
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Cutting off the upstream and downstream costs for CO2 electroreduction by upcycling fermentation emissions into ethanol†
Ruofan
Sun‡
ab,
Jiwu
Zhao‡
ab and
Xu
Lu
 *ab
*ab
aCCRC, Division of Physical Science and Engineering (PSE), King Abdullah University of Science and Technology (KAUST), Thuwal, 23955-6900, Kingdom of Saudi Arabia. E-mail: xu.lu@kaust.edu.sa
bKAUST Solar Center (KSC), PSE, KAUST, Kingdom of Saudi Arabia
First published on 22nd February 2024
Abstract
Electrochemical reduction of CO2 (CO2RR), when powered by renewables, opens up a new avenue to mitigate the greenhouse gas while producing value sustainably. Nevertheless, this technology has been largely limited by the high costs of the upstream CO2 feed and downstream product separation. Here we report a hybrid bio-electrochemical system, integrating yeast fermentation with CO2RR in one single cell, that upcycles the fermentation-emitted CO2 into ethanol. We engineer a CuO–Ag tandem electrocatalyst with rationally designed CuO–Ag interfaces that pose minimal impact on the yeast, while efficiently converting CO2 into ethanol against side reactions, such as hydrogen evolution and glucose reduction. We showcase the win–win model enabled by this hybrid system—the CO2RR cost can be cut by 17.8% because the fermentation process provides a free, high-purity CO2 source and free ethanol distillation and in return, the CO2RR reduces the CO2 emissions of fermentation and increases the final ethanol product concentration. This proof-of-concept procedure sheds light on a tempting possibility for a cost-effective CO2 value chain.
 Xu Lu | Dr Xu Lu obtained his BS and PhD degrees from the Department of Mechanical Engineering, University of Hong Kong in 2012 and 2017, respectively. He was then trained as a postdoctoral fellow in the Energy Sciences Institute, Yale University. Dr Lu joined King Abdullah University of Science and Technology (KAUST) as an Assistant Professor in Mechanical Engineering in March 2021. He established the Low-carbon Energy Conversion and Storage (LECS) Laboratory, which focuses on electrochemical upcycling of industrial high-pressure CO2. So far, the LECS Laboratory has published original research articles in Nature Communications (3), Journal of the American Chemical Society, Angewandte Chemie, Chemical Engineering Journal, Journal of Energy Chemistry, etc., and generated two US provisional patents. The LECS Laboratory is also developing kilowatt-scale electrolyzers with industrial partners such as ACWA Power and Saudi Aramco. |
Introduction
Renewable-driven production of value-added chemicals via electrochemical reduction of CO2 (CO2RR) holds promise to simultaneously benefit environmental sustainability and realize a low carbon footprint in manufacturing industry.1–3 To date, the field has demonstrated high-yield and efficient production of multiple CO2RR products, including carbon monoxide (CO), formic acid/formate (HCOOH or HCOO−), methane (CH4), methanol (CH3OH), ethylene (C2H4), acetic acid/acetate (CH3COOH or CH3COO−), and ethanol (CH3CH2OH).4–6 Converting CO2 to ethanol is of particular importance because ethanol is a high-energy-density fuel additive that can facilitate cleaner combustion compared to conventional gasoline, such as the E85 ethanol fuel.7–9 Ethanol also