
Advanced electrolyte with high stability and low-temperature resistance for zinc-ion batteries†
Qixian
Bai
a,
Qi
Meng
a,
Weiping
Liu
b,
Wenjun
Lin
b,
Pengfei
Yi
b,
Jingjing
Tang
 a,
Guilin
Zhang
a,
Penghui
Cao
*c and
Juan
Yang
*a
a,
Guilin
Zhang
a,
Penghui
Cao
*c and
Juan
Yang
*a
aSchool of Metallurgy and Environment, Central South University, Changsha 410083, P. R. China. E-mail: j-yang@csu.edu.cn
bZhuzhou Smelter Group Co. Ltd., Zhuzhou 412000, P. R. China
cCollege of Energy and Power Engineering, Changsha University of Science & Technology, Changsha 410114, P. R. China. E-mail: CaoPH@csust.edu.cn
First published on 22nd November 2023
Abstract
Aqueous zinc-ion batteries (AZIBs) are considered to be a green and safe energy storage system. However, electrolyte leakage, zinc dendrite growth and side reactions are still barriers to their practical application. A quasi-solid sodium alginate gel electrolyte (GE) was designed to alleviate these pain points. To further stabilize the water molecules in this GE and extend its application to subzero temperatures, a disaccharide called trehalose (TreH) was introduced as a multifunctional additive. The rational introduction of TreH notably improved the strength (186.6 kPa) of the sodium alginate GE while retaining a high ionic conductivity (22.4 mS cm−1) at 25 °C. The zinc–polyaniline full battery assembled with this composite GE exhibited a capacity retention of 70.4% after 500 cycles at 25 °C, and still delivered a reversible capacity of 120.6 mA h g−1 at −20 °C. These results show that the sodium alginate GE with trehalose as an additive has great potential for development in AZIBs.
1. Introduction
The rising demand for wearable electronic devices in the era of big data requires rechargeable batteries that are green, safe and flexible.1 Batteries also need to ensure that devices can function properly under extreme conditions, such as dehydration and low temperatures. With the advantages of low cost, high safety and environmental friendliness, aqueous zinc-ion batteries (AZIBs) have become a rising star in the field of electrochemical energy storage in recent years.2 However, conventional aqueous electrolyte systems have exposed several problems that urgently need to be addressed, such as liquid leakage, crazy growth of zinc dendrites, and severe side reactions.3–8 The tendency for aqueous electrolytes to freeze also limits the application of AZIBs at low temperatures.Gel polymer electrolytes, especially hydrogel electrolytes (HEs), are a feasible option for solving the above issues. Three-dimensional network structures of hydrogels retain a large amount of water and prohibit contact between free water molecules and the zinc anode, thus alleviating the problem of side reactions on the anode side. Besides, if a hydrogel has high adhesion and strength, it can also effectively inhibit the growth of zinc dendrites. Importantly, a hydrogel can be endowed with excellent flexibility, dehydration resistance and frost resistance through compounding.9 Most of the recent work about HEs has been based on synthetic polymers that are rich in hydrophilic groups (amide group and hydroxyl group). These groups not only give polymer matrixes a high water absorption capacity, but also provide abundant sites for ion migration. Through chemical or physical crosslinking, the chains of these polymers tend to form a lattice-like cross-linked network, which improves the strength. Due to the wide spacing between polymer chains in the lattice structure, zinc ions primarily migrate along single chains in jumps. For example, a polyacrylamide (PAM) base compound hydrogel with excellent mechanical properties and ionic conductivity (13.9 mS cm−1 at 25 °C) was suggested. The Zn–V2O5 full battery using the PAM hydrogel electrolyte (HE) achieved an extremely long life (>10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 cycles) and high reversible capacity (185 mA h g−1) at 5 A g−1.10 A polyvinyl alcohol HE composite with TiO2 nanosheets fabricated by freeze–thawing showed highly reversible zinc plating/stripping, and the full battery assembled with a zinc anode and a V2O5 cathode still exhibited a reversible capacity of 216 mA h g−1 after 115 cycles at 0.2 A g−1 (25 °C).11 Therefore, compared to liquid electrolytes, the HEs display better performance by using these matrix materials with polar groups that can guide zinc-ion migration.
000 cycles) and high reversible capacity (185 mA h g−1) at 5 A g−1.10 A polyvinyl alcohol HE composite with TiO2 nanosheets fabricated by freeze–thawing showed highly reversible zinc plating/stripping, and the full battery assembled with a zinc anode and a V2O5 cathode still exhibited a reversible capacity of 216 mA h g−1 after 115 cycles at 0.2 A g−1 (25 °C).11 Therefore, compared to liquid electrolytes, the HEs display better performance by using these matrix materials with polar groups that can guide zinc-ion migration.
The ionic conductivity through single-chain ion transfer is not enough to meet the anti-freezing demand, but based on this special structure with two-by-two lateral crosslinking and an ordered laminar arrangement it is possible to obtain higher ionic conductivity. Sodium alginate (SA) is a natural polysaccharide derived from brown algae species and composed of a linear copolymer of (1→4)-linked α-L-guluronic acid (G) and β-D-mannuronic acid (M) residues,12,13 where the G–G segments of the adjacent SA chains can be coordinated with multivalent metal cations (Ca2+, Zn2+, Fe3+etc.) to form eggshell-like microstructures.14 Therefore, SA as a matrix material for HEs in the energy field has also attained wide attention. For instance, a SA HE with a hierarchical structure was gained by coordination crosslinking between zinc ions and SA chains. It achieved high ionic conductivity (18.3 mS cm−1) and excellent mechanical performance at room temperature, which is due to the fact that constrained guidance of zinc ions by carboxyl groups with a stepwise arrangement accomplishes ultra-fast and uniformly ordered ion migration.15 Inspired by the above work, researchers obtained a composite SA HE (TA–SA) by introducing tannic acid (TA), which enhances the ion confinement effect.16 Hence, TA–SA has higher ionic conductivity (24.2 mS cm−1) and ionic transference number (t+ = 0.75) at room temperature, and the Zn/TA–SA/NH4V4O10 full battery exhibited a great capacity retention of 94.51% after 900 cycles at 2 A g−1. Although the SA hydrogels produced in these works hold superior strength, their fragility is evident. Unfortunately, the works have not extended the use of SA to low temperatures, which is ascribed to the functional characteristics of the additives. Therefore, introducing a multifunctional additive that can repair the structure and tolerate low temperatures into the SA matrix is a simple and effective way to address these inadequacies.
Trehalose (TreH), which has both tissue repair capacity and resistance to drought and cold, is formed by the condensation of two glucose molecules through a hemiacetal hydroxyl group.17,18 As a non-reducing disaccharide without free aldehyde or ketone groups, the physicochemical properties of TreH are stable enough to assist creatures in maintaining normal vital activities under extreme conditions.19 It may also exhibit great resilience to localized overheating, dehydration and extreme acid–base conditions in batteries when used as the electrolyte additive. Moreover, the low viscosity of TreH is crucial in preventing significant reductions in the matrix's ionic conductivity. The highly biofriendly nature of TreH has made it popular in biomedicine, food, health care, cosmetics and agricultural breeding, and it has also been used in the field of biofuel batteries in recent years.20,21 Importantly, TreH is rich in hydroxyl groups, which appear to hold promise for repairing the internal defects of the SA HE.
