
Hexagonal 2D covalent organic frameworks from nonpolar and symmetric electron-accepting substituents for electron transport layers in near-infrared PeLEDs†
Lili
Xu
a,
Lei
Zheng
a,
Yu
Jing
 b,
Xiangyu
Guo
c,
Xuemin
Hu
*d,
Bo
Xu
a and
Shengli
Zhang
*a
b,
Xiangyu
Guo
c,
Xuemin
Hu
*d,
Bo
Xu
a and
Shengli
Zhang
*a
aMIIT Key Laboratory of Advanced Display Materials and Devices, School of Materials Science and Engineering, Nanjing University of Science and Technology, Nanjing 210094, Jiangsu, China. E-mail: zhangslvip@njust.edu.cn
bJiangsu Co-Innovation Centre of Efficient Processing and Utilization of Forest Resources College of Chemical Engineering, Nanjing Forestry University, Nanjing 210037, China
cSchool of Science, Constructor University, Bremen 28759, Germany
dSchool of Material Engineering, Jinling Institute of Technology, Nanjing 211169, China. E-mail: huxuemvip@126.com
First published on 28th November 2023
Abstract
We conducted high-throughput calculations on the structure library of two-dimensional covalent organic frameworks (2D COFs), with the aim of finding new candidate materials as electron transport layers (ETLs) for the high-performance perovskite light-emitting diodes (PeLEDs). Starting with nearly 400 unique 2D COFs with hexagonal (hcb) topology, 23 candidates for β-ketoenamine linked 2D COFs composed of 1,3,5-triformylphloroglucinol (Tp) and linear diamines are identified by band structures. Interestingly, 2 β-ketoenamine linked 2D Tp-DAAQ and 2D Tp-DABDA with electron-accepting substituents, are the most promising candidates due to the optimal band edge and effective mass. Further, the nonpolarity of the Tp-DAAQ molecule makes it a more favorable choice for the ETL, as confirmed by nonadiabatic molecular dynamics that 2D Tp-DAAQ with anthraquinone diamine linker has a longer e–h nonradiative recombination time. This work provides a paradigm for the design of high-performance ETLs.
1. Introduction
Perovskite light-emitting diodes (PeLEDs) have experienced rapid development in recent years and are approaching the efficiency of organic light-emitting diodes (OLEDs), showing great potential for commercialization.1–7 However, nonradiative recombination is encountered in carrier transport, resulting in energy loss.8,9 Currently, the development of ideal electron transport layers (ETLs) for PeLEDs is an important research direction to improve device performance,10,11 in terms of its important role in lowering the potential barrier for electron injection from the cathode into the emitting layer (EML).12,13 Presently, ZnO is commonly used as an ETL material due to its ease of preparation and processing, and its relatively low cost.14,15 However, the energy level mismatch and mismatched mobility with hole transport layer (HTL) cause electron and hole transport imbalance and nonradiative recombination. Many efforts have been devoted to finding ETLs with suitable energy level and mobility.To improve the device performance, the ETLs for PeLEDs need to meet the following requirements: (1) possess similar mobility as HTL; (2) the conduction band minimum (CBM) is higher than or close to the CBM of EML, while the valence band maximums (VBM) is much lower than that of EML; (3) have stable physicochemical properties. Two-dimensional covalent organic frameworks (2D COFs) have a regular, conductive π–π stacking structure, leading to higher carrier mobility.16 In addition, through the interfacial modulation, the chemical or physical properties of the interface of 2D COFs can be modified, and thus have potential to achieve better band alignment, and display superior charge transfer pathways. These advantages make 2D COFs as the promising candidates for the ETLs. Previous studies have shown that five possible topologies in the case of 2D covalent plane, namely hexagonal (hcb),17 kagome (kgm),18 tetragonal (sql),19 rhombille (kgd),20 and triangular (hxl).21 In particular, 2D COFs with hcb topology usually have good electrical conductivity compared to other topologies due to their graphene-like planar structure, in which carbon atoms are arranged in hexagonal shape to form highly conjugated π orbitals.22–24 What new properties would these materials with hcb topology have? To our knowledge, attempts in this area have never been reported systematically.
Here, we screened close to 400 2D COFs with hcb topology in the core-cof/CoRE-COF-Database25,26 to find potential candidates for ETLs in PeLEDs. After systematic studies, β-ketoenamine linked candidates 2D Tp-DAAQ (2,6-diaminoanthraquinone)27 and 2D Tp-DABDA (2,5-diaminobenzene-1,4-disulfonic acid),28 consisting of Tp (1,3,5-triformylphloroglucinol) and different linear diamine linkers, stood out among nearly 400 materials. In both 2D Tp-DAAQ and 2D Tp-DABDA, the linear diamine linkers have electron-accepting substituents ![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O and –SO3H, with a low VBM and a suitable CBM. Furthermore, the dipole moment (EDM) of the monomer molecule Tp-DAAQ is zero making it a more favorable material for the ETL. Nonadiabatic molecular dynamics (NAMD) confirms that 2D Tp-DAAQ with anthraquinone linear diamine linker has a longer e–h nonradiative recombination time scale compared to 2D Tp-DABDA and is a good candidate for ETL in PeLEDs.
O and –SO3H, with a low VBM and a suitable CBM. Furthermore, the dipole moment (EDM) of the monomer molecule Tp-DAAQ is zero making it a more favorable material for the ETL. Nonadiabatic molecular dynamics (NAMD) confirms that 2D Tp-DAAQ with anthraquinone linear diamine linker has a longer e–h nonradiative recombination time scale compared to 2D Tp-DABDA and is a good candidate for ETL in PeLEDs.