serves as the key chemical in pharmaceutical and cosmetic industries.10,11 According to a report by GlobeNewswire, ethanol consumption in the European Union was estimated at approximately 4.8 million tons in 2021.12Despite major advances, CO2RR technology has been greatly challenged by its costly upstream and downstream processes.13–15 On the one hand, the upstream feed of CO2RR should be high-purity CO2, due to the substantial energy and capital expenses required for direct electroreduction of impure CO2.16–18 The prevailing way to generate pure CO2 gas is to capture CO2 from point sources or air. Recent studies have described the possibility of integrating CO2 capture with CO2RR.19–21 However, for now at least, CO2 capture and recycling still incurs a high cost, which accounts to 9.3% of the capital expenditure (CapEx) and 8.3% of the operating expense (OpEx).22–24 On the other hand, residue CO2 and the electrolyte solution hold a large portion of the CO2RR effluent, and this necessitates downstream separation of products, which is another constraint of CO2RR. Taking ethanol as an example again, the commonly used distillation apparatus not only leads to significant energy compensation, but also jeopardizes the cost effectiveness of the process—contributing approximately 3.9% CapEx and 2.3% OpEx of the overall CO2RR process.13,24,25
Bio-ethanol, produced from biomass through yeast fermentation, is the world's most popular ethanol supply, with a market size of $46.18 billion in 2022.26,27 However, a typical bio-fermentation process produces bio-ethanol via reaction in an anaerobic environment:
 | (1) |
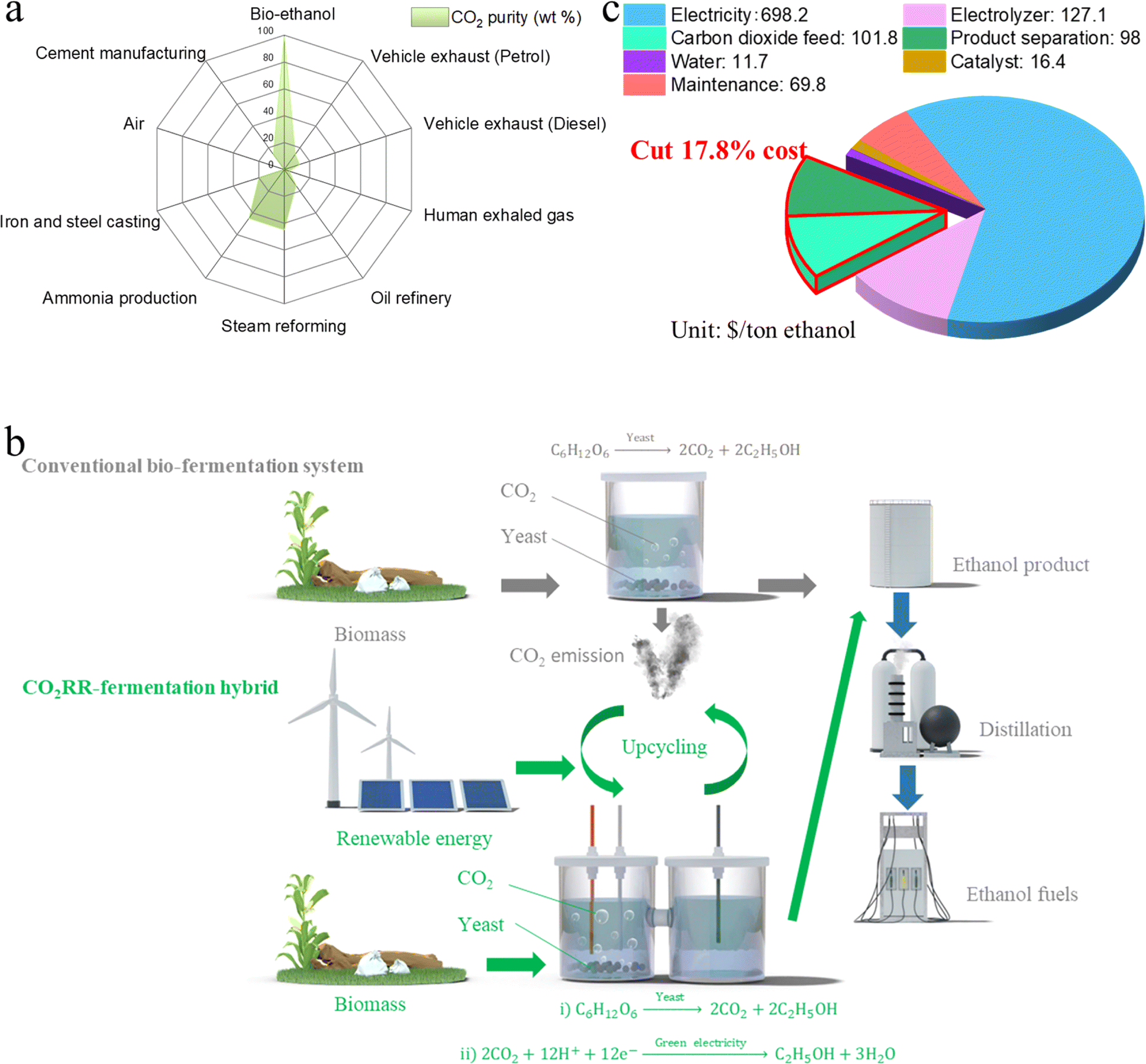 | ||
| Fig. 1 Schematics of TEA and the proposed hybrid system. (a) CO2 purity (wt%) of various point sources and ambient air. (b) Schematic illustration of the fermentation–CO2RR hybrid system. Bio-fermentation emits CO2, which is upcycled into ethanol via in situ electrochemical conversion. This ethanol produced by the CO2RR is then distilled together with the fermentation-generated bio-ethanol without extra cost. (c) Techno-economic analysis (TEA) of the proposed bio-electrochemical system vs. the conventional CO2RR process for the production of ethanol. Our system saves on the costs of (i) acquiring high-purity CO2 feed and (ii) separating the final product, thereby cutting the CO2RR cost by at least 17.8%. | ||
Here we propose a fermentation–CO2RR hybrid that electrochemically converts high-purity fermentation-generated CO2 into ethanol, which is then distilled together with the fermentation-produced bio-ethanol without extra cost. We tailor a CuO–Ag tandem catalyst with CuO–Ag interfaces that pose minimal influence on yeast activity while maintaining a reasonable faradaic efficiency (FE) for CO2-to-ethanol conversion. In situ Raman studies show that the role of the CO intermediate on the CuO–Ag surface accelerates the C–C coupling process, reversibly suppressing side reactions such as hydrogen evolution and glucose reduction. Techno-economic analysis (TEA) indicates that our system not only cuts off the upstream and downstream expenses of CO2RR (that is, around 17.8% of the total CO2RR cost), but also benefits the fermentation by reducing CO2 emissions. This proof-of-concept system provides the possibility to address major bottlenecks for both CO2RR and fermentation.