In this work, we selected SA as a matrix and TreH as an additive to obtain a composite hydrogel electrolyte (CHE) for AZIBs. Attributed to the intrinsic high ionic conductivity of the SA HE and the effective repair of its internal defects by TreH, the CHE exhibits a perfect balance of mechanical (186.6 kPa) and electrochemical properties (22.4 mS cm−1) at 25 °C. These values are higher than those from most of the reported works. The average coulombic efficiency (CE) of the Zn–Cu asymmetric battery over 300 cycles is 98.9%, and the zinc–polyaniline (Zn–PANI) full battery still has a reversible specific capacity of 63.5 mA h g−1 at 2 A g−1 (25 °C). Notably, through hydrogen bonding, TreH not only improves the stability of water molecules within the SA hydrogel network, but also inhibits the tendency of water to freeze at low temperatures. As a result, the electrochemical stability window (ESW) of the SA HE is broadened to 2.47 V by the synergistic effect between SA and TreH. Even at −20 °C, the Zn–PANI full battery equipped with CHE still retains 104.9 mA h g−1 after 250 cycles at 0.2 A g−1.
2. Experimental
2.1. Materials
Sodium alginate (SA, MW = 100000–170000, AR 90%, M/G = 1) was provided by Macklin, while trehalose (TreH, 99%) and aniline (AR ≥99.5%) were obtained from Aladdin. Zinc sulfate heptahydrate (ZnSO4·7H2O, AR ≥99.5%) and ammonium persulfate (APS, AR 98%) were supplied by Sinopharm Chemical Reagent Co., Ltd. (Shanghai). Carbon cloth (CC, thickness ∼0.36 mm) was obtained from Taiwan Carbon Energy Corporation. Potassium permanganate (KMnO4, AR ≥99.5%) was purchased from Xilong Scientific Co., Ltd. (Shantou). Hydrochloric acid (HCl, AR 36–38%) was purchased from Kelong Chemical Co., Ltd. (Chengdu). ZnSO4 solution (2 M) was prepared with ZnSO4·7H2O and deionized water (ρ = 18.25 MΩ cm at 30 °C, self-made in the lab). Commercial zinc foil of 0.1 mm thickness (99.9%, Sheng Zhengyang Industrial Co., Ltd.) was cut into 14 mm diameter discs for the anodes.
2.2. Preparation of hydrogels
First, 2 g of TreH was dissolved in 10 mL of deionized water by agitation, then 0.5 g of SA was added and stirred vigorously for 6 h to obtain a well-mixed viscous solution, followed by ultrasonic defoaming, then a 1 mm liquid film was attained by coating on a glass plate. Finally, a small amount of ZnSO4 solution (2 M) was added dropwise to cover the film, and the composite hydrogel (called SA–TreH-2) was achieved by fully crosslinking. Likewise, SA gel, SA–TreH-1 and SA–TreH-4 were obtained by adjusting the amount of TreH added. These hydrogels were cut into 19 mm diameter discs for the HEs, and ZnSO4 solution (2 M) was used as the liquid electrolyte for performance comparison.2.3. Preparation of the PANI@CC cathode
The cathode (called PANI@CC) was prepared by growing conductive polyaniline (PANI) on the surface of CC.22 Significantly, CC was soaked in an aqueous solution of KMnO4 (5 wt%) in advance to improve the hydrophilicity.23 Briefly, 0.365 mL of aniline was dissolved in 15 mL of 1 M HCl and CC was added to the mixture; another beaker was used to dissolve 0.228 g of APS into 5 mL of 1 M HCl. The two beakers were transferred to an ice bath at 0 °C and stirred for 1 h, then the HCl solution containing APS was slowly dropped into the other mixed solution containing CC. After 1 h of in situ polymerization, CC was washed three times with deionized water and finally dried overnight at 60 °C to obtain PANI@CC.2.4. Material characterization
Stress–strain curves of the various hydrogels were acquired by tensile testing (Instron 5982) at a strain rate of 0.2 s−1. The phase structure of the zinc foils and hydrogels was ascertained by X-ray diffraction (XRD) (PANalytical Empyrean 2, Cu Kα, 2θ from 5° to 80°). The morphology of the zinc foils, hydrogels, CC and PANI@CC was identified by scanning electron microscopy (SEM) (JSM-6360LV and JSM-7900F). Fourier transform infrared (FTIR) spectroscopy (Thermo Scientific Nicolet iS20, wave number: 400–4000 cm−1) and Raman spectra (Horiba LabRAM HR Evolution, wave number: 200–4000 cm−1) were used to confirm the interactions between the hydrogel components and the existence of PANI on the CC. An energy-dispersive spectrometer (EDS) was also utilized to detect PANI.2.5. Electrochemical measurements
An electrochemical workstation (PARSTAT 4000) was utilized to determine the electrochemical properties of the hydrogels, such as ionic conductivity, Zn2+ transfer number and electrochemical stability window (ESW). The ionic conductivity σ (mS cm−1) of electrolytes was measured using electrochemical impedance spectroscopy (EIS) (1 Hz to 1 MHz). The ionic conductivity, σ, was calculated according to the following equation:where d (μm), A (cm2) and R (Ω) are the thickness, electrode contact area and bulk resistance, which is determined by the intercept with the x-axis in the Nyquist plot. In order to calculate the Zn2+ transfer number, it is necessary to measure the I–t curve of the Zn–Zn symmetric battery with a constant voltage of 25 mV. The resistance of the battery before and after the polarization should be measured respectively. The Zn2+ transfer number t+ was calculated according to the following equation:
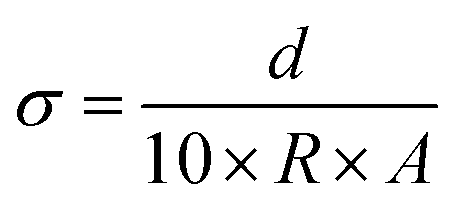
where I0 (mA) and Is (mA) refer to the initial and steady-state current, respectively, ΔV (25 mV) is the voltage applied across the battery, and R0 (Ω) and Rs (Ω) are, respectively, the resistance of the battery before and after the polarization. Linear sweep voltammetry (LSV) was tested for ESW at a scan rate of 0.5 mV s−1 from 1 to −0.3 V and 1 to 3 V.
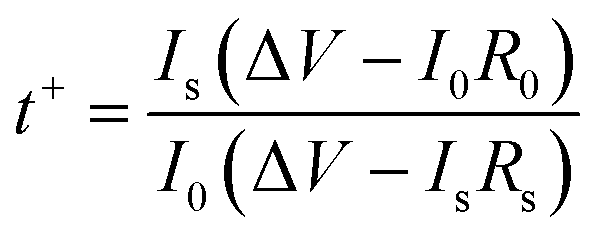
Zn–Zn symmetric, Zn–Cu asymmetric and Zn–PANI full batteries were tested using a battery test system (LAND CT2001A). The resistance of symmetric batteries was determined at an amplitude of 10 mV over the frequency range of 0.01–1 MHz (EIS). Cyclic voltammetry (CV) analysis of the Zn–PANI full batteries was carried out at a scan rate of 0.5 mV s−1 in the voltage range of 0.5–1.5 V.