2. Computational details
All the ground state properties are calculated using the PWmat29 and Vienna Ab Initio Simulation Package (VASP).30 Electron–ion interactions are described by using the all electron projector augmented wave (PAW) method.31 The generalized gradient approximation (GGA) with the Perdew–Burke–Ernzerhof (PBE) exchange–correlation functional is used to approximate correlation of electrons within the many-body wave function.32 Since the PBE generalization will underestimate the band gap, the Heyd–Scuseria–Ernzerhof (HSE06) functional is used to calculate the more accurate energy band structures.33 The Hefei-NAMD software package is used to calculate excited state processes.34 For more details on the calculation, see the ESI.†3. Result and discussion
As a transport layer material in PeLEDs, a suitable band structure and matching energy levels with EML are essential. We screened close to 400 2D COFs with hcb topology connected by five different linkages, namely boronate anhydride, boronate ester, nitrile cyclotrimerization, imine, and hydrazone,35 as shown in Fig. 1. Among them, hydrazone-linked 2D COF materials undergo spontaneous structure change during the DFT optimization, which implies that these are unstable, and some examples are listed in Fig. S1.† For the other four modes, the VBM and CBM of some 2D COFs are flat bands, which lead to an infinite effective mass of carriers and cannot be used as transport layer materials in PeLEDs. Several representatives of each linked mode are selected as examples (Fig. S2–S5†). There is an unexpected finding in the imine-linked 2D COFs that the CBMs of β-ketoenamine linked 2D COFs36 consisting of linear diamine linkers with the Tp are not flat bands, which have potential as ETL materials, as displayed in Fig. S6–S9.†27,28,37–51 Eventually, we screened out 23 2D COFs with different diamine linkers (Fig. 1). For the convenience of the later discussion, we use Tp + linker to name the 2D COF materials, and the full names of linkers are given in Table S1.† The lattice constants of these 23 2D COFs are listed in Table S2.† In addition to the above 2D COFs, another class of more recently studied materials are sp2 carbon conjugated 2D COFs, as shown in Fig. S10.† The calculated lattice parameters are shown in Table S3.† Band structure plots in Fig. S11† indicate that the VBMs and CBMs of these kinds of materials are flat bands with infinite effective mass, and are therefore unsuitable as ETLs in PeLEDs.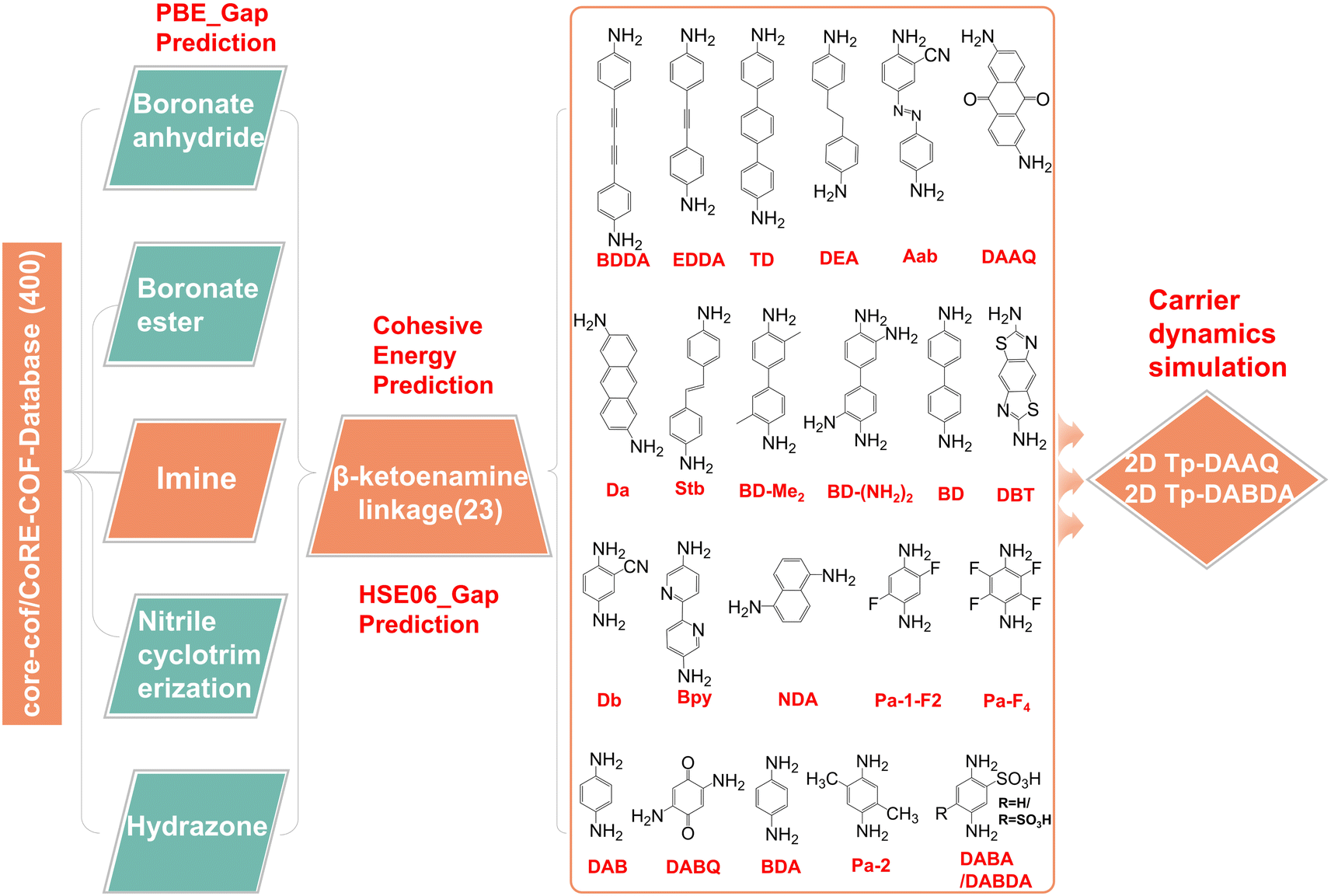 | ||
| Fig. 1 High-throughput computational screening model. Structures downloaded from the experimental library are sequentially passed through multiple computational filters, such as cohesive energies, energy bands, and carrier dynamics, in order to remove materials unsuitable for use as ETLs in PeLEDs and ultimately screen out 2 potential candidates that could be ETLs. | ||
Before studying their electronic properties, we first evaluate the stability of these 23 2D COFs by calculating the cohesive energy (Ecoh). Taking 2D Tp-DAAQ (C60H30N6O12) as an example, the equation is Ecoh = (60EC + 30EH + 6EN + 12EO − EC60H30N6O12)/108, where EC, EH, EN, EO, and EC60H30N6O12 are the total energies of a single C atom, a single H atom, a single N atom, a single O atom, and EC60H30N6O12 monolayer, respectively. According to the definition, a more positive value of Ecoh indicates a stronger bonding. The cohesive energies of 23 β-ketoenamine linked 2D COFs are calculated within 6.5–7.5 eV per atom, as shown in the insert of Fig. 2, which is greater than the 3.71 eV per atom for silicon.52 Although the relative stability of these materials cannot be directly determined by comparing their cohesive energies, the more positive Ecoh clearly demonstrate that these 2D COFs are strongly bonded network.