Results
Pathway design
CO2RR suffers from high costs of the (i) upstream CO2 feed and (ii) downstream product separation, making it challenging for commercialization. We therefore propose a fermentation–CO2RR hybrid for CO2RR to take a free ride on the bio-fermentation process, which affords the possibility of cutting the two major costs of CO2RR, by supplying high-purity CO2 and providing free ethanol distillation. The proposed bio-electrochemical system (Fig. 1b) realizes a greener production of ethanol via a three-step pathway as below.(i) Biomass fermentation as catalyzed by yeast, where ethanol is generated with high-purity CO2 emission:
 | (2) |
(ii) CO2RR powered by renewables, where the CO2 emission is upcycled into ethanol:
 | (3) |
(iii) Ethanol distillation for the delivery of final product.
To confirm the cost effectiveness of the fermentation–CO2RR hybrid system, we performed TEA for CO2 electroreduction to ethanol, assuming a base electricity cost of 0.02 $ per kW h, in line with the target set by US Department of Energy (DOE) for the year 2030.33 TEA revealed that the CO2 feed and the ethanol distillation accounted for 9.1% and 8.7% of the total CO2RR cost with an operating current density of 200 mA cm−2 and ethanol FE of 50%—that is, 17.8% of the expense can be saved by our proposed system compared to a stand-alone CO2RR device (Fig. 1c).
To realize this proof-of-concept system, we first need to validate the CO2 output from fermentation. We used 0.1 M glucose solution, as is commonly used in literature, as the fermentation feedstock for yeast to generate ethanol.34,35 Despite the fact that liquids such as glycerol and organic acids may exist as side products, CO2 was the only gas product from anaerobic bio-ethanol fermentation, as analyzed by gas chromatography (GC)—that is, almost 100% CO2 purity (Fig. 2a).36 After 25 h of fermentation, CO2 became saturated, and the saturation state lasted for at least 500 h (Fig. 2b). This indicated the capability of the fermentation to supply sufficient CO2 to CO2RR over a prolonged time. The pH value of the fermentation broth remained stable at 5.83 (Fig. 2c), which provides a weakly acidic environment that is beneficial to a high-carbon utilization efficiency during CO2RR because the otherwise neutral or alkaline environment at the cathode–electrolyte interface may lead to carbonate formation.37–39 These observations authenticated the idea of feeding the fermentation CO2 emissions to the CO2RR, and motivated us to conduct the subsequent electrochemical experiments.
 | ||
| Fig. 2 Fermentation emissions and broth condition. (a) Gas chromatography of fermentation-emitted CO2. CO2 is the only gas product. (b) Amount of fermentation-emitted CO2 and (c) pH value of the fermentation broth over time. | ||
CuO–Ag tandem catalyst for the fermentation–CO2RR hybrid
Catalyst design strategies for CO2RR to ethanol have been extensively studied, such as surface control, oxide modulation, oxophilicity engineering, etc.40–42 Recently, tandem catalysts, especially Cu–Ag tandems, have demonstrated the capability of sequentially catalyzing CO2-to-CO and CO-to-C2 at reaction rates approaching industrial relevance.43,44 Other homogeneously alloyed Cu-based bimetallic nanoparticles, as well as segmented tandem electrodes for increased CO coverage, can also enhance the CO2-to-ethanol conversion.45,46 For instance, CoPc@HC/Cu tandem electrode in acid CO2RR exhibited a C2+ FE of 90% and single-pass CO2 conversion efficiency of 76%.47 We therefore sought to explore the possibility of designing a CuO–Ag tandem catalyst for our system (Fig. 3a). In brief, an Ag layer was first deposited on a carbon paper, followed by shadow mask-based Cu deposition to create 40 CuO–Ag interfaces in a 0.5 cm × 2 cm area. Of note, interface numbers higher than 40 were not prepared due to instrumentation constraints. Then, the electrode underwent galvanostatic anodic oxidation to form the final CuO–Ag tandem (Fig. 3b and S1†). The CuO–Ag interfaces were confirmed by transmission electron microscopy (TEM), electron energy loss spectroscopy (EELS), and energy-dispersive spectroscopy (EDS). Cu(111) and Ag(111) facets were delineated (Fig. 3c). EELS revealed the interface structure at the nanoscale (Fig. 3d), while EDS uncovered the CuO–Ag at microscale (Fig. S2†). X-ray diffraction (XRD) patterns of the Cu segment of the as-prepared tandem electrode showed that CuO(110), CuO(111), and CuO (022) peaks increased during electrochemical oxidation, while those of Cu(111) and Cu(200) decreased over time (Fig. 3e), which implies the formation of CuO.48 This was confirmed using Raman spectroscopy, which showed the characteristic peaks of CuO at 282, 330, and 616 cm−1. Consistent with the literature, CuO exhibited a nanoplate morphology, while Ag appeared as nanoparticles (Fig. 3g and S3†).49 | ||
| Fig. 3 CuO–Ag tandem catalyst. Schematic of (a) the CuO–Ag tandem electrode synthesis and (b) the CuO–Ag interfaces. (c) HR-TEM and (d) EELS elemental mapping of the CuO–Ag interface. (e) XRD patterns of CuO–Ag over the electrochemical oxidation time. (f) Raman spectroscopy and (g) SEM of the CuO segment after oxidation. | ||
