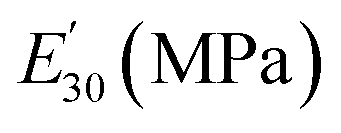Multi-functional syringol based epoxides and properties of their thermoset polymers
Received
21st June 2024
, Accepted 26th July 2024
First published on 13th August 2024
Abstract
Lignin-based syringol monomer, 2,6-dimethoxypropylphenol (DMPP), can be upgraded through two synthetic routes to multi-functional propylpyrogallol DMPPO. With three hydroxyl groups, DMPPO can serve as a renewable polymer building block. In this study, we describe the synthesis and characterization of DMPPO epoxide mixtures based on their synthesis methodology; one route uses HBr, another a greener methodology with Nb2O5 catalyst in water. Epoxy thermoset polymers using the two different epoxide preparations are synthesized, characterized, and contrasted. Optimization of reaction conditions, including temperature and catalyst concentration, to enhance the performance of epoxy thermosets is discussed. Through dynamic mechanical analysis (DMA) and thermogravimetric analysis (TGA), the thermo-mechanical behavior and thermal stability of the resulting epoxy thermoset networks are evaluated, shedding light on how structural differences impact performance.
Sustainability spotlight
The urgency of transitioning to a sustainable chemical industry has become increasingly evident. For instance, the petrochemical origins and toxic health impacts of bisphenol A (BPA) highlight the need for safer and more sustainable alternatives. Consequently, there is growing interest in using lignin-based aromatic monomers to develop bio-based building blocks and polymers with enhanced properties. This study presents two synthesis routes to lignin-based syringol monomer and tri-epoxide which allows for further customization in polymer applications, specifically epoxy thermosets. This work aligns with the UN's Sustainable Development Goals: 12 (responsible consumption and production), 13 (climate action), and 15 (life on land).
|
Introduction
Epoxy thermosets are a class of polymers that are widely used in various industrial applications such as adhesives, automotive body parts, and coatings.1 Epoxies are known for their excellent mechanical properties, chemical resistance, and thermal stability.1,2 Up to 90% of epoxy thermosets are made from petroleum-based bisphenol A (BPA); however, because of toxicity concerns and the goal of independence from fossil fuel feedstocks, there is increasing interest in BPA alternatives.3,4 Efforts to create bio-based materials led to the use of vegetable oils such as soybean or palm and sucrose ester fatty acids as renewable building blocks.5,6 Another valuable feedstock that has gained attention is lignocellulosic biomass, which is composed of three bio-polymers: cellulose, hemicellulose, and lignin. The carbohydrates can be upgraded to chemical building blocks such as furfural and 5-hydroxymethylfurfural (HMF).7–10 Lignin, the second most abundant bio-polymer on earth, is made from aromatic building blocks. It is also a byproduct of the pulp and paper industry and is underutilized.11–13 Lignin is an amorphous 3D-dimensional polyphenolic polymer composed of three primary units: sinapyl (3,5-dimethoxy-4-hydroxy-cinnamyl), coniferyl (3-methoxy-4-hydroxy-cinnamyl), and p-coumaryl (4-hydroxycinnamyl) alcohols, linked by ether and/or C–C bonds.14,15 Although, lignin's complexity and heterogeneity has made its valorization difficult, lignin depolymerization, albeit in moderate yields, affords bio-based phenolic compounds that can act as building blocks for polymeric materials.16,17
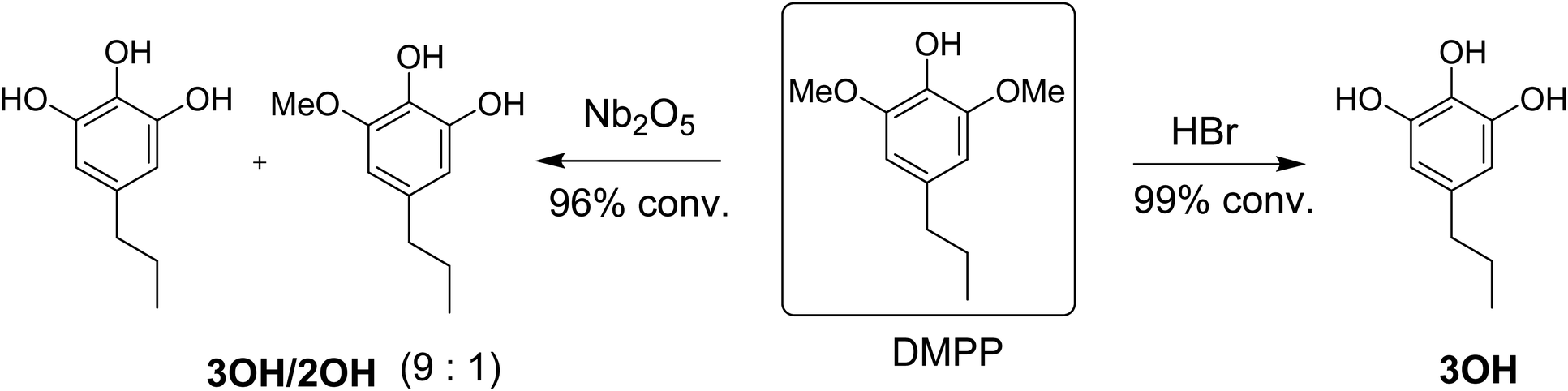