3. Results and discussion
A series of SA hydrogels with different TreH contents were prepared by a simple coating and ion-crosslinking method (Fig. 1a). Their thicknesses were around 460 μm (Fig. S1†). The surface morphology of the four SA hydrogels was observed with SEM after freeze-drying for 24 h. The pure SA hydrogel (SA gel) exhibits a dense but uneven surface, while the introduction of more TreH results in a smoother and flatter hydrogel surface (Fig. 1b–e). Since HEs may sometimes be exposed to dry conditions, evaluating their anti-drying capability is crucial. Fig. 1f displays images of these hydrogels and their changes over time at 30 °C. Initially, all fresh hydrogels are colorless and transparent. As time progresses, the SA gel shrinks and hardens quickly, while the dimensional stability of the TreH-containing hydrogels increases with the added weight of TreH. This phenomenon suggests that TreH may enhance SA gel's ability to prevent water evaporation. Besides, strength is a key index for assessing the aptitude of HEs to resist zinc dendrite formation. Hydrogel specimens for tensile testing were prepared, measuring 10 cm in length, 2 cm in width and 1 mm in thickness (Fig. S2b†). Compared to SA gel, the addition of TreH improves the strength and plasticity (Fig. S2a†), indicating that TreH probably has a significant repairing and strengthening effect on the internal network of SA gel.19 | ||
| Fig. 1 (a) Schematic diagram of the fabrication process of the SA hydrogels. (b–e) Surface morphology of the four SA hydrogels. (f) Resistance to dehydration test of the four SA hydrogels. | ||
To investigate the causes of the manifestations above, the composition and structure of the four SA hydrogels were characterized using XRD and FTIR, respectively. Fig. 2a reveals the phase structure of SA, TreH powder and the hydrogels. The two powder samples required pre-drying, while the four hydrogels were ground after freeze-drying for sample preparation. Firstly, in conjunction with the local magnification of Fig. 2a (Fig. 2b), it can be seen that the SA powder has two characteristic peaks located at 13.3° and 21.7°. As zinc alginate is the principal component of SA gel after drying, its XRD pattern only exhibits a broad peak between 14° and 30°.24,25 Due to the considerably stronger diffraction peak intensity of TreH powder compared to zinc alginate, SA–TreH-1's pattern consists of a broad peak for zinc alginate and sharp peaks for TreH. Besides, the other two hydrogels also contain more TreH after drying, resulting in similar patterns to TreH powder.26Fig. 2c presents the FTIR result for dried SA powder and the four hydrogels in their original state. The strong, broad peaks in the high-wavenumber region (∼3200 cm−1) indicate that all hydrogels are rich in hydrogen bonds, including O–H stretching vibrations of hydrogen bonds in SA chains.27 Taking SA gel as an example, the peaks at 1628 cm−1 and 1418 cm−1 respectively correspond to the asymmetric and symmetric stretching vibrations of –COO− groups on SA chains,15 while the peak at 1092 cm−1 is probably due to C–O stretching vibrations.28 Zinc ions coordinate firmly with –COO− groups in the G regions of SA chains, which is responsible for the blue shift of asymmetric stretching vibrations of –COO− groups compared to SA powder.29 Strikingly, this characteristic peak gradually shifts towards lower wavenumbers as the amount of TreH added increases, confirming the formation of intermolecular hydrogen bonds between TreH and SA chains.19 In addition, the Raman results for the four HEs are displayed in Fig. S3.† It is obvious that the tendency of water molecules to exist independently or to bind with each other is suppressed, which is shown in the figure as a gradual decrease in the area share of the medium- and high-wavenumber subspace of the hydroxyl group. At the same time, the area of the hydroxyl group of water molecules with low wavenumber subspace is increasing.2 These patterns of change reveal that TreH can stabilize free water molecules through hydrogen bonding.
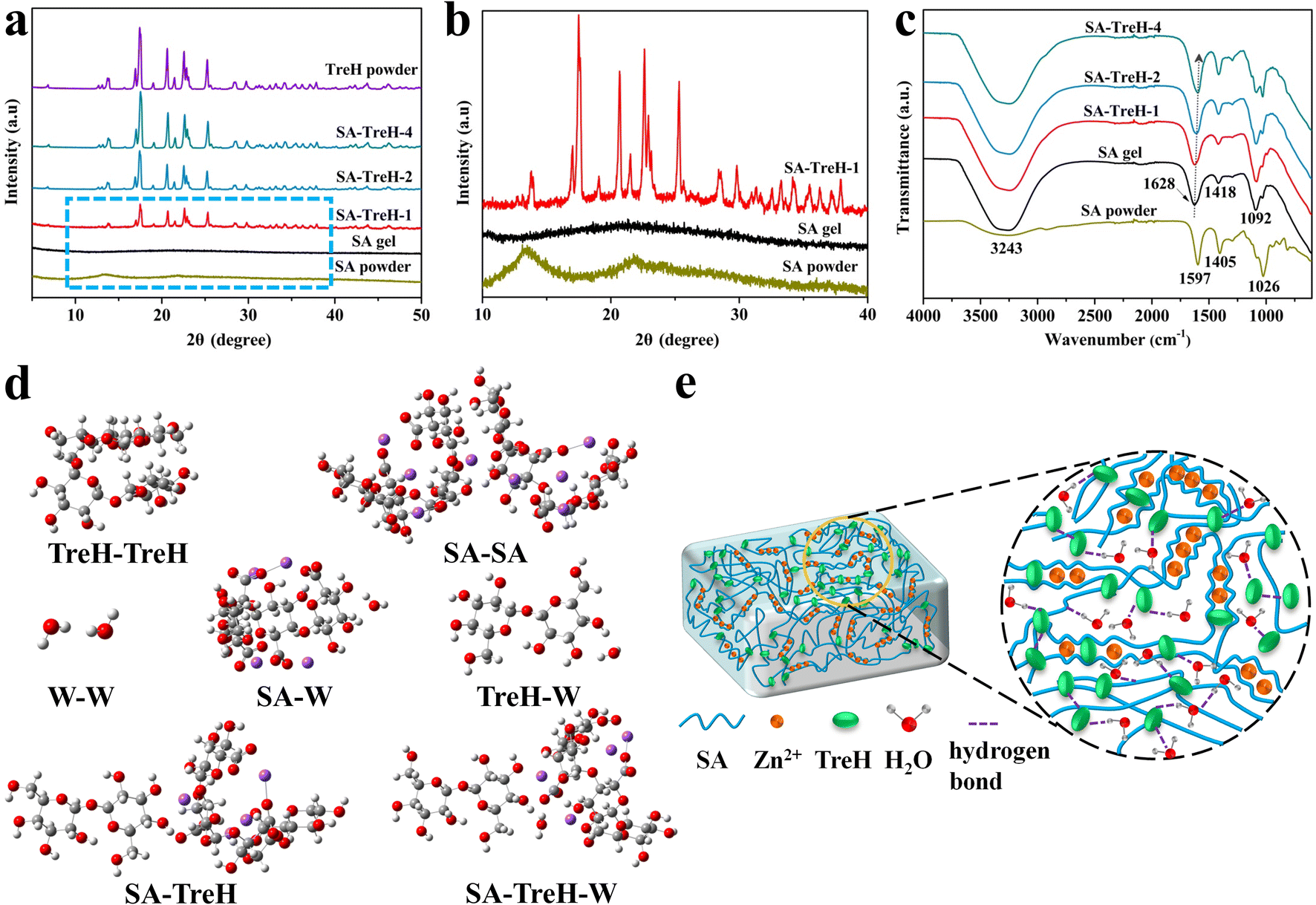 | ||
| Fig. 2 (a) XRD patterns of SA, TreH powder and four SA hydrogels. (b) Partial enlarged view of XRD patterns. (c) FTIR spectra of SA powder and four SA hydrogels. (d) Molecular models used to compute interaction energies among water molecules, TreH molecules and SA molecules. (e) Schematic diagram of the internal structure of the composite hydrogels. | ||
To further quantify the interaction strengths of the components in the composite hydrogels, Gaussian 16 was used to simulate molecular structures and compute interaction energies based on density functional theory (DFT) (ESI text†).30,31 In the molecular models (Fig. 2d), the white, grey, red and purple spheres represent hydrogen, carbon, oxygen and sodium atoms, respectively. The results of the DFT calculations are shown in Table S1.† The interaction energies of TreH–W and SA–W are smaller than that of W–W, indicating that both sugar molecules can inhibit the formation of hydrogen bonds between water molecules. In addition, the interaction energy of SA–TreH is smaller than that of SA–W, suggesting that SA prefers to bond with TreH through hydrogen bonds. Therefore, TreH can repair various imperfections in the SA gel's network, such as dangling chains and loop defects.19 Moreover, it is noteworthy that the interaction energy of SA–TreH–W is minimal, approaching that of SA–SA. This binding mode is able to bind water molecules more effectively, which accounts for the higher resistance to dehydration of the composite hydrogels. Finally, based on the characterization results and above analysis, we created an internal structure diagram for the composite hydrogels (Fig. 2e).
