
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceOrigin of electrocatalytic nitrogen reduction activity over transition metal disulfides: critical role of in situ generation of S vacancy†
Tianyi
Wang
a,
Zhongyuan
Guo
 b,
Hirofumi
Oka
a,
Akichika
Kumatani
*acde,
Chuangwei
Liu
*f and
Hao
Li
*a
b,
Hirofumi
Oka
a,
Akichika
Kumatani
*acde,
Chuangwei
Liu
*f and
Hao
Li
*a
aAdvanced Institute for Materials Research (WPI-AIMR), Tohoku University, Sendai, 980-8577, Japan. E-mail: li.hao.b8@tohoku.ac.jp
bCollege of Environmental and Resource Sciences, Zhejiang University, Hangzhou, 310058, China
cInstitute of Engineering Innovation (IEI), School of Engineering, The University of Tokyo, Tokyo, 113-8656, Japan. E-mail: kumatani@g.ecc.u-tokyo.ac.jp
dPrecursory Research for Embryonic Science and Technology (PRESTO), Japan Science and Technology Agency (JST), Saitama 332-0012, Japan
eGraduate School of Environmental Studies, Center for Science and Innovation in Spintronics (CSIS), Tohoku University, Sendai, Miyagi 980-8579, Japan
fKey Lab for Anisotropy and Texture of Materials, School of Materials Science and Engineering, Northeastern University, Shenyang, 110819, China. E-mail: liucw@mail.neu.edu.cn
First published on 22nd February 2024
Abstract
The electrochemical nitrogen reduction reaction (ENRR) is a promising and sustainable alternative to conventional Haber–Bosch ammonia (NH3) synthesis. Pursuing high-performance and cost-effective ENRR catalysts is an open challenge for achieving commercial-scale ambient NH3 production. Less-precious transition metal disulfides (TMS2) are a class of promising catalysts that can be highly active for ENRR. However, the origin of their high ENRR performance is not well understood. Herein, we analyze the origin of their activity by probing their electrochemistry-induced surface states. Starting with a typical ENRR TMS2 catalyst, iron disulfide (FeS2), from our calculated surface Pourbaix diagrams we found that S-vacancies can be easily generated under an ENRR potential. Our subsequent spin-polarized density functional theory (DFT) calculations show that this electrochemistry-driven “in situ” generation of S-vacancies shows significantly higher ENRR activity than a stoichiometric pristine FeS2 surface due to the stronger N–N adsorption and activation capacity of a lower-coordination-number S-vacancy site. This finding is in excellent agreement with experimental observations published in recent years regarding potential windows reaching the maximum faradaic efficiency. We then expanded our analysis to other typical TMS2 that had shown promising ENRR performance in recent experimental literature (SnS2, MoS2, NiS2, and VS2), and found that such an “in situ” S-vacancy generation phenomenon is universal under ENRR potentials, with results in good agreement with many experimental observations reported to date. We conclude that, though S-vacancy engineering during synthesis is a promising strategy to enhance the ENRR performance on TMS2 catalysts, the “in situ” generation of S-vacancies will also endow pristine TMS2 with a measurable ENRR performance. This study shows that the surface states of ENRR catalysts should not be dismissed before analyzing the activity of an ENRR catalyst. Most importantly, we found that when designing a promising TMS2 catalyst for ENRR, its capacity to form S-vacancies is a key performance indicator that needs to be analyzed.
Introduction
Ammonia (NH3), an indispensable chemical feedstock, plays a pivotal role in facilitating global economic development and demographic expansion. Its versatile utility spans pharmaceutical synthesis, fertilizer production, and compound fabrication.1 NH3 also emerges as a prospective carbon-neutral medium for realizing the hydrogen economy, functioning as a liquid hydrogen carrier without carbon emissions.2 Currently, the Haber–Bosch (H–B) process prevails as the industrial framework for NH3 synthesis, but harsh reaction conditions (c.a. 450 °C and 300 bar) demand substantial energy consumption and emit enormous amounts of greenhouse gases.3 As an alternative, the electrochemical nitrogen reduction reaction (ENRR) offers an ecologically benign route to synthesize NH3 under ambient reaction conditions. In this context, the N2 is fixed and hydrogenated by protons and electrons in an aqueous solution instead of gaseous H2. However, the competing hydrogen evaluation reaction (HER) and insertion of N2 fixation are two significant challenges for industrial-scale ENRR.4–6 Specifically, optimal catalysts maintain a delicate equilibrium between N2 adsorption and NH3 desorption, reducing formidable energy barriers. Additionally, HER remains the key obstacle to improving the faradaic efficiency (FE). Comparatively, HER is characterized by a two-step hydrogenation pathway, which is thermodynamically more favorable than the complicated six-step hydrogenation ENRR process.7,8Some noble-metal-based catalysts, such as those based on Ru, Au, Pd, and Rh, have demonstrated exceptional activities and FEs in ENRR. However, their prohibitive price and shortage impede their widespread industrial implementation.3,9–11 Therefore, the pursuit of cost-effective and high-performance catalysts has become an intensified research focus in recent years. Fe-based catalysts have garnered significant attention as promising candidates to get close to or even surpass existing benchmarks in ENRR. For example, Fe-related proteins can serve as intrinsic active sites within enzymes in biological N2 fixation.12 Fe is also the central constituent within catalysts deployed in the H–B method.13 Drawing inspiration from biological and H–B approaches, FeS2, as a typical transition metal disulfide (TMS2), has emerged as a promising candidate for ENRR with structural alignment with the Fe–S ligand arrangement characteristic of nitrogenase.14 Additionally, S-containing Fe materials exhibit notable hydrophobic properties attributed to the inherent hydrophobic nature of S, thereby mitigating the adverse effect of the competing HER.15–17 In recent studies, FeS2 nanoparticles embedded in graphene oxide (FeS2@GO) could generate low-coordination Fe active sites to facilitate N2 fixation and ENRR, exhibiting an FE of 6.80% and an NH3 yield of 27.9 μg h−1 mgcat−1 at −0.3 V vs. reversible hydrogen electrode (VRHE) under acidic and neutral conditions.18 In addition, doping is a common strategy to improve the catalytic performance of FeS2-based catalysts. For example, the synergy between Cr and S-vacancies (SV) of FeS2 can achieve an NH3 yield of 11.5 μg h−1 mgcat−1 and an FE of 14.6% at −0.2 VRHE.19 The synergistic effects among FeS2, MoS2, and reduced GO may also expedite the kinetics of N2 hydrogenation in pH-universal aqueous solutions, elevating the NH3 yield and FE to 41.1 μg h−1 mgcat−1 and 38.6% at −0.2 VRHE, respectively.20 Moreover, FeS2–MoS2 supported on Fe foam exhibited a considerable FE of 4.6% at −0.5 VRHE and an NH3 production rate of 7.1 × 10−10 mol s−1 cm−2 at −0.3 VRHE. This was attributed to the synergistic effects and interface interaction between FeS2 and MoS2 phases, which furnish favourable electron transfer pathways with abundant active sites.21 Doping Mo into FeS2 also achieved an FE of 14.41% at −0.2 VRHE, which was attributed to the joint factors of the superior activity of Mo and HER suppression of FeS2.22 Besides, other TMS2, such as VS2, NiS2, and SnS2, show great promise in ENRR.23–25Fig. 1 and Table S1† summarize the ENRR performance in terms of FE of typical TMS2 reported after 2018. It can be seen that, in general, these materials preferentially achieve the highest FE or experience a rapid growth in FE at a relatively “early” potential (c.a. −0.4 VRHE). For the better design and modification of TMS2 catalysts for ENRR, it is particularly important to provide a deep understanding of the superior ENRR performance of TMS2. Furthermore, it is worth noting that electrochemical nitrate reduction is another efficient approach for NH3 synthesis. The synergy achieved through the formation of the Sn–Fe pair site, where Sn atoms are dispersed on FeS2, can also promote the NO3 protonation process, reaching a maximum FE of 96.7% and an NH3 yield of 15.8 mg h−1 cm−2 at −0.5 VRHE.26 All above pioneering experimental results suggest that TMS2 catalysts, especially FeS2, are promising materials for electrocatalytic NH3 synthesis.
