CAS.
- L. Li, G. Hu, Y. Cao, D. Gong, Q. Fu, Z. Peng and K. Du, Electrochim. Acta, 2022, 435, 141386 CrossRef CAS.
- Y. Li, Y. Zheng, K. Guo, J. Zhao and C. Li, Energy Mater. Adv., 2022, 2022, 9840837 Search PubMed.
- J. Tian, X. Zhou, Q. Wu and C. Li, Energy Storage Mater., 2019, 22, 218–227 CrossRef.
- Z. Yao, Y. Yu, Q. Wu, M. Cui, X. Zhou, J. Liu and C. Li, Small, 2021, 17, 2102168 CrossRef CAS.
- M. Wang and S. Yagi, J. Alloys Compd., 2020, 820, 153135 CrossRef CAS.
- J. Dai, S. F. Li, K. Siong Siow and Z. Gao, Electrochim. Acta, 2000, 45, 2211–2217 CrossRef CAS.
- C. S. Johnson, D. W. Dees, M. F. Mansuetto, M. M. Thackeray, D. R. Vissers, D. Argyriou, C.-K. Loong and L. Christensen, J. Power Sources, 1997, 68, 570–577 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4ta00659c |
| This journal is © The Royal Society of Chemistry 2024 |