For a preliminary analysis of the feasibility of using these hydrogels as electrolytes for AZIBs, their electrochemical properties were tested. First, the ionic conductivity of the four hydrogels was assessed using a battery with the structure shown in Fig. 3a. Apparently, the addition of TreH reduces the ionic conductivity of the SA gel (Fig. 3b), which is attributed to the fact that the hydrogen bonding energy of SA–TreH is large enough to compete with the SA–Zn2+ coordination bond.32 As TreH takes up some of the Zn2+ ligand sites, it impedes the carboxyl transfer process of Zn2+ (Fig. 2e). This obstruction intensifies with the increase in TreH concentration, naturally leading to a decrease in ionic conductivity. In addition, an increase in the viscosity inside the electrolyte is also responsible for the decrease in ionic conductivity. The Nyquist plots of the four HEs are the basis for calculating the ionic conductivity (Fig. 3c). The measurement process of the ionic conductivity involves only the directional migration of ions and the swift formation of the electrical double layer (EDL), excluding any Faraday processes. The equivalent circuit (in the bottom right corner) corresponding to the EIS data in Fig. 3c is a simple series configuration of a resistor and a capacitor. Here, the resistor (R1) symbolizes the inherent impedance of the SA HEs and the stainless steel electrodes. The constant phase element (CPE1) approximates the capacitance of the non-ideal EDL, which forms between the electrode surface and the inactive ions in electrolytes. Next, the galvanostatic cycling of Zn–Zn symmetric batteries was measured to evaluate the compatibility of these HEs with a zinc anode (Fig. 3d). As the strength and surface flatness of SA–TreH-1 differ little from those of SA gel, its cycle life and form of invalidation are similar to those of SA gel. They are due to the failure to tenaciously bind the internal water molecules, coupled with the fact that the strength is still at a poor level, and is prone to the formation of zinc dendrites (Fig. S4†) and by-products (Fig. 3g) during cycling. In contrast, SA–TreH-2 and SA–TreH-4 achieve high cycle stability owing to their superior strength and surface flatness, combined with strong internal hydrogen bonding. While the initial impedance of the symmetric battery increases with increasing TreH content, the progressively decreasing impedance increment after 50 cycles reveals that the introduction of TreH is effective in mitigating the deterioration of the electrode/electrolyte interface (Fig. 3e and f). The Zn deposition/dissolution reaction on the zinc foil's surface is governed by a charge transfer process; the bulk phase diffusion process of zinc ions within the electrodes does not exist. In addition to R1 and CPE1, the equivalent circuit corresponding to the EIS data in Fig. 3e and f includes another resistor (R2), representing the charge transfer impedance, in parallel with CPE1. Obviously, there is no Warburg impedance in the equivalent circuit. Furthermore, a comparison of the CE of the Zn–Cu batteries assembled with the four HEs shows high zinc plating/stripping reversibility for both SA–TreH-2 and SA–TreH-4 (Fig. 3h). As indicated in Fig. S5† (voltage profiles of Fig. 3h), the Zn–Cu asymmetric cells associated with both SA gel and SA–TreH-1 exhibit a notable decrease in CE after 200 cycles. On the other hand, the cells assembled with the remaining two electrolytes demonstrate a sustained high CE for over 300 cycles.
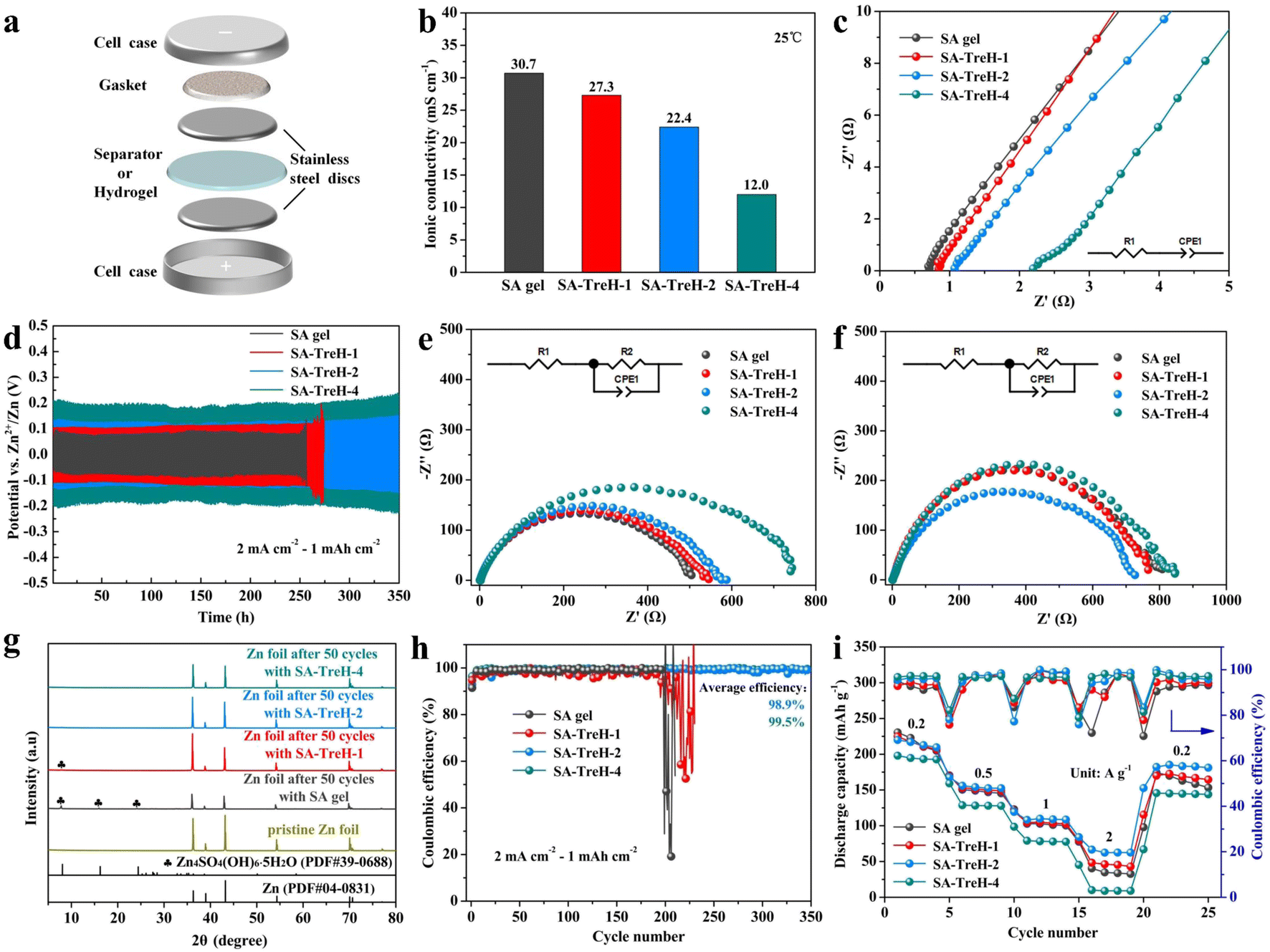 | ||
| Fig. 3 (a) Schematic diagram of the battery structure for testing ionic conductivity. (b) Ionic conductivity and (c) Nyquist plots of four HEs at 25 °C. Zn–Zn symmetric battery performance with four HEs at 25 °C: (d) zinc plating/stripping performance (2 mA cm−2 to 1 mA h cm−2); (e) initial impedance; (f) impedance after 50 cycles; (g) XRD pattern of zinc anodes after 50 cycles. (h) CE of the Zn–Cu asymmetric batteries assembled with four HEs at 25 °C; and (i) rate performance of the Zn–PANI full batteries assembled with four HEs at 25 °C. | ||