One approach for making lignin based material is to blend epoxy-lignin with resorcinol diglycidyl ether, keratin, and vegetable oils.18,19 However, composite materials incorporating technical lignin have limitations because of its structural heterogeneity and irregularity.20 Another category of lignin utilization for epoxy thermosets is to make epoxies from lignin monomers. For example, Zhao et al. investigated several modifications for lignin based dihydroeugenol (DHE) epoxy networks, including o-demethylation and phenol-formaldehyde reactions.21–24 Modifying lignin monomers can lead to enhanced and consistent materials properties with high renewable content.21,25 Recently, our research group reported on reductive catalytic fractionation (RCF) of high-S poplar using Pd–Zn/C catalyst with Pd![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :Zn ratio of 1:10.26 The reaction gives high yields of bio-phenol monomers, up to 60 wt% of the lignin component in poplar wood. The major bio-phenol product DMPP can be upgraded to DMPPO by reaction with either (1) HBr or (2) a greener, halogen-free Nb2O5 catalyst in water (Scheme 1). The reaction with HBr produces almost exclusively DMPPO (>95% selectivity), referred to from hereon as 3OH, while the reaction with Nb2O5 in water produces a mixture of DMPPO and MeO-DHEO in 9:1 ratio, referred to herein as 3OH/2OH (Scheme 1). With its chemical structure and untapped potential, DMPPO is a high-functional compound well suited for use as a polymer building block. The three hydroxyl groups offer a unique platform for enabling customization in polymer applications. In this report, we describe the use of this platform for making epoxy thermosets and investigate how the method of DMPPO preparation without extensive purification (3OHversus3OH/2OH in Scheme 1) impact the mechanical properties of the resulting polymer networks.
:Zn ratio of 1:10.26 The reaction gives high yields of bio-phenol monomers, up to 60 wt% of the lignin component in poplar wood. The major bio-phenol product DMPP can be upgraded to DMPPO by reaction with either (1) HBr or (2) a greener, halogen-free Nb2O5 catalyst in water (Scheme 1). The reaction with HBr produces almost exclusively DMPPO (>95% selectivity), referred to from hereon as 3OH, while the reaction with Nb2O5 in water produces a mixture of DMPPO and MeO-DHEO in 9:1 ratio, referred to herein as 3OH/2OH (Scheme 1). With its chemical structure and untapped potential, DMPPO is a high-functional compound well suited for use as a polymer building block. The three hydroxyl groups offer a unique platform for enabling customization in polymer applications. In this report, we describe the use of this platform for making epoxy thermosets and investigate how the method of DMPPO preparation without extensive purification (3OHversus3OH/2OH in Scheme 1) impact the mechanical properties of the resulting polymer networks.
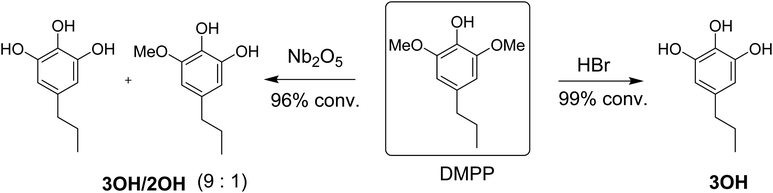 |
| | Scheme 1 Demethylation of DMPPvia HBr to produce DMPPO (3OH) versus Nb2O5 catalyzed reaction in water to make a mixture of DMPPO and DHEO (3OH/2OH) in 9:1 ratio. | |
Experimental
Materials
All commercial chemicals were purchased and used as received. 4-Allyl-2,6-dimethoxyphenol, palladium (5 wt%) on activated carbon, epichlorohydrin (ECH), tetrabutylammonium bromide, and diethylenetriamine (DETA), were purchased from Sigma-Aldrich. Methanol (ACS reagent grade) and ethyl acetate (ACS regent grade) were purchased from Fisher Chemical. Cetyltrimethyl ammonium bromide (CTAB 98%) was purchased from Alfa Aesar. Niobium(V) chloride (≥99%) was purchased from Strem Chemicals. Water was obtained from a A10 Milli-Q water purification system by Millipore.