We then retrofitted a conventional fermentation system into an H-type hybrid cell that is compatible with CO2RR—the cathode compartment contained 0.1 M glucose (a typical fermentation broth) and the anode compartment was filled with 0.5 M KHCO3 aqueous solution (Fig. 4a). Nevertheless, the fermentation broth cannot provide the necessary electrochemical reaction conditions—the low electrolyte concentration resulted in sluggish adsorption of OH− on the catalyst surface, and increased both the charge transfer resistance (Rct) and the electrical double layer (EDL) thickness.50,51 Consequently, the CO2RR current density was limited below 8.85 mA cm−2 at cathode potentials as negative as −1.6 V vs. the reversible hydrogen electrode (RHE) (Fig. 4b). To accommodate a reasonably high CO2RR reactivity, we attempted to add the most used cation, K+, into the fermentation broth, such as K2CO3, KCl, and KHCO3.52–54 At a cathode potential of −1.6 V vs. RHE, the current density reached −181, −152, and −161 mA cm−2 in 0.5 M K2CO3, 0.5 M KCl, and 0.5 M KHCO3 glucose solutions, respectively, which is significantly higher than those in 0.1 M glucose, as suggested by linear sweep voltammetry (LSV) curves (Fig. 4b). However, the addition of carbonate and bicarbonate anions resulted in an adverse impact on fermentation by tuning the pH towards alkaline, which was unfavorable for yeast activity,55 whereas adding KCl maintained the fermentation broth as a weak acid to facilitate the fermentation process (Fig. S4†).56 This was further confirmed by weighing the fermentation CO2 emissions, where 0.5 M KCl merely changed the amount of yeast-generated CO2, while in the case of K2CO3 and KHCO3 obvious degradation of yeast activity was shown (Fig. 4c).57 Therefore, we selected 0.1 M glucose aqueous solution (0.5 M KCl) as the catholyte for the fermentation–CO2RR hybrid cell.
 | ||
| Fig. 4 CO2RR performance in the fermentation–CO2RR hybrid cell. (a) Photo of the fermentation–CO2RR hybrid cell. (b) LSV curves of the CuO–Ag catalyst in different solutions. (c) Amount of fermentation CO2 emissions in various glucose solutions. (d) Ethanol FEs and partial current densities with different CuO–Ag interface numbers (0, 10, 20, 30, and 40) in 0.1 M glucose (0.5 M KCl). | ||
Rationally, more CuO–Ag interfaces in a fixed area should benefit the CO2-to-CO and CO-to-ethanol tandem reaction.58,59 To confirm this, we compared CuO–Ag interface numbers of 0 (i.e. CuO), 10, 20, 30, and 40. It was observed that increasing the interface number resulted in higher ethanol selectivity; at −0.87 V vs. RHE, the ethanol FE was improved from 14.4% on CuO to 27.7% on CuO–Ag (20), and eventually reached 42.7% on CuO–Ag (40). Similar phenomena hold for the ethanol productivity; at −1.27 V vs. RHE, the partial current density of ethanol was −8.4 mA cm−2 on CuO, whereas it gradually increased to −19.6 mA cm−2 on CuO–Ag (40) under the same conditions (Fig. 4d).
Understanding the enhanced ethanol production for proof-of-concept
To gain insights into the improved ethanol yield at higher CuO–Ag interface numbers, in situ studies and density functional theory (DFT) calculations were conducted. Time-resolved X-ray diffractograms (Fig. S5†) revealed that CuO was reduced to Cu(111) and Cu(200) under CO2RR condition, whereas the diffraction peaks of Ag barely changed, which is consistent with the X-ray photoelectron spectroscopy (XPS) results (Fig. S6†). This indicated that metallic Cu and Ag served as the dominant active sites during electrolysis, and prepared us to probe the key species distributions on the catalyst surface using in situ Raman spectroscopy. As mentioned above, the conversion of CO2 to ethanol on a tandem catalyst proceeded via two steps: (i) CO formation, as catalyzed by Ag (Fig. S7 and S8†) and (ii) C–C coupling, as catalyzed by Cu. Three regions in the Raman spectra were associated with the CO intermediates on the surface at negative potentials—the band at around 280 cm−1 referred to the Cu–CO frustrated rotation mode,58,60,61 the band at 360 cm−1 was related to the Cu–CO stretch mode,60,62 and the band at approximately 2075 cm−1 can be ascribed to the C![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) O stretch of the top-bound CO (Fig. 5a).63,64 Compared to CuO, the band of the Cu–CO stretch was blue-shifted on the CuO–Ag interface at −1.3, −1.5, and −1.7 V vs. RHE (Fig. 5b and S9†), indicating a stronger binding of CO to the CuO–Ag interface.65–67 The enhanced CO binding implied an enriched CO environment, which would promote the subsequent C–C coupling step, thereby resulting in a higher ethanol productivity.68 DFT calculations confirm that the interface of Cu–Ag is more effective in ethanol generation (Fig. S10†). Compared to bare Cu, the free energy difference on the Cu–Ag interface is more gradual. For instance, the energy difference for adsorption of the second CO2 is lower on the Cu–Ag interface (0.27 eV vs. 0.79 eV). Additionally, for C–C coupling, the Cu–Ag interface exhibits lower energy (1.09 eV vs. 1.46 eV).