To further ascertain the optimal quantity of TreH to be introduced, PANI is used as an organic cathode to assemble the full battery.33,34 Morphological and compositional determination of CC and PANI@CC are displayed in Fig. S6.† As can be seen from optical images, the pure CC is grey-black and it turns dark green after loading PANI (Fig. S6a and b†). High-magnification SEM (×2000) (Fig. S6d and e†) and EDS mapping images (Fig. S6g–i†) confirm the successful polymerization of PANI on CC. Besides, the FTIR and Raman spectra of PANI@CC also confirmed the existence of PANI. The FTIR results are interpreted as follows: the peaks at 807 and 1122 cm−1 are attributed to the C–H bending vibrations, while the peaks at 1294, 1485 and 1562 cm−1 are ascribed to the C–N stretching vibration, benzenoid and quinoid ring vibrations of PANI, respectively (Fig. S6c†). Next comes the analysis of Raman spectra: the peaks at 1165 and 1481 cm−1 are attributed to the C–H bending vibrations and C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C stretching vibrations of the quinoid ring, while the peaks at 1220 and 1595 cm−1 are ascribed to the C–H bending vibrations and CC stretching vibrations of the benzenoid ring. The characteristic peak of the C–N+ stretching vibration is observed at 1330 cm−1 (Fig. S6f†).22 The rate performance of the Zn–PANI full batteries assembled with four HEs is presented in Fig. 3i. Although batteries with composite HEs have inferior capacities at 0.2 A g−1 compared to the SA gel counterpart, they behave differently at higher currents. Thanks to the developed reversibility of zinc-ion transport, the batteries with SA–TreH-1 and SA–TreH-2 are able to maintain higher capacities at high currents. However, the battery with SA–TreH-4 demonstrates the worst rate performance due to the poor ionic conductivity of SA–TreH-4 (12.0 mS cm−1). Considering all factors, SA–TreH-2 is the most suitable composite hydrogel for use as an electrolyte in AZIBs, warranting further exploration of its electrochemical properties.
C stretching vibrations of the quinoid ring, while the peaks at 1220 and 1595 cm−1 are ascribed to the C–H bending vibrations and CC stretching vibrations of the benzenoid ring. The characteristic peak of the C–N+ stretching vibration is observed at 1330 cm−1 (Fig. S6f†).22 The rate performance of the Zn–PANI full batteries assembled with four HEs is presented in Fig. 3i. Although batteries with composite HEs have inferior capacities at 0.2 A g−1 compared to the SA gel counterpart, they behave differently at higher currents. Thanks to the developed reversibility of zinc-ion transport, the batteries with SA–TreH-1 and SA–TreH-2 are able to maintain higher capacities at high currents. However, the battery with SA–TreH-4 demonstrates the worst rate performance due to the poor ionic conductivity of SA–TreH-4 (12.0 mS cm−1). Considering all factors, SA–TreH-2 is the most suitable composite hydrogel for use as an electrolyte in AZIBs, warranting further exploration of its electrochemical properties.
After further comparing the ionic conductivity of the liquid electrolyte, SA gel and SA–TreH-2 at multiple temperatures, it was found that the ionic conductivity of SA–TreH-2 has the least tendency to decrease with decreasing temperature, retaining 12.0 mS cm−1 at −20 °C (Fig. 4a). The excellent temperature adaptation of SA–TreH-2 is attributed to the strong hydrogen bonding constraint of water molecules by TreH, which prevents dehydration in dry environments and inhibits ice crystal formation at low temperatures.35–37 Nyquist plots for calculating the ionic conductivity are shown in Fig. S7.† The ion transfer number test reaffirms the efficacy of Zn2+ transport (Fig. S8†). When compared to the preceding two cells, the Zn–Zn symmetric cell with SA–TreH-2 exhibits the least impedance alteration before and after polarization (∼250 Ω). Additionally, the current swiftly stabilizes at the lowest level (∼15 μA). These two factors contribute to a high Zn2+ transfer number of 0.72 at 25 °C, which is significantly higher than that of the liquid electrolyte (0.38) and SA gel (0.57). The electrochemical stability window (ESW) is an important parameter reflecting the stability of an electrolyte. The ESW values for three electrolytes are 2.05 V, 2.36 V and 2.47 V, respectively (Fig. 4b), indicating that HEs are more effective than the liquid electrolyte in stabilizing water molecules and delaying the decomposition reactions of electrolytes,38 especially for SA–TreH-2. The broader electrochemical window of gel electrolytes, compared to the liquid electrolyte, can be attributed to the firm binding of water molecules by SA chains and TreH. This hydrogen bonding not only inhibits the dissociation process of water molecules but also curbs the ion (H+/OH−) pairing redox process to varying extents.38–40