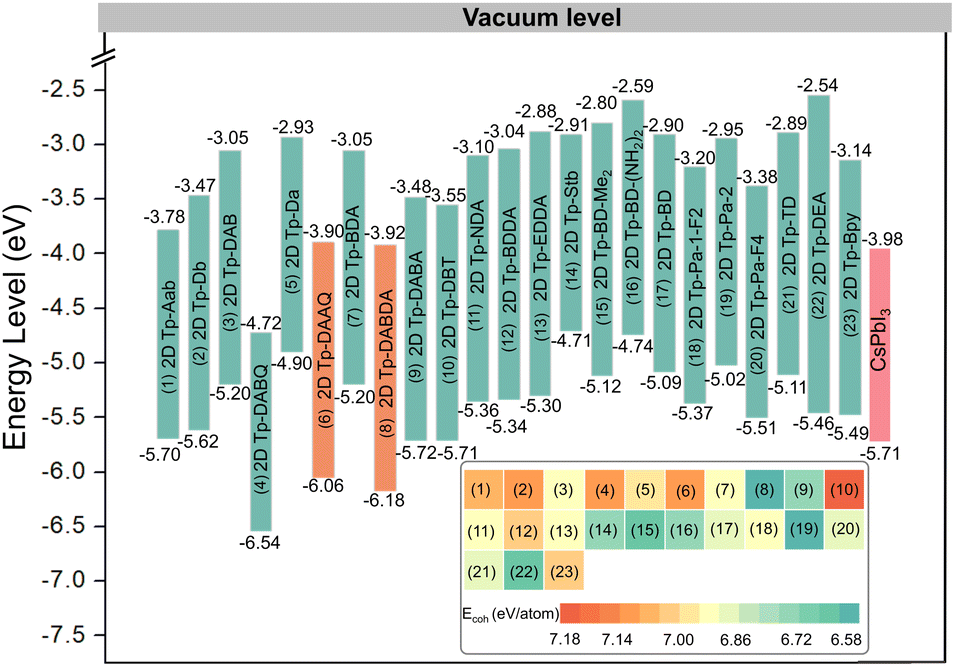 | ||
| Fig. 2 The energy levels of different materials acting as ETLs (23 β-ketoenamine linked 2D COFs) and EML (CsPbI3) in PeLEDs. The inset shows the cohesion energies of 23 β-ketoenamine linked 2D COFs. | ||
In PeLEDs, the band edges affect carrier transport and recombination significantly. To achieve efficient electron transport, the CBM of the ETL should be higher or closer to that of the EML, while its VBM must be much deeper than that of the EML to prevent holes from entering the ETL through the EML. Thus, the band edge is the second criterion of screening. PBE functional will underestimate the band gap, so the more accurate HSE06 is used to calculate the band edges, as shown in Fig. 2. The screening results show that 2D Tp-DAAQ and 2D Tp-DABDA, consisting of diamine linkers with symmetrical electron-accepting substituents, not only have the suitable CBMs to transport electrons, but also the quite deep VBMs can effectively block the passage of holes, which is comparable to that of ZnO (VBM of −7.39 eV, CBM of −4.2 eV).53 As stated above, the β-ketoenamine linked 2D COFs consisting of diamine linkers with symmetrical electron-accepting substituents can effectively reduce both VBM and CBM, and have better matching energy levels with the EML.
What kind of interesting electronic properties would this particular structure have? To answer this question, we calculated their highest occupied molecular orbital (HOMO) and lowest unoccupied molecular orbital (LUMO) in terms of the minimum construction unit, with the HOMO–LUMO gaps of 2.92 and 3.66 eV in Tp-DAAQ and Tp-DABDA, respectively (Fig. 3a), and band edges enclosing the corresponding 2D COFs, as displayed in Fig. 3b and c (left panel). The electrons of Tp-DABDA on HOMO and LUMO are distributed throughout the molecule. In Tp-DAAQ, electrons are more localized in the center at HOMO, while electrons are dispersed along the chain at LUMO, and this large difference between the HOMO and LUMO wave functions can facilitate the generation of intramolecular charge transfer.54 We further calculated the EDMs and obtained values of 0 and 2.32 Debye in Tp-DAAQ and Tp-DABDA, respectively, indicating that Tp-DAAQ is a nonpolar molecule while Tp-DABDA is a polar molecule (Fig. 3a). Nonpolar molecules have no significant charge difference in chemical bonding, so they are not strongly affected by the electric field during the transport between electrons and holes, which can reduce the scattering of carriers and improve the efficiency of electron and hole transport. To gain more insight into the electronic properties of 2D Tp-DAAQ and 2D Tp-DABDA, we calculated their projected density of states (PDOS). As can be seen from the Fig. 3b and c (right panel), the VBM of 2D Tp-DAAQ is contributed by C-2p, O-2p, and N-2p, and the CBM is provided by C-2p and O-2p. In 2D Tp-DABDA, the VBM and CBM are both provided by C-2p, O-2p, and N-2p. These results are consistent with the calculation of the projected energy bands (Fig. S12 and S13†).