O stretch of the top-bound CO (Fig. 5a).63,64 Compared to CuO, the band of the Cu–CO stretch was blue-shifted on the CuO–Ag interface at −1.3, −1.5, and −1.7 V vs. RHE (Fig. 5b and S9†), indicating a stronger binding of CO to the CuO–Ag interface.65–67 The enhanced CO binding implied an enriched CO environment, which would promote the subsequent C–C coupling step, thereby resulting in a higher ethanol productivity.68 DFT calculations confirm that the interface of Cu–Ag is more effective in ethanol generation (Fig. S10†). Compared to bare Cu, the free energy difference on the Cu–Ag interface is more gradual. For instance, the energy difference for adsorption of the second CO2 is lower on the Cu–Ag interface (0.27 eV vs. 0.79 eV). Additionally, for C–C coupling, the Cu–Ag interface exhibits lower energy (1.09 eV vs. 1.46 eV).
 | ||
| Fig. 5 Enhanced ethanol production and proof-of-concept demonstration. (a) In situ Raman spectra acquired on CuO–Ag (40) under different potentials. (b) Comparison of in situ Raman spectra on CuO–Ag (40) and CuO catalysts at −1.3, −1.5, and −1.7 V vs. RHE. (c) FEs and current densities toward CO2RR products, H2, and sorbitol. (d) Prolonged operation of the CO2RR–fermentation hybrid at −0.87 V vs. RHE. Pt foil was used as the anode. 0.1 M glucose (0.5 M KCl) solution was the electrolyte, and the CuO–Ag interface number was 40 unless otherwise stated. | ||
To further rationalize our choice of a high CuO–Ag interface number, we examined the side reactions that occurred simultaneously with CO2RR. In a typical CO2RR cell, the hydrogen evolution reaction (HER) is known to be the major competing reaction,69–73 and it was slightly impacted by the CuO–Ag interface number because the active sites for the HER were essentially unchanged. In our CO2RR–fermentation hybrid system, however, another competing reaction existed, namely, the glucose reduction reaction (GRR), where glucose is electrochemically reduced to sorbitol.74 Compared to the HER, the GRR may pose a greater threat to CO2RR because both Cu and Ag have been reported to be active GRR catalysts.75 Strikingly, our CuO–Ag (40) catalyst can effectively suppress the GRR: the GRR FE was 2.68% at −1.27 V vs. RHE, and this number even lower at 2.37% at −0.87 V vs. RHE (Fig. 5c). By stark contrast, the GRR FE increased to 2.91%, 3.51%, 3.71%, and 4.86% when the CuO–Ag interface number was 30, 20, 10, and 0, respectively (Fig. S11†). These observations verified our tandem catalyst design strategy, and, to the best of our knowledge, developing a CO2RR catalyst that can simultaneously suppress the GRR has never been reported. Finally, we sought to stably operate the proof-of-concept system. Our CO2RR–fermentation hybrid exhibited a maximal ethanol FE of 42.7% at −0.87 V vs. RHE, with an overall current density of 27 mA cm−2 (Fig. 5c). The partial current density towards ethanol reached 20 mA cm−2, with an FE of 18.9% at −1.27 V vs. RHE. The current density and ethanol FE were well retained over the course of 12 h of chronoamperometric operation at −0.87 V vs. RHE (Fig. 5d). Ethanol was detected as the predominant liquid product (Fig. S12†).
Conclusions
In summary, we have demonstrated a compelling bio-electrochemical system, combining yeast fermentation and CO2RR, to upcycle the fermentation-emitted CO2 into ethanol. Taking a free ride from the high-purity CO2 emissions and ethanol distillation during fermentation, the CO2RR–fermentation hybrid cuts the total CO2RR cost by 17.8%. A CuO–Ag tandem electrocatalyst with minimal impact on yeast was rationally designed, efficiently converting CO2 to ethanol while suppressing side reactions, such as hydrogen evolution and glucose reduction. These results illustrate the possibility of a cost-effective CO2 value chain for ethanol production. Further improvements are expected by optimizing the ethanol efficiency and yield through rational tandem electrode design, yeast selection, and system integration.Author contributions
X. L. supervised the project. X. L. and R. S. conceived the idea. R. S. synthesized and characterized the catalysts, and conducted electrochemical measurements. J. Z. made the DFT calculations. R. S. performed the fermentation experiments. R. S. and J. Z. carried out in situ Raman spectroscopy. R. S. conducted the TEA calculation. R. S. and X. L. wrote the manuscript. All authors discussed the results and assisted with the manuscript preparation.Conflicts of interest
The authors declare no competing interests.Acknowledgements
This work was financially supported by the Baseline Fund (BAS/1/1413-01-01) to X. L. from King Abdullah University of Science and Technology (KAUST).References
- O. S. Bushuyev, P. De Luna, C. T. Dinh, L. Tao, G. Saur, J. van de Lagemaat, S. O. Kelley and E. H. Sargent, Joule, 2018, 2, 825–832 CrossRef CAS.
- Y. Wu, Z. Jiang, X. Lu, Y. Liang and H. Wang, Nature, 2019, 575, 639–642 CrossRef CAS.