 | ||
| Fig. 1 Statistics of ENRR faradaic efficiencies (FEs) of transition metal disulfides (TMS2) with varying applied potentials. Data were extracted from typical experimental reports after 2018.18–20,22–25,27,28 | ||
Due to the electrochemistry-driven water-adsorbate equilibrium,29 the surface states (e.g., the electrochemistry-induced surface coverage and vacancy formation) of an electrocatalyst should be considered before the analysis of electrocatalytic activity. Recent combined experimental and theoretical studies found that under a moderate or high potential (e.g., oxygen reduction and evolution potentials), many transition metal X-ide (TMX) surfaces are precovered by O* or HO*, resulting in a very different electronic structure of a surface or the poisoning of too-reactive sites.30–32 This makes TMXs generally behave very differently from a stoichiometric pristine surface under electrocatalytic conditions. However, surface state analysis is a largely dismissed part of many previous theoretical analyses for electrocatalysis.33 Very recently, we found that only when we consider a more realistic surface coverage of ZrN can we fully understand its superior electrocatalytic oxygen reduction performance with good agreement with experiments.34 Therefore, for ENRR analysis, it is reasonable to hypothesize that in the presence of a low potential, anion vacancies may be directly formed on some TMXs (e.g., TMS2) because protons and electrons can easily combine with the surface and lead to the leaching out of the anions of the material. This may lead to the formation of vacancies, which would significantly change the type of active sites and electronic structures of an ENRR catalyst. Unfortunately, this phenomenon was usually dismissed in previous analyses of either TMX materials or ENRR processes.
Motivated by current stages, herein, we analyze the ENRR performance of TMS2 starting from probing electrochemistry-induced surface states, based on spin-polarized ab initio calculations with van der Waals corrections. Using FeS2 as an initial example, we found an interesting “in situ” generation of S-vacancies under ENRR operating potentials. This potential window is in good agreement with the potential where the highest experimental ENRR FE is located. This suggests that, even if starting with a stoichiometric FeS2 for ENRR, it is still more likely to result in an S-vacancy-containing surface. Furthermore, we found that these “in situ” generated S-vacancies are more active than an original FeS2 surface due to the lower coordination number of an Fe site that leads to stronger N–N adsorption and activation capacity. Finally, we extended this analysis to other TMS2 catalysts (SnS2, MoS2, NiS2, and VS2) and found a similar conclusion. All these analyses lead to good agreement with the experimentally highest ENRR FE-windows of TMS2. Therefore, a comprehensive assessment of the more realistic surface states of TMS2-based catalysts should be considered before theoretical and experimental studies of ENRR. Most importantly, this study proposes that, when designing a promising TMS2 catalyst for ENRR, whether it can form an S-vacancy easily is a key performance indicator that should be carefully evaluated.
Results and discussion
Surface properties are of paramount importance in the comprehension and design of catalysts, yielding characteristics distinctive to those of bulk materials. Grazing-incidence X-ray diffraction verified the prevalence of the FeS2 crystallographic phase (namely, (111), (200), (210), (211), and (311)) exhibiting higher peak intensities (Fig. S1†).35 Furthermore, we analysed the surface energies of these facets. Table S2† shows a comparison of their surface energies, highlighting that the (111) facet has higher thermodynamic stability owing to its lower surface energy, in good agreement with previous relevant experimental observations.27,36,37The calculated surface Pourbaix diagram is a key tool for simulating and describing catalytic surface states under electrocatalytic operating conditions as a function of pH and potential, which indicates the thermodynamic equilibrium surface structure in a certain aqueous environment.38 Under electrocatalytic conditions, pre-coverage with H*, O*, or HO* species generated through water activation can possibly occur on the surface under an oxidizing potential. These coverages exert a substantial influence on the reaction overpotential and surface configuration.39,45 The calculated surface Pourbaix diagram provides a visual representation of thermodynamically stable FeS2(111) (Fig. 2), and the lowest-energy line represents the surface with the lowest free energy under given operating conditions.38 It illustrates the diverse configurations of H*, HO*, and O* coverages across a range of pH and applied potentials, which also includes a stability assessment of various SV coverages. The observed trend reveals the transformation of pristine FeS2(111) into an O-terminated state at highly positive potentials exceeding 1.10 VRHE. Additionally, the single S-vacancy decorated FeS2 has the lowest free energy in the ENRR-preferred potential window (e.g., around and above −0.5 VRHE), indicating that S-vacancies are prone to occur spontaneously. Therefore, electrochemistry can drive the formation of S-vacancies during ENRR. Based on experimental data extracted from previous reports (Fig. 2b), it is easy to find that these experimentally identified high-FE potentials are located in the S-vacancy formation potential range, which means that in situ formed S-vacancy generation is prone to occur on the surface in experiments. The S-vacancy formation window and high activity of ENRR identified on the S-vacancy sites are linked to the experimental potential where a high ENRR FE is located.9,10,12,16,17 The H-coverage subsequently occurs at a potential more negative than −0.5 VRHE, suggesting that the competing HER or H*-poisoning may gradually predominate when the potential becomes too negative. This is also in good agreement with the experimental observation that ENRR-FEs of FeS2-based catalysts will gradually drop when the potential becomes more negative than −0.5 VRHE. These results suggest that the SV-containing surface is more favourable than a pristine FeS2 surface during ENRR, and therefore, it is vital to explore electrocatalytic surface states for guiding and designing superior catalysts. As illustrated in Fig. S2,† AIMD simulations further suggest a high stability of FeS2−x(111)–1SV without significant structural deformation.