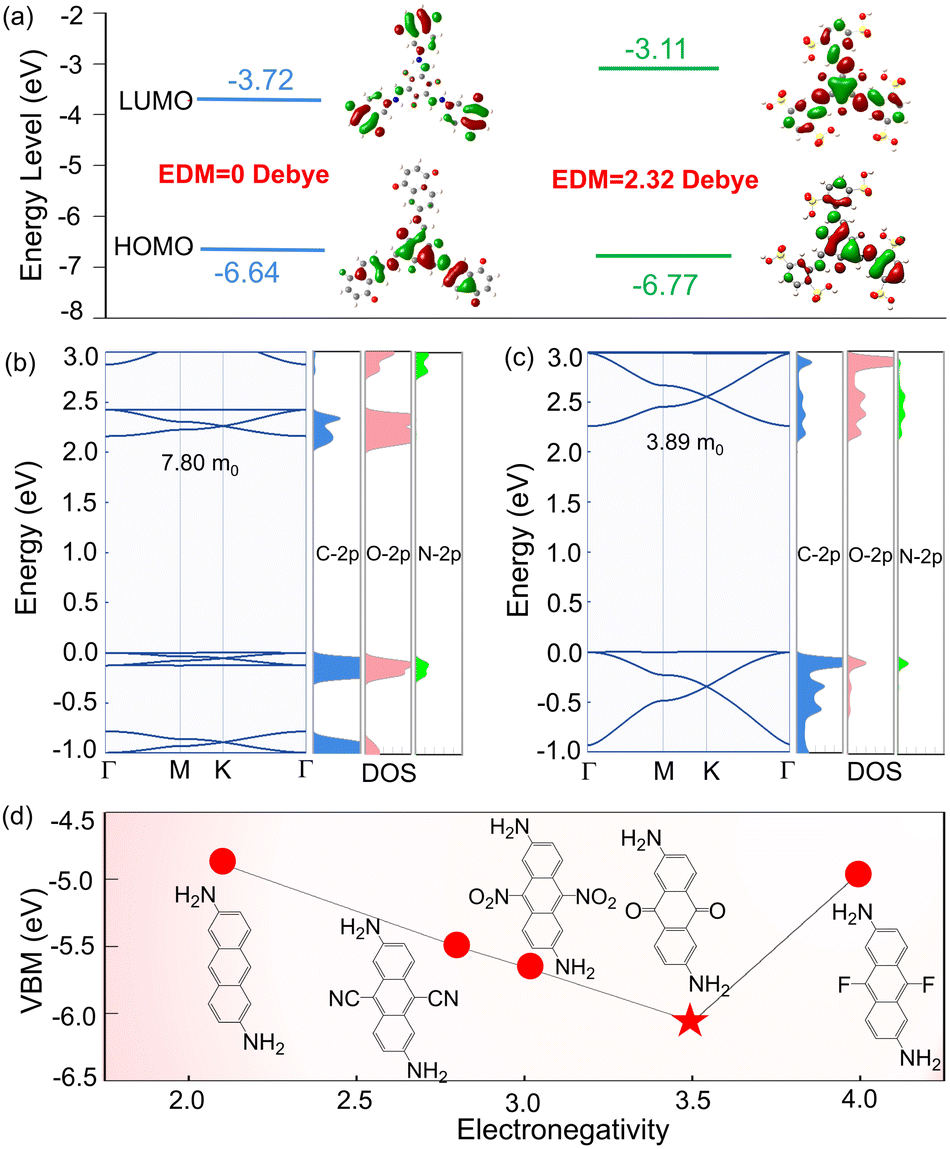 | ||
| Fig. 3 (a) Calculated HOMO/LUMO of Tp-DAAQ (left panel) and Tp-DABDA (right panel) molecules. Red values represent dipole moments (EDMs). (b) Band structure (left panel) and density of states (right panel) in 2D Tp-DAAQ. (c) Band structure (left panel) and density of states (right panel) in 2D Tp-DABDA. VBM is set to zero. (d) Calculated VBMs in 2D Tp-DAAQ with different electron-accepting substituents. | ||
The other question that arises is whether all β-ketoenamine linked 2D COFs with symmetric electron-accepting substituents in the linear diamine linkers are likely to be suitable band edges for PeLEDs? To address this query, we calculated their band edges by replacing O with substituents –CN, –NO2, and –F in 2D Tp-DAAQ, as shown in Fig. 3d. This presents an inverted volcanic relationship, where the VBM is the lowest and the band gap is the largest when the electron-accepting substituent is O. While the systems with substituents –CN, –NO2, and –F, the positions of VBMs and CBMs are not suitable for ETLs (Table S4†). We have studied the effect of electron-accepting substituents in the diamine linkers on the band edge, so how about the band edges of 2D COFs of the diamine linkers with electron-donating substituents? To this end, –OH and –CH3 are used to replace O in the diamine linker in 2D Tp-DAAQ, thus calculating their band edges to answer this question. The calculated VBMs and CBMs are listed in Table S4,† and excessively high VBMs make them unsuitable for ETLs. The above results demonstrate that β-ketoenamine linked symmetric 2D Tp-DAAQ and 2D Tp-DABDA are potential candidates for ETLs in PeLEDs.
Mobility is also a factor that affects transport efficiency.10 Excessive mobility can cause excess electrons to aggregate at the interface of ETL/EML, while low mobility cannot transport electrons. Because the systems are large, calculating their mobility is expensive. Accordingly, we evaluate their mobility by calculating the effective mass. The larger the effective mass, the smaller the mobility. Calculations show that the effective masses of 2D Tp-DAAQ and 2D Tp-DABDA are 7.80 and 3.89 m0, respectively, which are greater than the widely used effective mass of 2.95 m0 of monolayer ZnO (Fig. S14†). The reduced mobility compensates for the high mobility of ZnO and reduces the carrier aggregation at the ETL/EML interface, resulting in an increase of carrier recombination.
In general, more donor is more favorable for transporting electrons in the donor–acceptor structures.55–58 However, if the donor–acceptor structure is used as ETLs in PeLEDs, two conditions need to be satisfied simultaneously: (1) the energy level should be matched with that of the perovskite; (2) the transport rate should be similar to that of the hole transport rate. The only ones that fulfill the above two conditions are 2D Tp-DAAQ and 2D Tp-DABDA of the donor–acceptor structures.