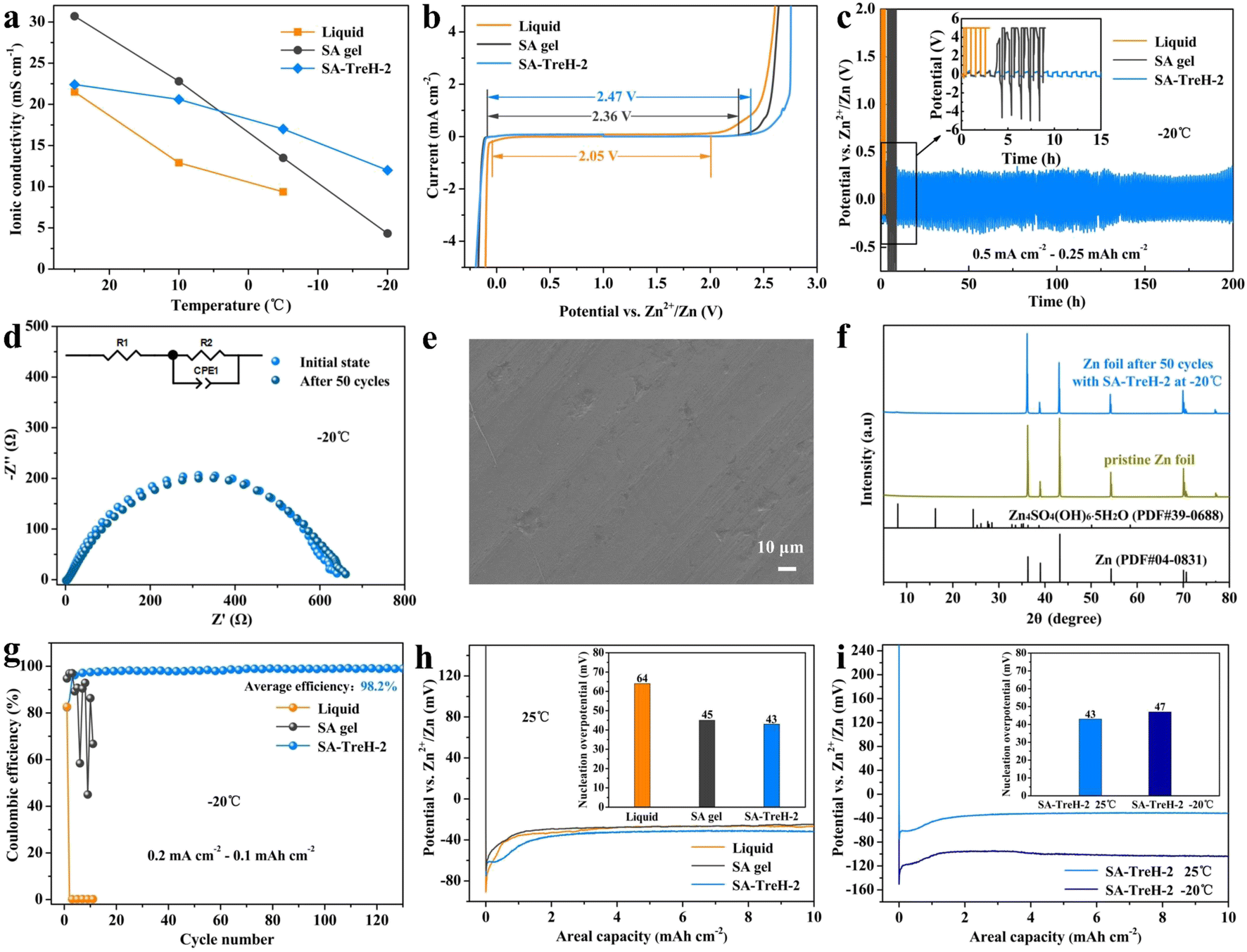 | ||
| Fig. 4 (a) Ionic conductivity at multiple temperatures and (b) ESW at 25 °C of the liquid electrolyte, SA gel and SA–TreH-2. Zn–Zn symmetric battery performance with three electrolytes at −20 °C: (c) zinc plating/stripping performance (0.5 mA cm−2 to 0.25 mA h cm−2); (d) initial impedance and impedance after 50 cycles; (e) SEM image of the zinc anode after 50 cycles; and (f) XRD pattern of the zinc anode after 50 cycles. (g) CE of the Zn–Cu asymmetric batteries assembled with three electrolytes at −20 °C. Nucleation overpotential of the Zn–Cu asymmetric batteries assembled with: (h) three electrolytes at 25 °C and (i) SA–TreH-2 at −20 °C. | ||
In the commonly used aqueous electrolyte systems, there are non-negligible problems with both the aqueous electrolyte and the glass fibre separator. The former has overactive water molecules that can easily corrode the zinc anode and cause side reactions (Fig. S9d†), while the accumulation of irreversible by-products on the anode will significantly increase the electrode/electrolyte interfacial impedance (Fig. S9b and f†).15 The latter exhibits uneven pore size and poor wet strength, which can effortlessly be punctured by zinc dendrites and bring about short circuits in batteries (Fig. S9a and c†).41 Notably, the growth of zinc dendrites and the occurrence of side reactions feed off each other, so they must be addressed simultaneously. Fortunately, SA–TreH-2 presents both low water molecule activity and high adhesive strength. The symmetric battery assembled with it maintains a steady voltage hysteresis of 180 mV at 5 mA cm−2 (Fig. S9e†). Even at −20 °C, the battery achieves a cycle life of up to 200 h, outperforming the liquid electrolyte and SA gel (Fig. 4c). In addition, the impedance of the battery after 50 cycles is virtually indistinguishable from the initial impedance (Fig. 4d), and neither zinc dendrites nor by-products form on the corresponding zinc anode (Fig. 4e and f). The results of the CE test at −20 °C show that SA–TreH-2 still ensures reversible transport of zinc ions (Fig. 4g). Compared to the freezing failure of liquid electrolyte and SA gel (Fig. S10†), CHE can work normally at low temperature for more than 100 cycles with an average CE of 98.2%. Meanwhile, the results of the nucleation overpotential test are shown in Fig. 4h and i, where the nucleation overpotentials of the Zn–Cu batteries assembled with the liquid electrolyte, SA gel and SA–TreH-2 are 64, 45 and 43 mV, respectively (25 °C). A high nucleation overpotential causes a low nucleation rate, resulting in non-uniform zinc deposition and accelerating the growth of dendrites.9 This explains, in another way, why SA–TreH-2 is effective in inhibiting zinc dendrite formation. Markedly, SA–TreH-2's nucleation overpotential increases to only 47 mV at −20 °C, which also demonstrates its exceptional low-temperature tolerance.
To further manifest the superiority of SA–TreH-2 as an electrolyte for AZIBs, the liquid electrolyte, SA gel and SA–TreH-2 were used to assemble Zn–PANI full batteries for performance comparison. Fig. 5a displays the rate performance of the three full batteries at 25 °C. Although the specific capacity of the battery with SA–TreH-2 is marginally lower than the batteries with the other two electrolytes at 0.2 A g−1, it has the best current robustness. Concretely, when the discharge current increases to 2 A g−1, its capacity remains at 63.5 mA h g−1, and upon returning to 0.2 A g−1, the capacity rebounds to 183.4 mA h g−1. Besides, the SA–TreH-2-based full battery achieved capacity retention of 70.4% after 500 cycles at 0.2 A g−1 (Fig. 5b), and still has a discharge specific capacity of 62.0 mA h g−1 after 200 cycles at 2 A g−1, a capacity retention of 91.0% (Fig. 5c). Although the performance vantage of SA–TreH-2 is not palpable when cycling at low currents, once cycled at high currents, the preponderance is enormously impressive. The CV curve of the SA–TreH-2-based battery also reveals the highest reversibility compared with the other two batteries (Fig. S11a–c†).