Catalyst preparation
The catalyst Nb2O5 was synthesized using a modified hydrothermal method following literature procedures.26,27 The precursor NbCl5 (5.4 g, 20 mmol) was dissolved in 20 mL ethanol and stirred for 10 min. The solution was added to an aqueous solution of CTAB (1 g in 15 mL distilled water) dropwise. The mixed solution was stirred for 0.5 h followed by adding 20 mL of aqueous HCl (pH 1), and stirring for another 1.5 h. The resulting solution was put into a Teflon-lined autoclave and aged at 160 °C for 24 h. The formed precipitate was separated and washed with distilled water and dried at 60 °C overnight. The sample was ground and packed for calcination in air in a Thermolyne F6020 1200C Muffle furnace. The ramping rate of the furnace was pre-set to 1 °C min−1. After 6 h calcination at 450 °C, the active Nb2O5 catalyst was collected at room temperature.
Synthesis of 2,6-dimethoxypropylphenol (DMPP)
4-Allyl-2,6-dimethoxyphenol i-DMPP (3 g) was added to a 75 mL stainless-steel reactor vessel with 5 wt% palladium on carbon (0.300 g) and ethanol (15 mL). The reactor vessel was sealed, purged, and pressurized with 10 bar hydrogen gas. The pressurized reactor vessel was heated to 50 °C and held for 5 h, after which the mixture was filtered and concentrated with a rotary evaporator. DMPP was obtained at 95% yield and used without further purification.
Synthesis of 5-propylpyrogallol (DMPPO)
Using HBr.
DMPP (3.0 g, 15 mmol) was added to 30 g of 48% aqueous hydrobromic acid. The reaction mixture was stirred at 115 °C for 20 h, cooled to room temperature, saturated with NaCl, and extracted three times with ethyl acetate. The organic layer was dried over Na2SO4 and concentrated using rotary evaporation. The o-demethylated product 3OH was obtained in 96% isolated yield with selectivity only to DMPPO and used without further purification.
Using Nb2O5.
The reaction with Nb2O5 was carried out in a 75 mL stainless-steel reactor vessel. DMPP (500 mg) was loaded into the vessel with 250 mg Nb2O5, 12 mL water, and 0.8 mL of MeOH. The reactor vessel was sealed, purged with N2, and pressurized with 10 bar nitrogen gas. The reactor vessel was heated to 230 °C for 20 h, after which the reaction mixture was cooled to room temperature and products in the liquid phase were extracted into ethyl acetate by centrifugation at 5000 rpm for 5 min. The organic layer was separated and concentrated by rotary evaporation. Analysis and yield of 3OH/2OH was done as described in our previous work.26 Due to time and scalability limitations, the DMPP used for the Nb2O5 route here is from purchased i-DMPP.
Synthesis of glycidyl ethers of DMPPO (DMPPO epoxides)
The reaction of 3OH or 3OH/2OH and epichlorohydrin can lead to side reactions like intramolecular cyclization between two adjacent oxiranes.3,28 Temperature and NaOH concentration can affect the yield and epoxy content of the product. Different temperatures (40, 60, 80, and 100 °C) and NaOH/phenolic hydroxyl molar ratios (0.5, 1, and 2), were investigated to determine optimal conditions. Mass ratio that reflected the conversion of DMPPO to epoxide products was obtained by measuring each weight. Epoxy equivalent weight (EEW) was determined by the HCl/acetone chemical titration method with the theoretical weight being 112 g per eq.29
The product 3OH (3.0 g, 18 mmol), epichlorohydrin (27 g, 292 mmol), and tetrabutylammonium bromide (0.29 g, 0.89 mmol) as a phase-transfer catalyst were heated at 80 °C for 3 h followed by a dropwise addition of 50% w/w NaOH solution. The reaction was stirred for another 5 h, and then washed with acetone, filtered to remove salt, and concentrated with a rotary evaporator to an 80% yield.