To understand the charge transfer between the ETL and EML interfaces, we built an ETL/EML heterojunction to calculate its charge transfer. CsPbI3, as a direct band gap material, possesses advantages such as low density of defect states and is a potential high-quality EML material in near-infrared PeLEDs (Fig. 2).59,60 Due to the large size (632 atoms) of 2D Tp-DAAQ/CsPbI3 (Fig. S15†), it is impractical to perform calculations on it. Accordingly, only 2D Tp-DABDA/CsPbI3 heterojunction is constructed as an example to study the transport efficiency. In 2D Tp-DABDA/CsPbI3 heterojunction, van der Waals interactions between the interfaces are found due to the Pb–O distance (3.69 Å) being larger than the covalent radii of Pb (1.46 Å) and O (0.66 Å)61 and approximately equal to the van der Waals radii of Pb (2.3 Å) and O (1.4 Å).62 See ESI† for computational details and stability discussions, including electron localization function (ELF) and deformation charge density (DCD) (Fig. S16a and b†).
To intuitively determine the charge transfer at the heterojunction interface, the plane-averaged charge-density difference (Δρ) is calculated along the z direction as Δρ = ρheterojunction − ρCsPbI3 − ρTp-DABDA, where ρheterojunction, ρCsPbI3, and ρTp-DABDA represent the plane-averaged charge-densities of heterojunction and isolated CsPbI3 and 2D Tp-DABDA, respectively. Δρ > 0 for electron accumulation; Δρ < 0 for electron depletion. Fig. S14d† shows the direction of electron transport from 2D Tp-DABDA to CsPbI3 at the 2D Tp-DABDA/CsPbI3 interface. The above analysis confirms that 2D Tp-DAAQ and 2D Tp-DABDA do have potential as ETLs in near-infrared PeLEDs.
Suppression of e–h nonradiative recombination is an effective way to obtain high-performance PeLEDs under the premise of carrier injection balance.63 When an electron or hole is excited to an excited state, the nonradiative recombination will occur by the action of a single particle such as a phonon. However, the process of nonradiative recombination is complex, including generalized recombination dynamics of the photo-generated carriers and interfacial induced dynamics.64 Understanding the origin of nonradiative recombination and suppressing it is key to achieving high-performance PeLEDs. Here, we investigated the interface-induced nonradiative recombination in 2D Tp-DAAQ and 2D Tp-DABDA by the NAMD method using Hefei-NAMD software. Since HSE06 has the same band gap trend as the PBE (Table S2†), excited state calculations and discussions are conducted using the PBE approach to minimize the computational cost. The time scales of e–h nonradiative recombination for 2D Tp-DAAQ and 2D Tp-DABDA are 8.24 and 0.88 ns, respectively, as shown in Fig. 4a. e–h recombination time scales (τ) are fitted using an exponential function P(t) = exp(−t/τ). In NAMD simulations, the main factor of e–h recombination time is the nonadiabatic coupling (NAC), the larger the NAC, the faster the excited state process.34 The equation is given by:
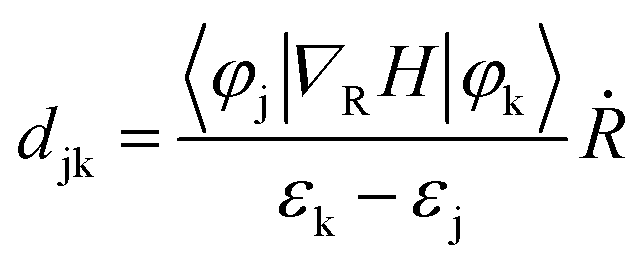
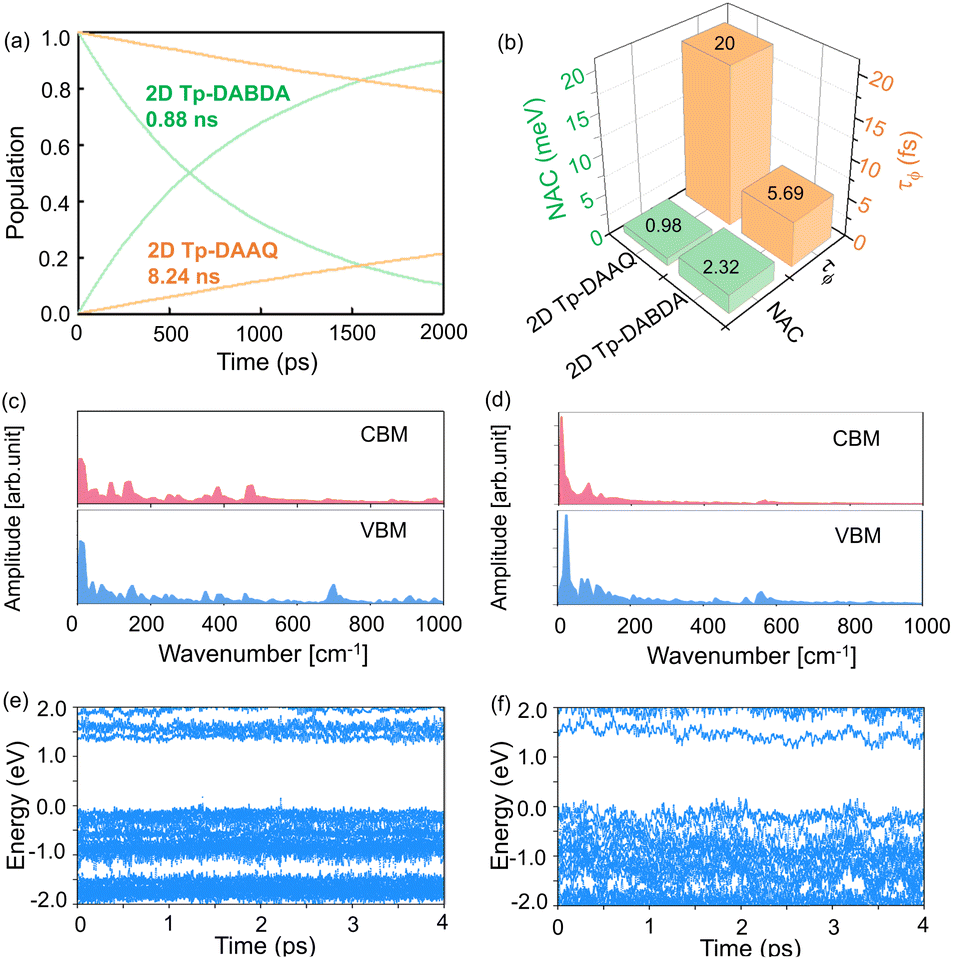 | ||
| Fig. 4 (a) e–h recombination time scale in 2D Tp-DAAQ and 2D Tp-DABDA. (b) Nonadiabatic coupling (NAC) and decoherence time (τϕ) in 2D Tp-DAAQ and 2D Tp-DABDA. Fourier transform for (c) 2D Tp-DAAQ and (d) 2D Tp-DABDA. Energy evolution of KS states in (e) 2D Tp-DAAQ and (f) 2D Tp-DABDA. | ||