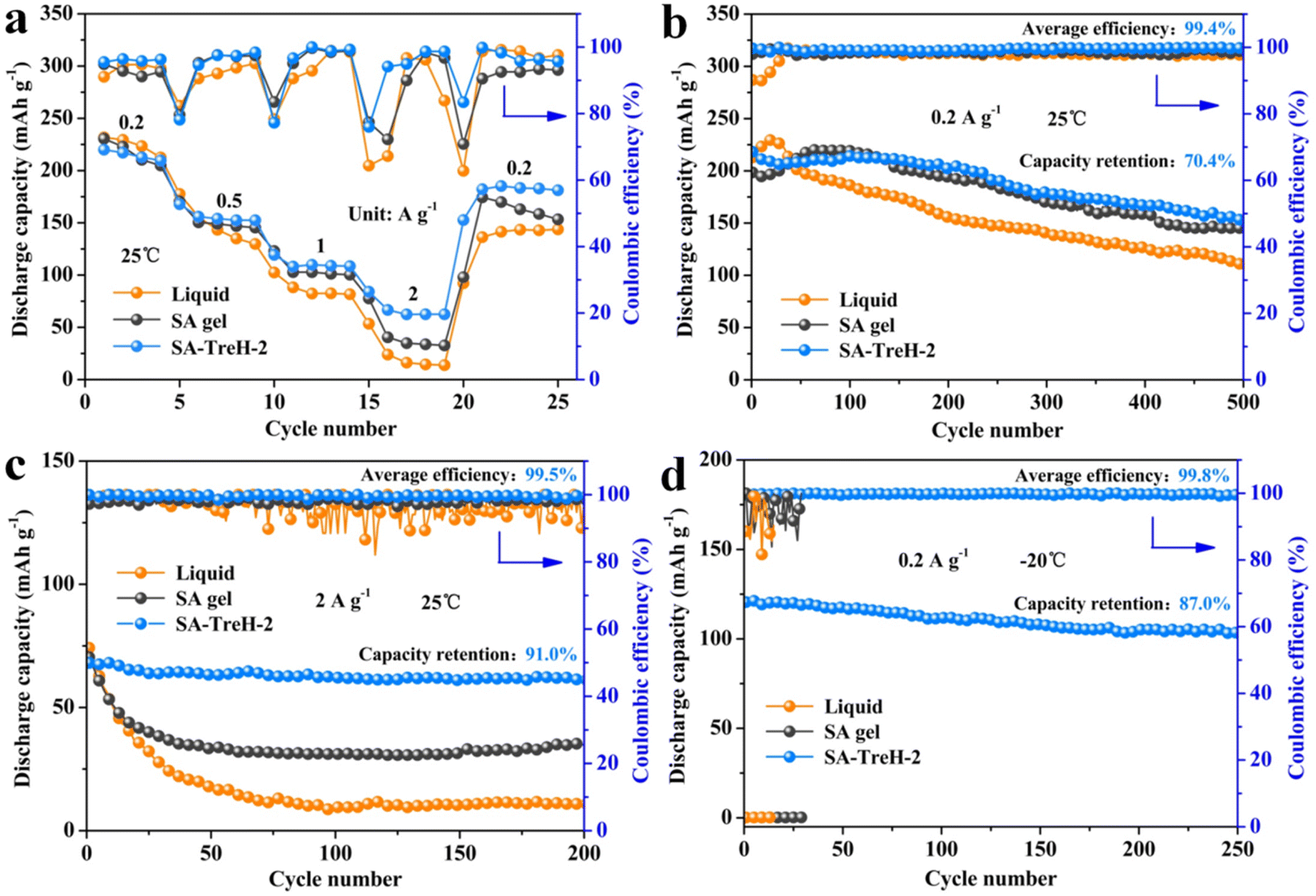 | ||
| Fig. 5 (a) Rate performance of the Zn–PANI batteries assembled with the liquid electrolyte, SA gel and SA–TreH-2. The cycling performance of three kinds of Zn–PANI batteries at: (b) 0.2 A g−1 and 25 °C; (c) 2 A g−1 and 25 °C; and (d) 0.2 A g−1 and −20 °C. | ||
The reason for the large difference in the performance of the three full batteries could be explicated through the energy storage mechanism of the PANI cathode. During the charge/discharge process, there is not only the insertion/extraction of zinc ions on the cathode side, but also a similar process involving hydrated anions.22 The hydration number of the sulfate is 6 and therefore the radius of the entire hydration structure is bulky. However, the larger the radius of hydration of the anion, the greater the volume change caused during insertion and deinsertion on the cathode. Especially at high currents, recurrent and marked volume changes will cause the active material to crack and fall off the current collector, which naturally leads to poor cycling stability. Conversely, reducing the hydration number of anions could mitigate structural changes in the PANI cathode, resulting in a more stable cycle.42–44 The cathode morphology of the three full batteries after 100 cycles at 2 A g−1 is shown in Fig. S12,† where Fig. S12a† presents the original morphology of the PANI@CC cathode. After cycling, little PANI is observed on the carbon fibres of the corresponding cathode of the aqueous battery (Fig. S12b†). A small amount of PANI is visible on the cathode of the SA gel counterpart, while the cathode of the SA–TreH-2-based battery is closest to its initial state, which intuitively supports the above analysis (Fig. S12c and d†). At −20 °C, the first two full batteries fail to work due to the freezing of the electrolytes, whereas SA–TreH-2, which retains a high ionic conductivity, still assists the full battery to accomplish a reversible capacity of 120.6 mA h g−1 and stably cycle 250 times with an average CE of 99.8% (Fig. 5d). The low reactivity of water molecules and the strong binding effect of SA–TreH-2 on them at low temperatures are responsible for this excellent performance.
4. Conclusions
A quasi-solid SA gel electrolyte was successfully prepared using a simple coating and solution crosslinking method. The integrated mechanical properties and electrochemical stability of this electrolyte are improved by the introduction of TreH. The ESW is further broadened (2.47 V) without any significant sacrifice in ionic conductivity (22.4 mS cm−1). Defects in the SA gel's network are repaired and water molecules are more strongly bound owing to the strong hydrogen bonding among TreH, SA chains and water molecules. This results in a composite hydrogel that not only has excellent resistance to dehydration, but is also stably matched to the PANI cathode and zinc anode to achieve a zinc-ion battery with high cycling stability. In addition, the anti-freezing property of TreH further facilitates the application of HEs in low-temperature batteries. Furthermore, the matrix material of the hydrogel can be replaced by other polysaccharides that can also be cross-linked by zinc ions, such as chitosan, pectin and gellan gum, while the disaccharide additive can also be sucrose, which also has a low viscosity.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We are greatly thankful for the financial support from the Natural Science Foundation of Hunan Province [No. 2023JJ10076] and Zhuzhou Smelter Group Co. Ltd. (ZYGFGH2307071500015).Notes and references
- L. Wen, J. Chen, J. Liang, F. Li and H.-M. Cheng, Natl. Sci. Rev., 2017, 4, 20–23 CrossRef CAS.
- Q. Meng, R. Zhao, P. Cao, Q. Bai, J. Tang, G. Liu, X. Zhou and J. Yang, Chem. Eng. J., 2022, 447, 137471 CrossRef CAS.
- P. Cao, H. Zhou, X. Zhou, Q. Du, J. Tang and J. Yang, ACS Sustain. Chem. Eng., 2022, 10, 8350–8359 CrossRef CAS.
- H. Dong, J. Li, J. Guo, F. Lai, F. Zhao, Y. Jiao, D. L. Brett, T. Liu, G. He and I. Parkin, Adv. Mater., 2021, 33, 2007548 CrossRef CAS PubMed.
- L. Ma, S. Chen, N. Li, Z. Liu, Z. Tang, J. A. Zapien, S. Chen, J. Fan and C. Zhi, Adv. Mater., 2020, 32, 1908121 CrossRef CAS PubMed.