The product mixture 3OH/2OH from the Nb2O5 catalytic reaction (1.0 g) was reacted with epichlorohydrin (7.0 g, 76 mmol). Tetrabutylammonium bromide (0.10 g, 0.34 mmol) was used as a phase-transfer catalyst. The mixture was heated at 80–100 °C for 3 h and followed by a dropwise addition of 50% w/w NaOH solution. The reaction was maintained at this temperature for another 5 h, and then the mixture was washed with acetone, filtered to remove salt, and concentrated by rotary evaporator. Epoxy equivalent weight (EEW) was determined by the HCl/acetone chemical titration method.29
Synthesis of thermoset polymer networks
The epoxide products made from DMPPO (3OH) or 3OH/2OH mixture were added to diethylenetriamine (DETA) in a 1:1 stoichiometric ratio of epoxy to –NH. The reaction mixture was stirred for 5 min, poured into a Teflon mold and cured at 30 °C for 2 h, 55 °C for 2 h, and 75 °C for 2 h.
Products analysis
Phenol and epoxide products were characterized by 1H NMR in chloroform-d1 on a Varian Unity Inova 500 MHZ spectrometer. For each analysis, 30 mg of sample was dissolved in 650 μL chloroform-d1.
ATR-FTIR was used to follow structural changes associated with deprotection of methoxy substituents on the phenol ring and installation of epoxide groups. Spectra were taken on a Nicolet iS10 FTIR instrument equipped with a diamond Attenuated Total Reflection (ATR) probe and scanned over 500–4000 cm−1.
Characterization of thermoset polymer networks
Thermal analysis.
Thermoset samples of 2–5 mg were placed into an Al2O3 crucible and suspended on a Pt hanging pan of a Discovery 5500 Thermo-Gravimetric Analyzer (TGA). The sample was heated from 50 °C to 500 °C at a heating rate of 20 °C min−1 under nitrogen flow of 40 mL min−1.
Dynamic mechanical analysis (DMA).
Mechanical properties of thermoset samples were characterized using a TA Instruments DMA850 with a tension clamp. DMA samples dimensions were 20 mm × 6 mm × 1 mm (length, width, thickness). Measurements were conducted from 0 °C to 130 °C at a ramp rate of 4 °C min−1 and a frequency of 1 Hz.
Results and discussion
Synthesis of epoxides
The synthesis of DMPP required hydrogenation of the allyl group in commercially purchased i-DMPP. The peaks in the 1H spectrum at δ 5.95, 5.07, and 3.31 ppm disappear (Panels a and b in Fig. 1). Signals at δ 2.51, 1.62, and 0.94 ppm are observed due to the propyl chain while the peak at 3.88 ppm is due to the –OCH3 substituents (singlet, 6H) of DMPP (panel b in Fig. 1). After the o-demethylation by HBr (Panel c, Fig. 1), the –OCH3 singlet disappears entirely, and a slight upfield shift is observed for the aromatic –CH and benzylic –CH2. The same major product (DMPPO) is observed in the o-demethylation over Nb2O5 (panel d), but the –OCH3 peak at 3.86 ppm does not entirely disappear as DHEO is left over as a side product. The mixture 3OH/2OH based on NMR integration represents DMPPO:DHEO in an approximate 9:1 ratio. Depending on the Nb2O5 catalyst batch, the o-demethylation of DMPP produces 3OH/2OH in the range 8.5–9.5:1.