 | ||
| Fig. 2 Calculated (a) 1D and (b) 2D surface Pourbaix diagrams of FeS2(111) considering different coverages of SV, O*, H*, and HO*. The experimental potentials at the highest faradaic efficiencies of reported FeS2-based catalysts are plotted for a direct comparison. These experimental data were extracted from ref. 18–20, 22 and 27. | ||
Next, we analyzed the free energy pathways of the ENRR reaction over pristine FeS2 and FeS2−x surfaces (Fig. 3 and S3†). The ENRR is commonly divided into associative and dissociative mechanisms. In the dissociative mechanism, N![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) N cleavage occurs first, followed by the subsequent hydrogenation of the N atom. The direct N2 dissociation is energetically unfavorable due to the N2 endoergicity of 1.73 eV, and thus, it is not discussed in this study.39Fig. 3 displays the ENRR elementary steps of pristine FeS2(111) and FeS2−x(111)–1SVvia the associative mechanism, including alternating and distal pathways. When ENRR follows an alternating pathway, the protons attack two N atoms alternately, and the reaction continuously releases two NH3 molecules. On stoichiometric pristine FeS2(111), Fe serves as the reaction site, and the first hydrogenation step
N cleavage occurs first, followed by the subsequent hydrogenation of the N atom. The direct N2 dissociation is energetically unfavorable due to the N2 endoergicity of 1.73 eV, and thus, it is not discussed in this study.39Fig. 3 displays the ENRR elementary steps of pristine FeS2(111) and FeS2−x(111)–1SVvia the associative mechanism, including alternating and distal pathways. When ENRR follows an alternating pathway, the protons attack two N atoms alternately, and the reaction continuously releases two NH3 molecules. On stoichiometric pristine FeS2(111), Fe serves as the reaction site, and the first hydrogenation step  is the potential-determining step (PDS) with a very endothermic energy (ΔGmax = 2.76 eV). For FeS2−x(111)–1SV, Fe and SV can potentially serve as active sites. The protonation of
is the potential-determining step (PDS) with a very endothermic energy (ΔGmax = 2.76 eV). For FeS2−x(111)–1SV, Fe and SV can potentially serve as active sites. The protonation of  to NNH* is also the PDS along the Fe site, while the energy barrier is much lower (ΔGmax = 1.02 eV). The transition state energy of the first coupled proton–electron transfer is 0.19 eV (Fig. S5†). Notably, the SV site has an excellent ability to fix N2
to NNH* is also the PDS along the Fe site, while the energy barrier is much lower (ΔGmax = 1.02 eV). The transition state energy of the first coupled proton–electron transfer is 0.19 eV (Fig. S5†). Notably, the SV site has an excellent ability to fix N2 but release of the second NH3 molecule is a significantly endothermic step (ΔGmax = 1.73 eV). In the distal pathway, protons are continuously added to the distal N atom of adsorbed N2, and later, the first NH3 molecule is released. Afterward, the other N atom is hydrogenated to generate the second NH3 molecule. For pristine FeS2(111) and FeS2−x(111)–1SV, the PDS and associated ΔGmax remain consistent with those identified in the alternating pathway. However, the reaction
but release of the second NH3 molecule is a significantly endothermic step (ΔGmax = 1.73 eV). In the distal pathway, protons are continuously added to the distal N atom of adsorbed N2, and later, the first NH3 molecule is released. Afterward, the other N atom is hydrogenated to generate the second NH3 molecule. For pristine FeS2(111) and FeS2−x(111)–1SV, the PDS and associated ΔGmax remain consistent with those identified in the alternating pathway. However, the reaction  occurs at the SV site of FeS2−x(111)–1SV, featuring a relatively higher ΔG (0.99 eV). Therefore, the Fe active site of FeS2−x(111)–1SV yields a smoother and lower energy diagram in the alternating pathway. Note that in this study, we followed the reaction mechanisms discussed in some previous studies (e.g., studies by Nørskov and colleagues)40 that the formation and desorption of
occurs at the SV site of FeS2−x(111)–1SV, featuring a relatively higher ΔG (0.99 eV). Therefore, the Fe active site of FeS2−x(111)–1SV yields a smoother and lower energy diagram in the alternating pathway. Note that in this study, we followed the reaction mechanisms discussed in some previous studies (e.g., studies by Nørskov and colleagues)40 that the formation and desorption of  may be coupled into one step. Therefore, based on this mechanism, the rate-determining step may still be the formation of NNH* instead of
may be coupled into one step. Therefore, based on this mechanism, the rate-determining step may still be the formation of NNH* instead of  desorption on SV. After the formation of NH3, NH4+ can be further formed easily, which will provide a further driving force to make NH3 less likely to get stuck on an SV. In addition, as a supplementary calculation, we also tabulated the NH3 desorption energy on the SV site (Table S3†), showing that the energetics of NH3 desorption from