- R. G. Grim, Z. Huang, M. T. Guarnieri, J. R. Ferrell, L. Tao and J. A. Schaidle, Energy Environ. Sci., 2020, 13, 472–494 RSC.
- G. Wang, J. Chen, Y. Ding, P. Cai, L. Yi, Y. Li, C. Tu, Y. Hou, Z. Wen and L. Dai, Chem. Soc. Rev., 2021, 50, 4993–5061 RSC.
- W. Ma, X. He, W. Wang, S. Xie, Q. Zhang and Y. Wang, Chem. Soc. Rev., 2021, 50, 12897–12914 RSC.
- D. Gao, R. M. Arán-Ais, H. S. Jeon and B. Roldan Cuenya, Nat. Catal., 2019, 2, 198–210 CrossRef CAS.
- A. Salvo and F. M. Geiger, Nat. Geosci., 2014, 7, 450–458 CrossRef CAS.
- P. Zhu and H. Wang, Nat. Catal., 2021, 4, 943–951 CrossRef CAS.
- C. E. Wyman and N. D. Hinman, Appl. Biochem. Biotechnol., 1990, 24, 735–753 CrossRef.
- A. Duereh, H. Guo, T. Honma, Y. Hiraga, Y. Sato, R. Lee Smith Jr and H. Inomata, Ind. Eng. Chem. Res., 2018, 57, 7331–7344 CrossRef CAS.
- D. W. Lachenmeier, J. Occup. Med. Toxicol., 2008, 3, 1–16 CrossRef.
- EU Ethanol Market Report: Suppliers, Prices, Trends and Forecast to 2030, IndexBox, 2022 Search PubMed.
- M. Jouny, W. Luc and F. Jiao, Ind. Eng. Chem. Res., 2018, 57, 2165–2177 CrossRef CAS.
- M. G. Kibria, J. P. Edwards, C. M. Gabardo, C. T. Dinh, A. Seifitokaldani, D. Sinton and E. H. Sargent, Adv. Mater., 2019, 31, 1807166 CrossRef PubMed.
- J. M. Spurgeon and B. Kumar, Energy Environ. Sci., 2018, 11, 1536–1551 RSC.
- P. Li, X. Lu, Z. Wu, Y. Wu, R. Malpass-Evans, N. B. McKeown, X. Sun and H. Wang, Angew. Chem., Int. Ed., 2020, 59, 10918–10923 CrossRef CAS.
- X. Lu, Z. Jiang, X. Yuan, Y. Wu, R. Malpass-Evans, Y. Zhong, Y. Liang, N. B. McKeown and H. Wang, Sci. Bull., 2019, 64, 1890–1895 CrossRef CAS.
- Y. Xu, J. P. Edwards, J. Zhong, C. P. O'Brien, C. M. Gabardo, C. McCallum, J. Li, C.-T. Dinh, E. H. Sargent and D. Sinton, Energy Environ. Sci., 2020, 13, 554–561 RSC.
- G. Lee, A. S. Rasouli, B.-H. Lee, J. Zhang, Y. C. Xiao, J. P. Edwards, M. G. Lee, E. D. Jung, F. Arabyarmohammadi, H. Liu and E. Sargent, Joule, 2023, 7, 1277–1288 CrossRef CAS.
- Y. Cheng, J. Hou and P. Kang, ACS Energy Lett., 2021, 6, 3352–3358 CrossRef CAS.
- A. J. Welch, E. Dunn, J. S. DuChene and H. A. Atwater, ACS Energy Lett., 2020, 5, 940–945 CrossRef CAS.
- M. T. Ho, G. W. Allinson and D. E. Wiley, Energy Procedia, 2009, 1, 763–770 CrossRef CAS.
- A. Raksajati, M. T. Ho and D. E. Wiley, Ind. Eng. Chem. Res., 2013, 52, 16887–16901 CrossRef CAS.
- M. Ramdin, B. De Mot, A. R. Morrison, T. Breugelmans, L. J. Van Den Broeke, J. M. Trusler, R. Kortlever, W. De Jong, O. A. Moultos and P. Xiao, Ind. Eng. Chem. Res., 2021, 60, 17862–17880 CrossRef CAS.
- D. R. Sánchez, K. Khalilpour and A. F. Hoadley, Sustainable Energy Fuels, 2021, 5, 5866–5880 RSC.
- Bioethanol Market (By Application: Transportation, Alcoholic Beverages, Cosmetics, Pharmaceuticals, Others; By Feedstock: Starch Based, Sugar Based, Cellulose-Based; By Fuel Generation: First Generation, Second Generation, Third Generation; By Blend: E5, E10, E15 to E70, E75 & E85, Others (E85 to E100)) – Global Industry Analysis, Size, Share, Growth, Trends, Regional Outlook, and Forecast 2022–2030, Report 2210, Precedence Research, 2022 Search PubMed.
- Ethanol fuel, can be found under https://en.wikipedia.org/wiki/Ethanol_fuel, accessed April 2023 Search PubMed.
- F. Rodrigues, P. Ludovico and C. Leão, Biodiversity and Eecophysiology of Yeasts, 2006, pp. 101–121 Search PubMed.