Since the studied e–h nonradiative recombination times are generally longer, the dephasing time (τϕ) can have a significant effect on them.65 The larger the τϕ, the faster the dynamics process and the faster the nonradiative recombination. There are many methods to calculate τϕ, here, the DISH method is used to calculate the quantum dephasing time in both systems. The calculated τϕ of 2D Tp-DAAQ and 2D Tp-DABDA are 20 and 5.69 fs, respectively (Fig. 4b). By comparing the results in Fig. 4a, it can be found that the determinant of NAC is greater than the effect of τϕ in this system. Calculations based on ground and excited states of 2D COFs confirm that β-ketoenamine linked 2D Tp-DAAQ with anthraquinone diamine linker is the most promising ETL candidate for near-infrared PeLEDs.
4. Conclusion
In summary, we propose a material design guide to discover the ETLs as near-infrared PeLEDs by high-throughput and NAMD simulations, as demonstrated in β-ketoenamine linked 2D COFs with an anthraquinone linear diamine linker. We first perform computational screening in the core-cof/Core-cof-Database, a large experimental structure library containing 400 2D COF crystal structures with hcb topology. By calculating the energy band structures, we identify a total of 23 possible ETL candidates consisting of Tp and linear diamines in near-infrared PeLEDs. Among them, 2D Tp-DAAQ and 2D Tp-DABDA with symmetric electron-accepting substituents can significantly reduce the VBMs and CBMs, resulting in them have more suitable band edges. Furthermore, the EDM of Tp-DAAQ molecule is 0 debye, while the EDM of Tp-DABDA molecule is 2.3 debye, making Tp-DAAQ more likely to become an ETL material. Finally, the NAMD calculations show that the e–h recombination time scales of 2D Tp-DAAQ and 2D Tp-DABAQ are 8.24 ns and 0.88 ns, respectively, which indicates that 2D Tp-DAAQ with anthraquinone diamine linker is the most promising candidate for ETL in near-infrared PeLEDs.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was financially supported by the National Natural Science Foundation of China (No. 52202247, 22279059) and the Training Program of the Major Research Plan of the National Natural Science Foundation of China (91964103), the Fundamental Research Funds for the Central Universities (No. 30921011106).Notes and references
- B. Guo, R. Lai, S. Jiang, L. Zhou, Z. Ren, Y. Lian, P. Li, X. Cao, S. Xing, Y. Wang, W. Li, C. Zou, M. Chen, Z. Hong, C. Li, B. Zhao and D. Di, Nat. Photonics, 2022, 16, 637–643 CrossRef CAS.
- W. Chen, Z. Huang, H. Yao, Y. Liu, Y. Zhang, Z. Li, H. Zhou, P. Xiao, T. Chen, H. Sun, J. Huang and Z. Xiao, Nat. Photonics, 2023, 17, 401–407 CrossRef CAS.
- S. Chu, W. Chen, Z. Fang, X. Xiao, Y. Liu, J. Chen, J. Huang and Z. Xiao, Nat. Commun., 2021, 12, 147 CrossRef CAS.
- M. Liu, Q. Wan, H. Wang, F. Carulli, X. Sun, W. Zheng, L. Kong, Q. Zhang, C. Zhang, Q. Zhang, S. Brovelli and L. Li, Nat. Photonics, 2021, 15, 379–385 CrossRef CAS.
- H. Tsai, S. Shrestha, R. A. Vilá, W. Huang, C. Liu, C.-H. Hou, H.-H. Huang, X. Wen, M. Li, G. Wiederrecht, Y. Cui, M. Cotlet, X. Zhang, X. Ma and W. Nie, Nat. Photonics, 2021, 15, 843–849 CrossRef.
- E. Jang and H. Jang, Chem. Rev., 2023, 123, 4663–4692 CrossRef CAS PubMed.
- S. O. Jeon, K. H. Lee, J. S. Kim, S.-G. Ihn, Y. S. Chung, J. W. Kim, H. Lee, S. Kim, H. Choi and J. Y. Lee, Nat. Photonics, 2021, 15, 208–215 CrossRef CAS.
- Z. Zhu, Y. Wu, Y. Shen, J. Tan, D. Shen, M.-F. Lo, M. Li, Y. Yuan, J.-X. Tang, W. Zhang, S.-W. Tsang, Z. Guan and C.-S. Lee, Chem. Mater., 2021, 33, 4154–4162 CrossRef CAS.
- Y. Jiang, M. Cui, S. Li, C. Sun, Y. Huang, J. Wei, L. Zhang, M. Lv, C. Qin, Y. Liu and M. Yuan, Nat. Commun., 2021, 12, 336 CrossRef CAS PubMed.
- X. Li, M. Haghshenas, L. Wang, J. Huang, E. Sheibani, S. Yuan, X. Luo, X. Chen, C. Wei, H. Xiang, G. Baryshnikov, L. Sun, H. Zeng and B. Xu, ACS Energy Lett., 2023, 8, 1445–1454 CrossRef CAS.