desorption on SV. After the formation of NH3, NH4+ can be further formed easily, which will provide a further driving force to make NH3 less likely to get stuck on an SV. In addition, as a supplementary calculation, we also tabulated the NH3 desorption energy on the SV site (Table S3†), showing that the energetics of NH3 desorption from  are lower than for the formation of NNH*. In addition, Table S4† directly compares the adsorption energies of N2 and NH3, elucidating the feasibility of NH3 desorption in the vicinity of the S-vacancy. Besides, the markedly positive ΔGH* value (1.16 eV) indicates the thermodynamic infeasibility of the competing HER. In addition, solvation effects were tested by developing an explicit model with three water molecules (Fig. S6†). After considering the solvation effect, the energy barrier of the PDS (NN* + H* → NNH*) became 0.98 eV, which is only slightly more negative than that without a solvent (1.02 eV). Therefore, we consider that the solvation effect has no significant influence on the ENRR activity on FeS2−x, which is consistent with a previous study.41 Previous studies considering explicit solvent effects on ENRR suggested that the solvation effects of ENRR may lower theoretical overpotentials by less than 0.1 eV.41 The PDS and energy profiles remain largely unchanged following the interactions exclusively involving H2O. Furthermore, an implicit model is unable to provide accurate results because it cannot consider the H-bonding effect between the adsorbate and solvent; therefore, it was not considered in our study. To further assess the Hubbard-U correction, U values were implemented to investigate the energy barriers to N2 adsorption and PDS (NN* + H* → NNH*) on the identified active site.42 As shown in Table S5†, standard RPBE and RPBE + U led to similar results. In addition, the Material Project database suggests that U correction is not necessary for FeS2.43,44
are lower than for the formation of NNH*. In addition, Table S4† directly compares the adsorption energies of N2 and NH3, elucidating the feasibility of NH3 desorption in the vicinity of the S-vacancy. Besides, the markedly positive ΔGH* value (1.16 eV) indicates the thermodynamic infeasibility of the competing HER. In addition, solvation effects were tested by developing an explicit model with three water molecules (Fig. S6†). After considering the solvation effect, the energy barrier of the PDS (NN* + H* → NNH*) became 0.98 eV, which is only slightly more negative than that without a solvent (1.02 eV). Therefore, we consider that the solvation effect has no significant influence on the ENRR activity on FeS2−x, which is consistent with a previous study.41 Previous studies considering explicit solvent effects on ENRR suggested that the solvation effects of ENRR may lower theoretical overpotentials by less than 0.1 eV.41 The PDS and energy profiles remain largely unchanged following the interactions exclusively involving H2O. Furthermore, an implicit model is unable to provide accurate results because it cannot consider the H-bonding effect between the adsorbate and solvent; therefore, it was not considered in our study. To further assess the Hubbard-U correction, U values were implemented to investigate the energy barriers to N2 adsorption and PDS (NN* + H* → NNH*) on the identified active site.42 As shown in Table S5†, standard RPBE and RPBE + U led to similar results. In addition, the Material Project database suggests that U correction is not necessary for FeS2.43,44
 | ||
| Fig. 3 Calculated Gibbs free energy diagrams of ENRR over FeS2(111) and FeS2−x(111)–1SV surfaces. (a) and (b) demonstrate the energy profiles through the alternating and distal pathways, respectively. The insets show the optimized geometries of the key reaction intermediates. Brown, yellow, blue, and white spheres represent Fe, S, N, and H, respectively. | ||
Based on Fig. 2 and 3, the electrochemical surface states (i.e., the electrochemistry-induced surface coverage) of FeS2 under different pH conditions and applied potentials indicate that highly active S-vacancies are formed under ENRR operating conditions (e.g., around and above −0.50 VRHE). Meanwhile, the experimental potentials of Fe-based catalysts in ENRR at their highest FEs are located in the S-vacancy formation potential range, which means that the in situ generation of S-vacancies is prone to occurring on the surface during ENRR experiments. The DFT-calculated energy profiles further proved that S-vacancies are highly active for ENRR, in contrast to the low theoretical activity on a stoichiometric pristine FeS2 surface. Because N2 fixation is the pivotal step in the entire ENRR process, herein, the projected density of states (PDOS) and charge density difference upon N2 adsorption were calculated to analyze the electronic structure of the  intermediate (Fig. 4).
intermediate (Fig. 4). 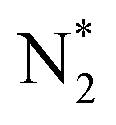 has a strong peak in the deep valence band of N2 on pristine FeS2(111), while the SV formation encourages electron transfer between N2 and FeS2−x(111)–1SV. The robust N2 bonding is weakened as it serves as an electron donor, which in turn can promote N2 fixation and hydrogenation.
has a strong peak in the deep valence band of N2 on pristine FeS2(111), while the SV formation encourages electron transfer between N2 and FeS2−x(111)–1SV. The robust N2 bonding is weakened as it serves as an electron donor, which in turn can promote N2 fixation and hydrogenation.
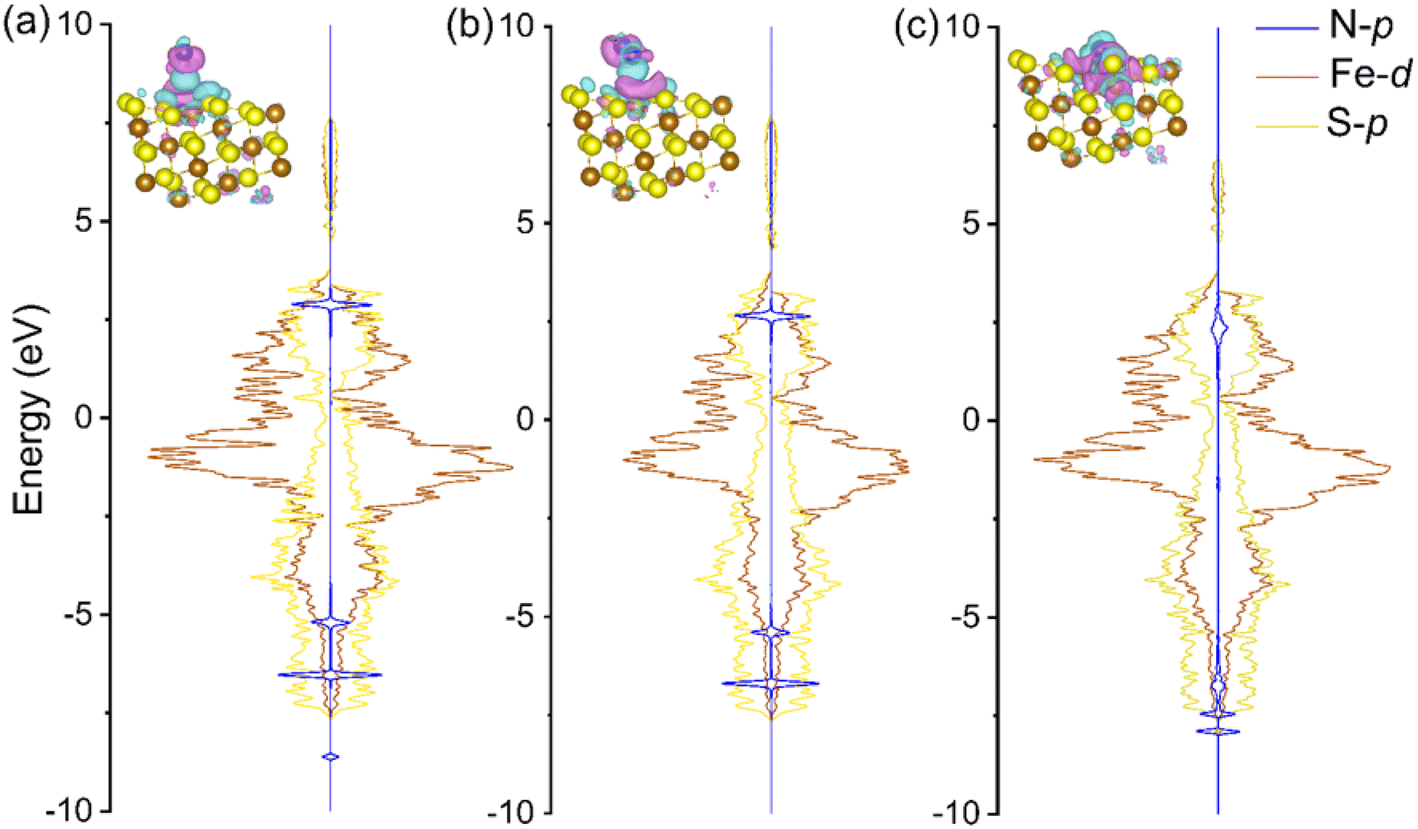 | ||
Fig. 4 Calculated projected density of states (PDOS) of  intermediates on (a) pristine FeS2(111), (b) the Fe site on FeS2−x(111)–1SV, and (c) the SV site on FeS2−x(111)–1SV. The charge density differences upon intermediates on (a) pristine FeS2(111), (b) the Fe site on FeS2−x(111)–1SV, and (c) the SV site on FeS2−x(111)–1SV. The charge density differences upon  adsorption are shown in insets. Brown, yellow, and blue spheres represent Fe, S, and N, respectively. Pink and cyan isosurfaces indicate electron charge accumulation and depletion, respectively. adsorption are shown in insets. Brown, yellow, and blue spheres represent Fe, S, and N, respectively. Pink and cyan isosurfaces indicate electron charge accumulation and depletion, respectively. | ||