- Q. Zhang, C.-L. Cheng, D. Nagarajan, J.-S. Chang, J. Hu and D.-J. Lee, Appl. Energy, 2017, 206, 364–371 CrossRef CAS.
- I. Edeh, Bioethanol Technologies, 2021, p. 1 Search PubMed.
- J. K. Stolaroff, S. H. Pang, W. Li, W. G. Kirkendall, H. M. Goldstein, R. D. Aines and S. E. Baker, Front. Energy Res., 2021, 9, 639943 CrossRef.
- H. Pilorgé, N. McQueen, D. Maynard, P. Psarras, J. He, T. Rufael and J. Wilcox, Environ. Sci. Technol., 2020, 54, 7524–7532 CrossRef.
- DOE, can be found under https://www.energy.gov/eere/solar/sunshot-2030, accessed: October 2018.
- A. F. Duro and R. Serrano, Curr. Microbiol., 1981, 6, 111–113 CrossRef CAS.
- A. Aminian and E. Motamedian, Sci. Rep., 2023, 13, 1165 CrossRef CAS.
- B. Maiorella, H. W. Blanch and C. R. Wilke, Biotechnol. Bioeng., 1983, 25, 103–121 CrossRef CAS.
- A. S. Varela, Curr. Opin. Green Sustainable Chem., 2020, 26, 100371 CrossRef.
- J. A. Rabinowitz and M. W. Kanan, Nat. Commun., 2020, 11, 5231 CrossRef CAS.
- Y. Wu, K. Kamiya, T. Hashimoto, R. Sugimoto, T. Harada, K. Fujii and S. Nakanishi, Electrochemistry, 2020, 88, 359–364 CrossRef CAS.
- Y. Zhao, L. Hao, A. Ozden, S. Liu, R. K. Miao, P. Ou, T. Alkayyali, S. Zhang, J. Ning, Y. Liang, Y. Xu, M. Fan, Y. Chen, J. E. Huang, K. Xie, J. Zhang, C. P. O'Brien, F. Li, E. H. Sargent and D. Sinton, Nat. Synth., 2023, 2, 403–412 CrossRef.
- J. Li, A. Ozden, M. Wan, Y. Hu, F. Li, Y. Wang, R. R. Zamani, D. Ren, Z. Wang, Y. Xu, D.-H. Nam, J. Wicks, B. Chen, X. Wang, M. Luo, M. Graetzel, F. Che, E. H. Sargent and D. Sinton, Nat. Commun., 2021, 12, 2808 CrossRef CAS.
- M. Li, N. Song, W. Luo, J. Chen, W. Jiang and J. Yang, Adv. Sci., 2023, 10, 2204579 CrossRef CAS.
- P.-C. Chen, C. Chen, Y. Yang, A. L. Maulana, J. Jin, J. Feijoo and P. Yang, J. Am. Chem. Soc., 2023, 145, 10116–10125 CrossRef CAS.
- D. Wei, Y. Wang, C. L. Dong, Z. Zhang, X. Wang, Y. C. Huang, Y. Shi, X. Zhao, J. Wang and R. Long, Angew. Chem., 2023, 135, e202217369 CrossRef.
- T. Zhang, J. C. Bui, Z. Li, A. T. Bell, A. Z. Weber and J. Wu, Nat. Catal., 2022, 5, 202–211 CrossRef CAS.
- L. Fan, C. Xia, F. Yang, J. Wang, H. Wang and Y. Lu, Sci. Adv., 2020, 6, eaay3111 CrossRef CAS.
- Y. Chen, X.-Y. Li, Z. Chen, A. Ozden, J. E. Huang, P. Ou, J. Dong, J. Zhang, C. Tian, B.-H. Lee, X. Wang, S. Liu, Q. Qu, S. Wang, Y. Xu, R. K. Miao, Y. Zhao, Y. Liu, C. Qiu, J. Abed, H. Liu, H. Shin, D. Wang, Y. Li, D. Sinton and E. H. Sargent, Nat. Nanotechnol., 2023, 1, 8 Search PubMed.
- Q. Lei, L. Huang, J. Yin, B. Davaasuren, Y. Yuan, X. Dong, Z.-P. Wu, X. Wang, K. X. Yao, X. Lu and Y. Han, Nat. Commun., 2022, 13, 4857 CrossRef CAS.
- W. Liu, P. Zhai, A. Li, B. Wei, K. Si, Y. Wei, X. Wang, G. Zhu, Q. Chen and X. Gu, Nat. Commun., 2022, 13, 1877 CrossRef CAS.
- S. S. Bhargava, F. Proietto, D. Azmoodeh, E. R. Cofell, D. A. Henckel, S. Verma, C. J. Brooks, A. A. Gewirth and P. J. Kenis, ChemElectroChem, 2020, 7, 2001–2011 CrossRef CAS.
- M. König, J. Vaes, E. Klemm and D. Pant, iscience, 2019, 19, 135–160 CrossRef.
- J.-J. Li and Z.-C. Zhang, Rare Met., 2022, 41, 723–725 CrossRef CAS.
- X. Zhang, J. Li, Y.-Y. Li, Y. Jung, Y. Kuang, G. Zhu, Y. Liang and H. Dai, J. Am. Chem. Soc., 2021, 143, 3245–3255 CrossRef CAS.