- W. Zhang, Y. Dai, R. Chen, Z. Xu, J. Li, W. Zong, H. Li, Z. Li, Z. Zhang, J. Zhu, F. Guo, X. Gao, Z. Du, J. Chen, T. Wang, G. He and I. Parkin, Angew. Chem., Int. Ed., 2023, 62, e202212695 CrossRef CAS PubMed.
- Y. Dai, C. Zhang, W. Zhang, L. Cui, C. Ye, X. Hong, J. Li, R. Chen, W. Zong, X. Gao, J. Zhu, P. Jiang, Q. An, D. L. Brett, I. Parkin, G. He and L. Mai, Angew. Chem., Int. Ed., 2023, 62, e202301192 CrossRef CAS PubMed.
- R. Chen, C. Zhang, J. Li, Z. Du, F. Guo, W. Zhang, Y. Dai, W. Zong, X. Gao, J. Zhu, Y. Zhao, X. Wang and G. He, Energy Environ. Sci., 2023, 16, 2540–2549 RSC.
- M. Chen, J. Chen, W. Zhou, X. Han, Y. Yao and C.-P. Wong, Adv. Mater., 2021, 33, 2007559 CrossRef CAS.
- T. Wei, Y. Ren, Z. Li, X. Zhang, D. Ji and L. Hu, Chem. Eng. J., 2022, 434, 134646 CrossRef CAS.
- C. Liu, Y. Tian, Y. An, Q. Yang, S. Xiong, J. Feng and Y. Qian, Chem. Eng. J., 2022, 430, 132748 CrossRef CAS.
- T. A. Davis, F. Llanes, B. Volesky and A. Mucci, Environ. Sci. Technol., 2003, 37, 261–267 CrossRef CAS PubMed.
- T. A. Fenoradosoa, G. Ali, C. Delattre, C. Laroche, E. Petit, A. Wadouachi and P. Michaud, J. Appl. Phycol., 2010, 22, 131–137 CrossRef CAS.
- N. Flórez-Fernández, H. Domínguez and M. D. Torres, Int. J. Biol. Macromol., 2019, 124, 451–459 CrossRef PubMed.
- Y. Tang, C. Liu, H. Zhu, X. Xie, J. Gao, C. Deng, M. Han, S. Liang and J. Zhou, Energy Storage Mater., 2020, 27, 109–116 CrossRef.
- B. Zhang, L. Qin, Y. Fang, Y. Chai, X. Xie, B. Lu, S. Liang and J. Zhou, Sci. Bull., 2022, 67, 955–962 CrossRef CAS PubMed.
- C. Ash, Science, 2017, 358, 1398–1399 CrossRef.
- A. K. Soper, M. A. Ricci, F. Bruni, N. H. Rhys and S. E. Mclain, J. Phys. Chem. B, 2018, 122, 7365–7374 CrossRef CAS PubMed.
- Z. Han, P. Wang, Y. Lu, Z. Jia, S. Qu and W. Yang, Sci. Adv., 2022, 8, eabl5066 CrossRef CAS PubMed.
- N. Kim, Y. Choi, S. Jung and S. Kim, Biotechnol. Bioeng., 2000, 70, 109–114 CrossRef CAS.
- M. Rasmussen, R. E. Ritzmann, I. Lee, A. J. Pollack and D. Scherson, J. Am. Chem. Soc., 2012, 134, 1458–1460 CrossRef CAS PubMed.
- F. Wan, L. Zhang, X. Wang, S. Bi, Z. Niu and J. Chen, Adv. Funct. Mater., 2018, 28, 1804975 CrossRef.
- X. Zeng, J. Liu, J. Mao, J. Hao, Z. Wang, S. Zhou, C. D. Ling and Z. Guo, Adv. Energy Mater., 2020, 10, 1904163 CrossRef CAS.
- L. Abi Nassif, S. Rioual, W. Farah, M. Fauchon, Y. Toueix, C. Hellio, M. Abboud and B. Lescop, J. Environ. Chem. Eng., 2020, 8, 104246 CrossRef CAS.
- C. Larosa, M. Salerno, J. S. de Lima, R. M. Meri, M. F. da Silva, L. B. de Carvalho and A. Converti, Int. J. Biol. Macromol., 2018, 115, 900–906 CrossRef CAS PubMed.
- T. Sekitoh, T. Okamoto, A. Fujioka, T. Yoshioka, S. Terui, H. Imanaka, N. Ishida and K. Imamura, J. Food Eng., 2021, 292, 110325 CrossRef CAS.
- Y. Wei, H. Wang, Q. Ding, Z. Wu, H. Zhang, K. Tao, X. Xie and J. Wu, Mater. Horiz., 2022, 9, 1921–1934 RSC.
- D. Leal, B. Matsuhiro, M. Rossi and F. Caruso, Carbohydr. Res., 2008, 343, 308–316 CrossRef CAS PubMed.
- J. Mirtič, J. Ilaš and J. Kristl, Carbohydr. Polym., 2018, 181, 93–102 CrossRef PubMed.
- S. Grimme, J. Antony, S. Ehrlich and H. Krieg, J. Chem. Phys., 2010, 132, 154104 CrossRef PubMed.
- S. Grimme, S. Ehrlich and L. Goerigk, J. Comput. Chem., 2011, 32, 1456–1465 CrossRef CAS PubMed.
- Z. J. Li, S. Srebnik and O. J. Rojas, Biomacromolecules, 2021, 22, 4027–4036 CrossRef CAS PubMed.
- A. G. MacDiarmid, L. S. Yang, W. S. Huang and B. D. Humphrey, Synth. Met., 1987, 18, 393–398 CrossRef CAS.
- L. S. Yang, Z. Q. Shan and Y. D. Liu, J. Power Sources, 1991, 34, 141–145 CrossRef CAS.
- D. Corradini, E. Strekalova, H. Stanley and P. Gallo, Sci. Rep., 2013, 3, 1218 CrossRef PubMed.
- E. C. A. Eleutherio, P. S. de Araujo and A. D. Panek, Biochim. Biophys. Acta, Gen. Subj., 1993, 1156, 263–266 CrossRef CAS PubMed.
- D. Koshland and H. Tapia, Mol. Biol. Cell, 2019, 30, 731–809 CrossRef PubMed.
- X. Li, Z. Xu, Y. Qian and Z. Hou, Energy Storage Mater., 2022, 53, 72–78 CrossRef.
- Y. Wang, T. Wang, D. Dong, J. Xie, Y. Guan, Y. Huang, J. Fan and Y.-C. Lu, Matter, 2022, 5, 162–179 CrossRef CAS.
- Y. Fang, Y. Fan, K. Xie, W. Ge, Y. Zhu, Z. Qi, Z. Song, H. Jiang and C. Li, Angew. Chem., Int. Ed., 2023, 62, e202304413 CrossRef CAS.
- K. Zhao, C. Wang, Y. Yu, M. Yan, Q. Wei, P. He, Y. Dong, Z. Zhang, X. Wang and L. Mai, Adv. Mater. Interfaces, 2018, 5, 1800848 CrossRef.
- Y. Zhang, L. Zhao, Y. Liang, X. Wang and Y. Yao, eScience, 2022, 2, 110–115 CrossRef.
- G. He, Y. Liu, D. E. Gray and J. Othon, Compos. Commun., 2021, 27, 100882 CrossRef.
- P. Cao, C. Chen, X. Zhou, J. Tang and J. Yang, J. Energy Storage, 2023, 74, 109529 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3ta05052a |
| This journal is © The Royal Society of Chemistry 2024 |