Currently, TMS2 catalysts have become a class of popular components for ENRR, due to their excellent electrochemistry and cost-effectiveness. In addition to FeS2(111), other TMS2 catalysts, including MoS2(100), NiS2(210), VS2(001), SnS2(111), and VS2(011), were observed in experiments as primary materials and facets in effective electrochemical NH3 synthesis.23–25,28 Therefore, we further expanded our analysis to these catalysts. Interestingly, as depicted in Fig. 5a, in situ SV generation on all these TMS2 catalysts is energetically favorable within the ENRR potential range, which may regulate the electronic structure and enhance catalytic activity. In either a direct or an indirect way, various theoretical and experimental studies can support the idea that SV sites of TMS2 generally show better ENRR performance than a pristine surface. For example, Chu et al.24 found that N2 adsorption is typically weak on pristine SnS2, while SV formation encourages Mo doping to form an Mo–Sn–Sn trimer site. Such an active site exhibits strong cleavage of the inert NN bond, and the energy barrier is conspicuously reduced with an optimal FE of 20.8% at −0.40 VRHE. Similarly, introducing SV on the VS2 basal plane can diminish the high N2 adsorption barrier on the pristine surface, reducing it from 0.25 to −0.07 eV. Additionally, B-doping could further elevate the activity of SV-enriched surfaces via a synergistic effect. The electron-deficient B-adjacent-unsaturated-V sites achieve an excellent NH3 yield of 55.7 μg h−1 mgcat−1 at −0.4 VRHE and an FE of 16.4% at −0.2 VRHE, with a reduced N2 adsorption energy of −0.59 eV.23 It should be noted that MoS2 is a prominent HER electrocatalyst, while its similar Mo–S linkages with nitrogenases are expected to be active for ENRR. Sun and co-workers showed that MoS2 has high ENRR activity and selectivity even under acid conditions;45 they further reported a remarkable study in which defect-MoS2 attains a significantly high FE of 8.34% and NH3 yield of 29.28 μg h−1 mgcat−1 at −0.4 VRHE, which are significantly higher than its less-defected counterpart under the same potential (FE: 2.18% and NH3 yield: 13.42 μg h−1 mgcat−1).28 Recent studies have further shown that SV-rich MoS2 can effectively facilitate N2 fixation and hydrogenation. For instance, the in-plane defect cluster of MoS2 obtained a remarkable performance, achieving optimal FE and NH3 yield of 16.8% and 13.42 μg h−1 mgcat−1 at −0.3 VRHE, respectively.46 Chen and co-workers also demonstrated a significant enhancement in ENRR performance in MoS2 through SV introduction (NH3 yield: 23.38 μg h−1 mgcat−1 and FE: 17.9% at −0.35 VRHE). These values represent an approximately twofold improvement compared to pristine MoS2.47
 | ||
| Fig. 5 (a) 1D surface Pourbaix diagrams of different transition metal disulfide (TMS2) surfaces. (b) 2D surface Pourbaix diagram of SV formation as a function of pH and potential. The color-coded segments demarcate the potential windows of SV generation on the respective instances of TMS2. The reported experimental operating potentials at their highest NH3 faradaic efficiencies are drawn on the diagram.23–25,28,46–54 | ||
Notably, Fig. 5b and Table S6† demonstrate the operating potentials of TMS2-based catalysts at their highest ENRR-FEs (with data extracted from previous experimental results23–25,28,46–54), which are all consistent with our predicted potential windows for SV formation (Fig. 5a). Note that six cited reports in Fig. 5b employed isotope labeling experiments to confirm the N-source in their NH3 synthesis processes.48 These findings can be further supported by the experimental evidence that SnS2 was significantly reduced after ENRR,50 and some experiments on metal sulfides showed an increased ENRR performance after the first few cycles.51,54,55 The occupations of H2O, HO, and O on the S-vacancy sites were further analyzed. Fig. S6† shows that SV is still the most stable surface state, which is less likely to be further occupied by H2O, OH, or O. Table S7† further confirms that the adsorption free energy of N2 is much more negative than the formation free energies of H2O*, HO*, and O* on FeS2−x, indicating the stronger adsorption capacity of N2 for ENRR. All in all, our analyses prove that the in situ generation of SV is facile on pristine TMS2 catalysts under ENRR operating conditions, which holds great promise for facilitating N2 fixation and hydrogenation in ENRR. Specifically, even if there are no S-vacancies after the synthesis of TMS2, these highly active S-vacancies will still be generated simultaneously under ENRR operating conditions. Based on our surface Pourbaix diagrams (Fig. 2 and 5), surface S-vacancies are formed on aforementioned TMSs uner operating conditions that are favorable for ENRR, which are more realistic surfaces under electrochemical conditions. Currently, most theoretical studies have either considered a pristine surface as the active site of TMS2 or have directly created artificial S-vacancies for theoretical activity analysis, which might lead to high uncertainty in the analysis because they generally dismiss the fact that S-vacancies can easily be generated under ENRR operating conditions. Therefore, it is crucial to investigate a more realistic surface state for a TMS2 ENRR catalyst, which will ensure a deeper understanding of catalytic performance and reaction mechanisms. Our study also provides guidance for designing promising TMS2 catalysts for ENRR, where the capacity to form S-vacancies is a key performance indicator. TMS2 surfaces are prone to forming surface S-vacancies spontaneously under ENRR operating conditions, and better N2 adsorption and reduction capacity is observed. However, it should be noted that, when designing a high-performance TMS2 catalyst for ENRR, the bonding strength of its surface-S should neither be too strong nor too weak. S-binding that is too strong will lead to difficulty in forming highly active S-vacancies, while S-binding that is too weak will mean the materials are not stable under the operating conditions.