- Z. Chen and S. Chen, Adv. Opt. Mater., 2022, 10, 2102404 CrossRef CAS.
- L. Xu, G. Liu, H. Xiang, R. Wang, Q. Shan, S. Yuan, B. Cai, Z. Li, W. Li, S. Zhang and H. Zeng, Appl. Phys. Rev., 2022, 9, 021308 CAS.
- L. Yang, X. Li, Q. Yang, S. Wang, H. Tian, J. Ding and L. Wang, Adv. Funct. Mater., 2020, 31, 2007686 CrossRef.
- K. Qasim, B. Wang, Y. Zhang, P. Li, Y. Wang, S. Li, S.-T. Lee, L.-S. Liao, W. Lei and Q. Bao, Adv. Funct. Mater., 2017, 27, 1606874 CrossRef.
- D. Chen, L. Ma, Y. Chen, X. Zhou, S. Xing, Y. Deng, Y. Hao, C. Pu, X. Kong and Y. Jin, Nano Lett., 2023, 23, 1061–1067 CrossRef CAS PubMed.
- C. S. Diercks and O. M. Yaghi, Science, 2017, 355, eaal1585 CrossRef PubMed.
- A. P. Cote, A. I. Benin, N. W. Ockwig, M. O'Keeffe, A. J. Matzger and O. M. Yaghi, Science, 2005, 310, 1166–1170 CrossRef CAS PubMed.
- T. Y. Zhou, S. Q. Xu, Q. Wen, Z. F. Pang and X. Zhao, J. Am. Chem. Soc., 2014, 136, 15885–15888 CrossRef CAS PubMed.
- L. Chen, C. Gong, X. Wang, F. Dai, M. Huang, X. Wu, C. Z. Lu and Y. Peng, J. Am. Chem. Soc., 2021, 143, 10243–10249 CrossRef CAS PubMed.
- L. Li, Q. Yun, C. Zhu, G. Sheng, J. Guo, B. Chen, M. Zhao, Z. Zhang, Z. Lai, X. Zhang, Y. Peng, Y. Zhu and H. Zhang, J. Am. Chem. Soc., 2022, 144, 6475–6482 CrossRef CAS PubMed.
- S. Dalapati, M. Addicoat, S. Jin, T. Sakurai, J. Gao, H. Xu, S. Irle, S. Seki and D. Jiang, Nat. Commun., 2015, 6, 7786 CrossRef CAS PubMed.
- M. Feng, Y. Zhang, X. Zhu, W. Chen, W. Lu and G. Wu, Angew Chem. Int. Ed. Engl., 2023, e202307195 CAS.
- D. Pakulski, A. Gorczynski, D. Brykczynska, V. Montes-Garcia, W. Czepa, I. Janica, M. Bielejewski, M. Kubicki, V. Patroniak, P. Samori and A. Ciesielski, Angew Chem. Int. Ed. Engl., 2023, e202305239 CAS.
- M. Yang, H. Hanayama, L. Fang, M. A. Addicoat, Y. Guo, R. Graf, K. Harano, J. Kikkawa, E. Jin, A. Narita and K. Mullen, J. Am. Chem. Soc., 2023, 145, 14417–14426 CrossRef CAS PubMed.
- M. Tong, Y. Lan, Q. Yang and C. Zhong, Chem. Eng. Sci., 2017, 168, 456–464 CrossRef CAS.
- M. Tong, Y. Lan, Z. Qin and C. Zhong, J. Phys. Chem. C, 2018, 122, 13009–13016 CrossRef CAS.
- C. R. DeBlase, K. E. Silberstein, T.-T. Truong, H. D. Abruña and W. R. Dichtel, J. Am. Chem. Soc., 2013, 135, 16821–16824 CrossRef CAS PubMed.
- Y. Peng, G. Xu, Z. Hu, Y. Cheng, C. Chi, D. Yuan, H. Cheng and D. Zhao, ACS Appl. Mater. Interfaces, 2016, 8, 18505–18512 CrossRef CAS PubMed.
- W. Jia, J. Fu, Z. Cao, L. Wang, X. Chi, W. Gao and L.-W. Wang, J. Comput. Phys., 2013, 251, 102–115 CrossRef.
- G. Kresse and J. Hafner, Phys. Rev. B: Condens. Matter Mater. Phys., 1993, 47, 558–561 CrossRef CAS.
- A. El-Ghayoury, C. Meziere, S. Simonov, L. Zorina, M. Cobian, E. Canadell, C. Rovira, B. Nafradi, B. Sipos, L. Forro and P. Batail, Chemistry, 2010, 16, 14051–14059 CrossRef CAS PubMed.
- J. P. Perdew, K. Burke and M. Ernzerhof, Phys. Rev. Lett., 1996, 77, 3865–3868 CrossRef CAS PubMed.
- J. Heyd, G. E. Scuseria and M. Ernzerhof, J. Chem. Phys., 2003, 118, 8207–8215 CrossRef CAS.
- Q. Zheng, W. Chu, C. Zhao, L. Zhang, H. Guo, Y. Wang, X. Jiang and J. Zhao, Wiley Interdiscip. Rev. Comput. Mol. Sci., 2019, 9, e1411 CrossRef CAS.
- S. Y. Ding and W. Wang, Chem. Soc. Rev., 2013, 42, 548–568 RSC.
- L. Cusin, H. Peng, A. Ciesielski and P. Samori, Angew Chem. Int. Ed. Engl., 2021, 60, 14236–14250 CrossRef CAS PubMed.
- P. Pachfule, A. Acharjya, J. Roeser, T. Langenhahn, M. Schwarze, R. Schomäcker, A. Thomas and J. Schmidt, J. Am. Chem. Soc., 2018, 140, 1423–1427 CrossRef CAS PubMed.
- P. Bhanja, S. Mishra, K. Manna, A. Mallick, K. Das Saha and A. Bhaumik, ACS Appl. Mater. Interfaces, 2017, 9, 31411–31423 CrossRef CAS PubMed.
- B. P. Biswal, S. Chandra, S. Kandambeth, B. Lukose, T. Heine and R. Banerjee, J. Am. Chem. Soc., 2013, 135, 5328–5331 CrossRef CAS PubMed.