- A. B. Moss, S. Garg, M. Mirolo, C. A. G. Rodriguez, R. Ilvonen, I. Chorkendorff, J. Drnec and B. Seger, Joule, 2023, 7, 350–365 CrossRef CAS.
- Y. Lin, W. Zhang, C. Li, K. Sakakibara, S. Tanaka and H. Kong, Biomass Bioenergy, 2012, 47, 395–401 CrossRef CAS.
- H. Zentou, Z. Z. Abidin, M. Zouanti and D. Greetham, Int. J. Appl. Eng. Res., 2017, 12, 5202–5506 Search PubMed.
- I. Jood, J. W. Hoff and M. E. Setati, World J. Microbiol. Biotechnol., 2017, 33, 1–11 CrossRef CAS.
- J. Li, H. Xiong, X. Liu, D. Wu, D. Su, B. Xu and Q. Lu, Nat. Commun., 2023, 14, 698 CrossRef CAS.
- C. Chen, Y. Li, S. Yu, S. Louisia, J. Jin, M. Li, M. B. Ross and P. Yang, Joule, 2020, 4, 1688–1699 CrossRef CAS.
- C. Chen, X. Yan, S. Liu, Y. Wu, Q. Wan, X. Sun, Q. Zhu, H. Liu, J. Ma and L. Zheng, Angew. Chem., 2020, 132, 16601–16606 CrossRef.
- J. Gao, H. Zhang, X. Guo, J. Luo, S. M. Zakeeruddin, D. Ren and M. Grätzel, J. Am. Chem. Soc., 2019, 141, 18704–18714 CrossRef CAS.
- J. Li, A. Ozden, M. Wan, Y. Hu, F. Li, Y. Wang, R. R. Zamani, D. Ren, Z. Wang and Y. Xu, Nat. Commun., 2021, 12, 2808 CrossRef CAS.
- F. Li, A. Thevenon, A. Rosas-Hernández, Z. Wang, Y. Li, C. M. Gabardo, A. Ozden, C. T. Dinh, J. Li and Y. Wang, Nature, 2020, 577, 509–513 CrossRef CAS PubMed.
- G. Zhang, Z.-J. Zhao, D. Cheng, H. Li, J. Yu, Q. Wang, H. Gao, J. Guo, H. Wang and G. A. Ozin, Nat. Commun., 2021, 12, 5745 CrossRef CAS.
- A. Fielicke, P. Gruene, G. Meijer and D. M. Rayner, Surf. Sci., 2009, 603, 1427–1433 CrossRef CAS.
- X. Wang, Z. Wang, F. P. García de Arquer, C.-T. Dinh, A. Ozden, Y. C. Li, D.-H. Nam, J. Li, Y.-S. Liu, J. Wicks, Z. Chen, M. Chi, B. Chen, Y. Wang, J. Tam, J. Y. Howe, A. Proppe, P. Todorović, F. Li, T.-T. Zhuang, C. M. Gabardo, A. R. Kirmani, C. McCallum, S.-F. Hung, Y. Lum, M. Luo, Y. Min, A. Xu, C. P. O'Brien, B. Stephen, B. Sun, A. H. Ip, L. J. Richter, S. O. Kelley, D. Sinton and E. H. Sargent, Nat. Energy, 2020, 5, 478–486 CrossRef CAS.
- C. Zhan, F. Dattila, C. Rettenmaier, A. Bergmann, S. Kühl, R. García-Muelas, N. r. López and B. R. Cuenya, ACS Catal., 2021, 11, 7694–7701 CrossRef CAS.
- R. B. Sandberg, J. H. Montoya, K. Chan and J. K. Nørskov, Surf. Sci., 2016, 654, 56–62 CrossRef CAS.
- A. Goyal, G. Marcandalli, V. A. Mints and M. T. Koper, J. Am. Chem. Soc., 2020, 142, 4154–4161 CrossRef CAS.
- X. Li, H. Zhang, Q. Hu, W. Zhou, J. Shao, X. Jiang, C. Feng, H. Yang and C. He, Angew. Chem., Int. Ed., 2023, 62, e202300478 CrossRef CAS.
- X. Li, C. Deng, Y. Kong, Q. Huo, L. Mi, J. Sun, J. Cao, J. Shao, X. Chen and W. Zhou, Angew. Chem., Int. Ed., 2023, 62, e202309732 CrossRef CAS.
- Q. Hu, K. Gao, X. Wang, H. Zheng, J. Cao, L. Mi, Q. Huo, H. Yang, J. Liu and C. He, Nat. Commun., 2022, 13, 3958 CrossRef CAS.
- C. Feng, M. Lv, J. Shao, H. Wu, W. Zhou, S. Qi, C. Deng, X. Chai, H. Yang and Q. Hu, Adv. Mater., 2023, 35, 2305598 CrossRef CAS.
- A. Bin Kassim, C. Rice and A. Kuhn, J. Appl. Electrochem., 1981, 11, 261–267 CrossRef.
- Y. Kwon and M. T. Koper, ChemSusChem, 2013, 6, 455–462 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3ta07558c |
| ‡ These authors contributed equally. |
| This journal is © The Royal Society of Chemistry 2024 |