Conclusion
In summary, using ab initio calculations, we have analyzed the origin of the ENRR activity of pristine TMS2 catalysts, starting by probing their electrochemistry-induced surface states. Our calculated surface Pourbaix diagrams found that S-vacancies can be generated directly over a pristine TMS2 surface under ENRR operating conditions. Using FeS2 as a typical example, we found that this electrochemistry-driven “in situ” generation of S-vacancies possesses a significantly higher ENRR activity than stoichiometric pristine FeS2 due to the stronger N–N adsorption and activation capacity of a lower-coordination-number S-vacancy site. This finding is in excellent agreement with the experimental observations published in recent years in terms of potential windows reaching the maximum FEs. Expanding the analysis to other typical TMS2 which had shown promising ENRR performance in recent experimental literature (e.g., SnS2, MoS2, NiS2, and VS2), we found that such an “in situ” S-vacancy generation phenomenon is universal under ENRR potentials, with the results generally in good agreement with the experimental observations reported to date. We concluded that, though S-vacancy engineering during synthesis is a promising strategy to enhance the ENRR performance of TMS2 catalysts, the “in situ” generation of S-vacancies will also endow pristine TMS2 with a measurable ENRR performance. This study shows that the electrochemistry-induced surface states of ENRR catalysts should not be dismissed before analyzing the activity of an ENRR catalyst. Most importantly, when designing a promising TMS2 catalyst for ENRR, its capacity to form S-vacancies is a key performance indicator that needs to be analyzed.Methods
Spin-polarized DFT calculations were employed to optimize structural geometries and calculate free energy changes via the Vienna ab initio simulation package.56 The generalized gradient approximation (GGA) method with the revised Perdew–Burke–Ernzerhof (RPBE) functional was used to treat exchange–correlation interactions.57 The projected augmented-wave method was applied to describe core electrons, and valence electrons were described via Kohn–Sham wave functions.58,59 A 400 eV energy cutoff was adopted, and k-points in the Brillouin zone were sampled with a 3 × 3 × 1 grid by the Monkhorst–Pack scheme.60 A 15 Å vacuum space was built to prohibit interactions between periodic units. All geometries were optimized when the residual force was lower than 0.02 eV Å−1, and the energy convergence was set to 1 × 10−4 eV. The binding interactions between nitrogen-containing species and the catalyst were corrected by the DFT-D3 method.61Ab initio molecular dynamics (AIMD) simulations were employed to analyse the stability of the vacancy-containing surface at 300 K with a time step of 0.2 fs and an overall time of 10 ps. The climbing-image nudged elastic band (CI-NEB) method was employed to calculate transition states.62Surface energy (Esurf) serves as a critical metric for stability assessment, where a lower Esurf is indicative of higher stability. The following equation was employed to calculate Esurf:63,64
 | (1) |
In this study, the computational hydrogen electrode (CHE) method was employed to establish the surface Pourbaix diagram as a function of pH and potential.57 The water dissociation equilibrium during electrochemical reaction conditions is described by eqn (2) because a pristine surface can be covered by O*, HO*, and H*:30,38,48,65,66
 | (2) |
For the surface Pourbaix diagrams, the free energy of each surface state was calculated viaeqn (3):
 | (3) |
The stability of the SV-containing surface was analyzed in the surface Pourbaix diagram viaeqn (4):
 | (4) |
The Gibbs free energy change of each hydrogenation step in ENRR was calculated via the following equation:
| ΔG = ΔEDFT + ΔEZPE − TΔS, | (5) |
Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the JSPS KAKENHI (No. JP23K13703, JP23KF0102, JP19H05814, and JP23H00301), AIMR Fusion Research & Advanced Target Project, Ensemble Grant for Early Career Researchers 2023, JST PRESTO Grant Number: JPMJPR2274, and Tanaka Kikinzoku Memorial Foundation. We acknowledge the Center for Computational Materials Science, Institute for Materials Research, Tohoku University for the use of MASAMUNE-IMR (202212-SCKXX-0204) and Institute for Solid State Physics (ISSP) at the University of Tokyo for the use of their supercomputers.References
- D. Bao, Q. Zhang, F. L. Meng, H. X. Zhong, M. M. Shi, Y. Zhang, J. M. Yan, Q. Jiang and X. B. Zhang, Adv. Mater., 2017, 29, 1604799 CrossRef.
- S. Wu, N. Salmon, M. M. J. Li, R. Bañares-Alcántara and S. C. E. Tsang, ACS Energy Lett., 2022, 7, 1021–1033 CrossRef CAS.
- D. Wang, L. M. Azofra, M. Harb, L. Cavallo, X. Zhang, B. H. R. Suryanto and D. R. MacFarlane, ChemSusChem, 2018, 11, 3416–3422 CrossRef CAS PubMed.