- S. Chandra, S. Kandambeth, B. P. Biswal, B. Lukose, S. M. Kunjir, M. Chaudhary, R. Babarao, T. Heine and R. Banerjee, J. Am. Chem. Soc., 2013, 135, 17853–17861 CrossRef CAS PubMed.
- S. Chandra, T. Kundu, S. Kandambeth, R. BabaRao, Y. Marathe, S. M. Kunjir and R. Banerjee, J. Am. Chem. Soc., 2014, 136, 6570–6573 CrossRef CAS PubMed.
- S. K. Das, K. Bhunia, A. Mallick, A. Pradhan, D. Pradhan and A. Bhaumik, Microporous Mesoporous Mater., 2018, 266, 109–116 CrossRef CAS.
- S. Kandambeth, B. P. Biswal, H. D. Chaudhari, K. C. Rout, S. Kunjattu H, S. Mitra, S. Karak, A. Das, R. Mukherjee, U. K. Kharul and R. Banerjee, Adv. Mater., 2016, 29, 1603945 CrossRef PubMed.
- S. Kandambeth, A. Mallick, B. Lukose, M. V. Mane, T. Heine and R. Banerjee, J. Am. Chem. Soc., 2012, 134, 19524–19527 CrossRef CAS PubMed.
- M. A. Khayum, S. Kandambeth, S. Mitra, S. B. Nair, A. Das, S. S. Nagane, R. Mukherjee and R. Banerjee, Angew. Chem., Int. Ed., 2016, 55, 15604–15608 CrossRef CAS.
- M. S. Lohse, T. Stassin, G. Naudin, S. Wuttke, R. Ameloot, D. De Vos, D. D. Medina and T. Bein, Chem. Mater., 2016, 28, 626–631 CrossRef CAS.
- D. B. Shinde, H. B. Aiyappa, M. Bhadra, B. P. Biswal, P. Wadge, S. Kandambeth, B. Garai, T. Kundu, S. Kurungot and R. Banerjee, J. Mater. Chem. A, 2016, 4, 2682–2690 RSC.
- Q. Sun, B. Aguila, L. D. Earl, C. W. Abney, L. Wojtas, P. K. Thallapally and S. Ma, Adv. Mater., 2018, 30, 1705479 CrossRef PubMed.
- H. Wang, C. Qian, J. Liu, Y. Zeng, D. Wang, W. Zhou, L. Gu, H. Wu, G. Liu and Y. Zhao, J. Am. Chem. Soc., 2020, 142, 4862–4871 CrossRef CAS PubMed.
- S. Wang, Q. Wang, P. Shao, Y. Han, X. Gao, L. Ma, S. Yuan, X. Ma, J. Zhou, X. Feng and B. Wang, J. Am. Chem. Soc., 2017, 139, 4258–4261 CrossRef CAS PubMed.
- H. Zhong, R. Sa, H. Lv, S. Yang, D. Yuan, X. Wang and R. Wang, Adv. Funct. Mater., 2020, 30, 2002654 CrossRef CAS.
- Y. Li, Y. Liao and Z. Chen, Angew Chem. Int. Ed. Engl., 2014, 53, 7248–7252 CrossRef CAS PubMed.
- S. Adjokatse, H.-H. Fang and M. A. Loi, Mater. Today, 2017, 20, 413–424 CrossRef CAS.
- S. W. Lee, H. J. Shin, B. Park, S. Shome, D. R. Whang, H. Bae, S. Chung, K. Cho, S. J. Ko, H. Choi and D. W. Chang, ACS Appl. Mater. Interfaces, 2022, 14, 19785–19794 CrossRef CAS PubMed.
- B. Albinsson, M. P. Eng, K. Pettersson and M. U. Winters, Phys. Chem. Chem. Phys., 2007, 9, 5847 RSC.
- L. G. Franca, A. Danos and A. Monkman, J. Phys. Chem. Lett., 2023, 14, 2764–2771 CrossRef CAS PubMed.
- K. Müllen and W. Pisula, J. Am. Chem. Soc., 2015, 137, 9503–9505 CrossRef PubMed.
- P. Pachfule, A. Acharjya, J. Roeser, R. P. Sivasankaran, M.-Y. Ye, A. Brückner, J. Schmidt and A. Thomas, Chem. Sci., 2019, 10, 8316–8322 RSC.
- J. Shi, F. Li, Y. Jin, C. Liu, B. Cohen-Kleinstein, S. Yuan, Y. Li, Z. K. Wang, J. Yuan and W. Ma, Angew Chem. Int. Ed. Engl., 2020, 59, 22230–22237 CrossRef CAS PubMed.
- B. Han, B. Cai, Q. Shan, J. Song, J. Li, F. Zhang, J. Chen, T. Fang, Q. Ji, X. Xu and H. Zeng, Adv. Funct. Mater., 2018, 28, 1804285 CrossRef.
- B. Cordero, V. Gómez, A. E. Platero-Prats, M. Revés, J. Echeverría, E. Cremades, F. Barragán and S. Alvarez, Dalton Trans., 2008, 2832–2838 RSC.
- A. Bondi, J. Phys. Chem., 2002, 68, 441–451 CrossRef.
- Y. Sun, L. Ge, L. Dai, C. Cho, J. Ferrer Orri, K. Ji, S. J. Zelewski, Y. Liu, A. J. Mirabelli, Y. Zhang, J. Y. Huang, Y. Wang, K. Gong, M. C. Lai, L. Zhang, D. Yang, J. Lin, E. M. Tennyson, C. Ducati, S. D. Stranks, L. S. Cui and N. C. Greenham, Nature, 2023, 615, 830–835 CrossRef CAS PubMed.
- D. Luo, R. Su, W. Zhang, Q. Gong and R. Zhu, Nat. Rev. Mater., 2019, 5, 44–60 CrossRef.
- Y. Wang and R. Long, J. Phys. Chem. Lett., 2020, 11, 1601–1608 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3ta05200a |
| This journal is © The Royal Society of Chemistry 2024 |