- C. Liu, Q. Li, J. Zhang, Y. Jin, D. R. MacFarlane and C. Sun, J. Phys. Chem. C, 2018, 122, 25268–25273 CrossRef CAS.
- B. Yu, H. Li, J. White, S. Donne, J. Yi, S. Xi, Y. Fu, G. Henkelman, H. Yu, Z. Chen and T. Ma, Adv. Funct. Mater., 2019, 30, 1905665 CrossRef.
- Q. Li, C. Liu, S. Qiu, F. Zhou, L. He, X. Zhang and C. Sun, J. Mater. Chem. A, 2019, 7, 21507–21513 RSC.
- T. Wang, Z. Guo, X. Zhang, Q. Li, A. Yu, C. Wu and C. Sun, J. Mater. Sci. Technol., 2023, 140, 121–134 CrossRef CAS.
- Y. Fu, Y. Liao, P. Li, H. Li, S. Jiang, H. Huang, W. Sun, T. Li, H. Yu, K. Li, H. Li, B. Jia and T. Ma, Coord. Chem. Rev., 2022, 460, 214468 CrossRef CAS.
- Y. Yao, S. Zhu, H. Wang, H. Li and M. Shao, J. Am. Chem. Soc., 2018, 140, 1496–1501 CrossRef CAS PubMed.
- J. Wang, L. Yu, L. Hu, G. Chen, H. Xin and X. Feng, Nat. Commun., 2018, 9, 1795 CrossRef PubMed.
- H. M. Liu, S. H. Han, Y. Zhao, Y. Y. Zhu, X. L. Tian, J. H. Zeng, J. X. Jiang, B. Y. Xia and Y. Chen, J. Mater. Chem. A, 2018, 6, 3211–3217 RSC.
- O. Einsle and D. C. Rees, Chem. Rev., 2020, 120, 4969–5004 CrossRef CAS PubMed.
- J. Humphreys, R. Lan and S. Tao, Adv. Energy Sustainability Res., 2020, 2, 2000043 CrossRef.
- M. I. Ahmed, L. J. Arachchige, Z. Su, D. B. Hibbert, C. Sun and C. Zhao, ACS Catal., 2022, 12, 1443–1451 CrossRef CAS.
- F. He, Z. Li, S. Shi, W. Xu, H. Sheng, Y. Gu, Y. Jiang and B. Xi, Environ. Sci. Technol., 2018, 52, 8627–8637 CrossRef CAS PubMed.
- J. Xu, Y. Wang, C. Weng, W. Bai, Y. Jiao, R. Kaegi and G. V. Lowry, Environ. Sci. Technol., 2019, 53, 5936–5945 CrossRef CAS.
- H. Li, W. Yang, C. Wu and J. Xu, Phys. Chem. Chem. Phys., 2021, 23, 13971–13976 RSC.
- L. Gao, C. Guo, M. Zhao, H. Yang, X. Ma, C. Liu, X. Liu, X. Sun and Q. Wei, ACS Appl. Mater. Interfaces, 2021, 13, 50027–50036 CrossRef CAS PubMed.
- D. Feng, X. Zhang, Y. Sun and T. Ma, Nano Mater. Sci., 2020, 2, 132–139 CrossRef.
- Z. Feng, G. Li, X. Wang, C. J. Gómez-García, J. Xin, H. Ma, H. Pang and K. Gao, Chem. Eng. J., 2022, 445, 136797 CrossRef CAS.
- M. Yang, Z. Jin, C. Wang, X. Cao, X. Wang, H. Ma, H. Pang, L. Tan and G. Yang, ACS Appl. Mater. Interfaces, 2021, 13, 55040–55050 CrossRef CAS.
- H. B. Wang, J. Q. Wang, R. Zhang, C. Q. Cheng, K. W. Qiu, Y. J. Yang, J. Mao, H. Liu, M. Du, C. K. Dong and X. W. Du, ACS Catal., 2020, 10, 4914–4921 CrossRef CAS.
- Q. Li, Y. Guo, Y. Tian, W. Liu and K. Chu, J. Mater. Chem. A, 2020, 8, 16195–16202 RSC.
- K. Chu, J. Wang, Y. P. Liu, Q. Q. Li and Y. L. Guo, J. Mater. Chem. A, 2020, 8, 7117–7124 RSC.
- M. Zhao, C. Guo, L. Gao, X. Kuang, H. Yang, X. Ma, C. Liu, X. Liu, X. Sun and Q. Wei, Inorg. Chem. Front., 2021, 8, 3266–3272 RSC.
- G. Zhang, F. Wang, K. Chen, J. Kang and K. Chu, Adv. Funct. Mater., 2023, 2305372 Search PubMed.
- H. Du, C. Yang, W. Pu, L. Zeng and J. Gong, ACS Sustain. Chem. Eng., 2020, 8, 10572–10580 CrossRef CAS.
- X. Li, T. Li, Y. Ma, Q. Wei, W. Qiu, H. Guo, X. Shi, P. Zhang, A. M. Asiri, L. Chen, B. Tang and X. Sun, Adv. Energy Mater., 2018, 8, 1801357 CrossRef.
- V. Sumaria, D. Krishnamurthy and V. Viswanathan, ACS Catal., 2018, 8, 9034–9042 CrossRef CAS.
- H. Liu, X. Jia, A. Cao, L. Wei, C. D'agostino and H. Li, J. Chem. Phys., 2023, 12, 158 Search PubMed.
- S. Pan, H. Li, D. Liu, R. Huang, X. Pan, D. Ren, J. Li, M. Shakouri, Q. Zhang, M. Wang, C. Wei, L. Mai, B. Zhang, Y. Zhao, Z. Wang, M. Graetzel and X. Zhang, Nat. Commun., 2022, 13, 2294 CrossRef CAS PubMed.
- R. Zhao, Z. Chen, Q. Li, X. Wang, Y. Tang, G. Fu, H. Li, J. M. Lee and S. Huang, Chem Catal., 2022, 2, 3590–3606 CrossRef CAS.
- W. Yang, Z. Jia, B. Zhou, L. Wei, Z. Gao and H. Li, Commun. Chem., 2023, 6, 6 CrossRef CAS PubMed.
- H. Liu, D. Zhang, S. M. Holmes, C. D'Agostino and H. Li, Chem. Sci., 2023, 14, 9000–9009 RSC.
- S. Kment, H. Kmentova, A. Sarkar, R. J. Soukup, N. J. Ianno, D. Sekora, J. Olejnicek, P. Ksirova, J. Krysa, Z. Remes and Z. Hubicka, J. Alloys Compd., 2014, 607, 169–176 CrossRef CAS.
- M. Cabán-Acevedo, D. Liang, K. S. Chew, J. P. DeGrave, N. S. Kaiser and S. Jin, ACS Nano, 2013, 7, 1731–1739 CrossRef PubMed.
- A. B. Puthirath, A. P. Balan, E. F. Oliveira, V. Sreepal, F. C. R. Hernandez, G. Gao, N. Chakingal, L. M. Sassi, P. Thibeorchews, G. Costin, R. Vajtai, D. S. Galvao, R. R. Nair and P. M. Ajayan, J. Phys. Chem. C, 2021, 125, 18927–18935 CrossRef CAS.
- H. A. Hansen, J. Rossmeisl and J. K. Nørskov, Phys. Chem. Chem. Phys., 2008, 10, 3722–3730 RSC.
- C. Yang, Y. Zhu, J. Liu, Y. Qin, H. Wang, H. Liu, Y. Chen, Z. Zhang and W. Hu, Nano Energy, 2020, 77, 105126 CrossRef CAS.
- A. R. Singh, B. A. Rohr, M. J. Statt, J. A. Schwalbe, M. Cargnello and J. K. Nørskov, ACS Catal., 2019, 9, 8316–8324 CrossRef CAS.
- J. H. Montoya, C. Tsai, A. Vojvodic and J. K. Norskov, ChemSusChem, 2015, 8, 2180–2186 CrossRef CAS PubMed.
- X. Wen, Y. Liang, P. Bai, B. Luo, T. Fang, L. Yue, T. An, W. Song and S. Zheng, Phys. B, 2017, 525, 119–126 CrossRef CAS.
- A. Kjekshus and T. Rakke, Acta Chem. Scand., Ser. A, 1975, 29, 443–452 CrossRef.
- P. Bayliss, Am. Mineral., 1977, 62, 1168–1172 CAS.
- L. Zhang, X. Ji, X. Ren, Y. Ma, X. Shi, Z. Tian, A. M. Asiri, L. Chen, B. Tang and X. Sun, Adv. Mater., 2018, 30, e1800191 CrossRef PubMed.
- W. Liao, K. Xie, L. Liu, X. Wang, Y. Luo, S. Liang, F. Liu and L. Jiang, J. Energy Chem., 2021, 62, 359–366 CrossRef CAS.
- M. You, S. Yi, X. Hou, Z. Wang, H. Ji, L. Zhang, Y. Wang, Z. Zhang and D. Chen, J. Colloid Interface Sci., 2021, 599, 849–856 CrossRef CAS PubMed.
- Y. Liu, M. Han, Q. Xiong, S. Zhang, C. Zhao, W. Gong, G. Wang, H. Zhang and H. Zhao, Adv. Energy Mater., 2019, 9, 1803935 CrossRef.
- K. Chu, Y. P. Liu, Y. B. Li, Y. L. Guo and Y. Tian, ACS Appl. Mater. Interfaces, 2020, 12, 7081–7090 CrossRef CAS PubMed.
- P. Li, W. Fu, P. Zhuang, Y. Cao, C. Tang, A. B. Watson, P. Dong, J. Shen and M. Ye, Small, 2019, 15, e1902535 CrossRef.
- X. Li, X. Ren, X. Liu, J. Zhao, X. Sun, Y. Zhang, X. Kuang, T. Yan, Q. Wei and D. Wu, J. Mater. Chem. A, 2019, 7, 2524–2528 RSC.
- X. W. Lv, X. L. Liu, Y. J. Suo, Y. P. Liu and Z. Y. Yuan, ACS Nano, 2021, 15, 12109–12118 CrossRef CAS PubMed.
- L. Niu, D. Wang, K. Xu, W. Hao, L. An, Z. Kang and Z. Sun, Nano Res., 2021, 14, 4093–4099 CrossRef CAS.
- L. Zhao, R. Zhao, Y. Zhou, X. Wang, X. Chi, Y. Xiong, C. Li, Y. Zhao, H. Wang, Z. Yang and Y. M. Yan, J. Mater. Chem. A, 2021, 9, 24985–24992 RSC.
- J. Wang, H. Nan, Y. Tian and K. Chu, ACS Sustain. Chem. Eng., 2020, 8, 12733–12740 CrossRef CAS.
- G. Kresse and J. Furthmüller, Phys. Rev. B: Condens. Matter Mater. Phys., 1996, 16, 11169 CrossRef PubMed.
- J. P. Perdew, K. Burke and M. Ernzerhof, Phys. Rev. Lett., 1996, 18, 3865 CrossRef PubMed.
- W. Kohn and L. J. Sham, Phys. Rev., 1965, 140, A1133–A1138 CrossRef.
- P. E. Blochl, Phys. Rev. B: Condens. Matter Mater. Phys., 1994, 50, 17953–17979 CrossRef PubMed.
- H. J. Monkhorst and J. D. Pack, Phys. Rev. B: Solid State, 1976, 13, 5188–5192 CrossRef.
- S. Grimme, J. Antony, S. Ehrlich and H. Krieg, J. Chem. Phys., 2010, 132, 154104 CrossRef PubMed.
- G. Henkelman, B. P. Uberuaga and H. Jónsson, J. Chem. Phys., 2000, 113, 9901–9904 CrossRef CAS.
- M. Li, H. Li, X. Jiang, M. Jiang, X. Zhan, G. Fu, J. M. Lee and Y. Tang, J. Mater. Chem. A, 2021, 9, 2999–3006 RSC.
- W. Xiong, M. Zhou, X. Huang, W. Yang, D. Zhang, Y. Lv and H. Li, Chemistry, 2022, 28, e202200779 CrossRef CAS PubMed.
- A. E. Russell, Phys. Chem. Chem. Phys., 2008, 10, 3607–3608 RSC.
- J. K. Nørskov, J. Rossmeisl, A. Logadottir, L. Lindqvist, J. R. Kitchin, T. Bligaard and H. Jonsson, J. Phys. Chem. B, 2004, 108, 17886–17892 CrossRef.
- R. D. Johnson III, NIST 101. Computational Chemistry Comparison and Benchmark Database, 1999 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4ta00307a |
| This journal is © The Royal Society of Chemistry 2024 |