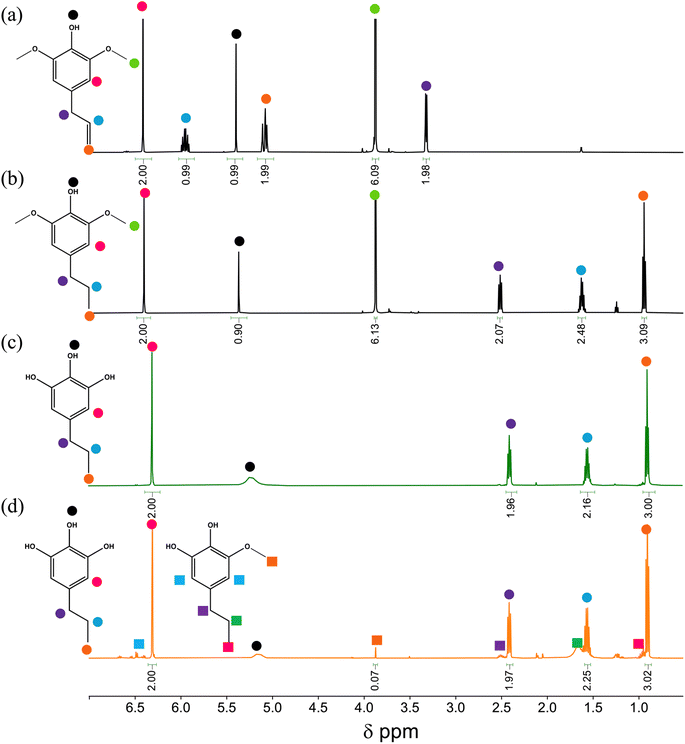 |
| | Fig. 1
1H NMR spectra of (a) i-DMPP (b) DMPP (c) 3OH and (d) 3OH/2OH. | |
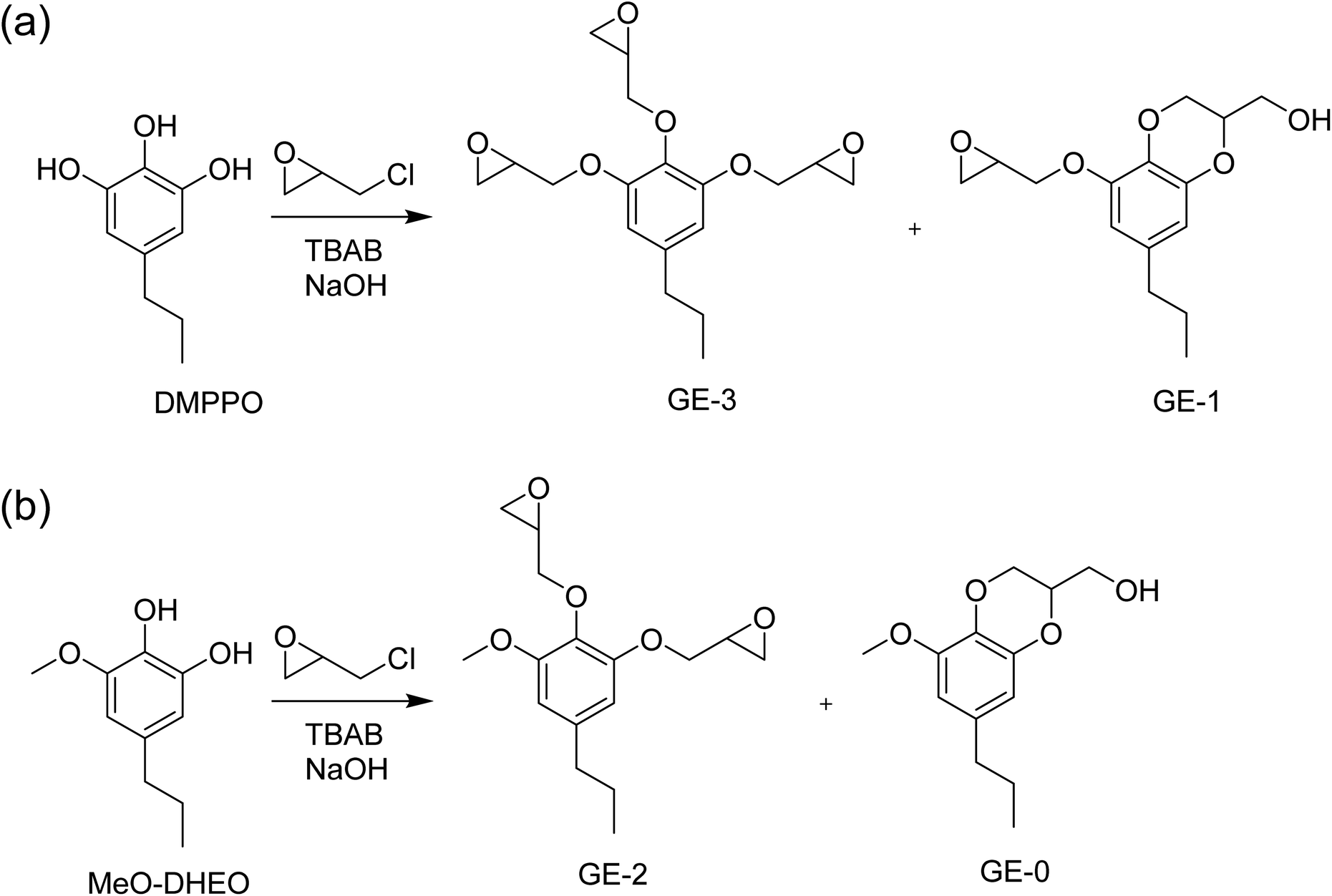
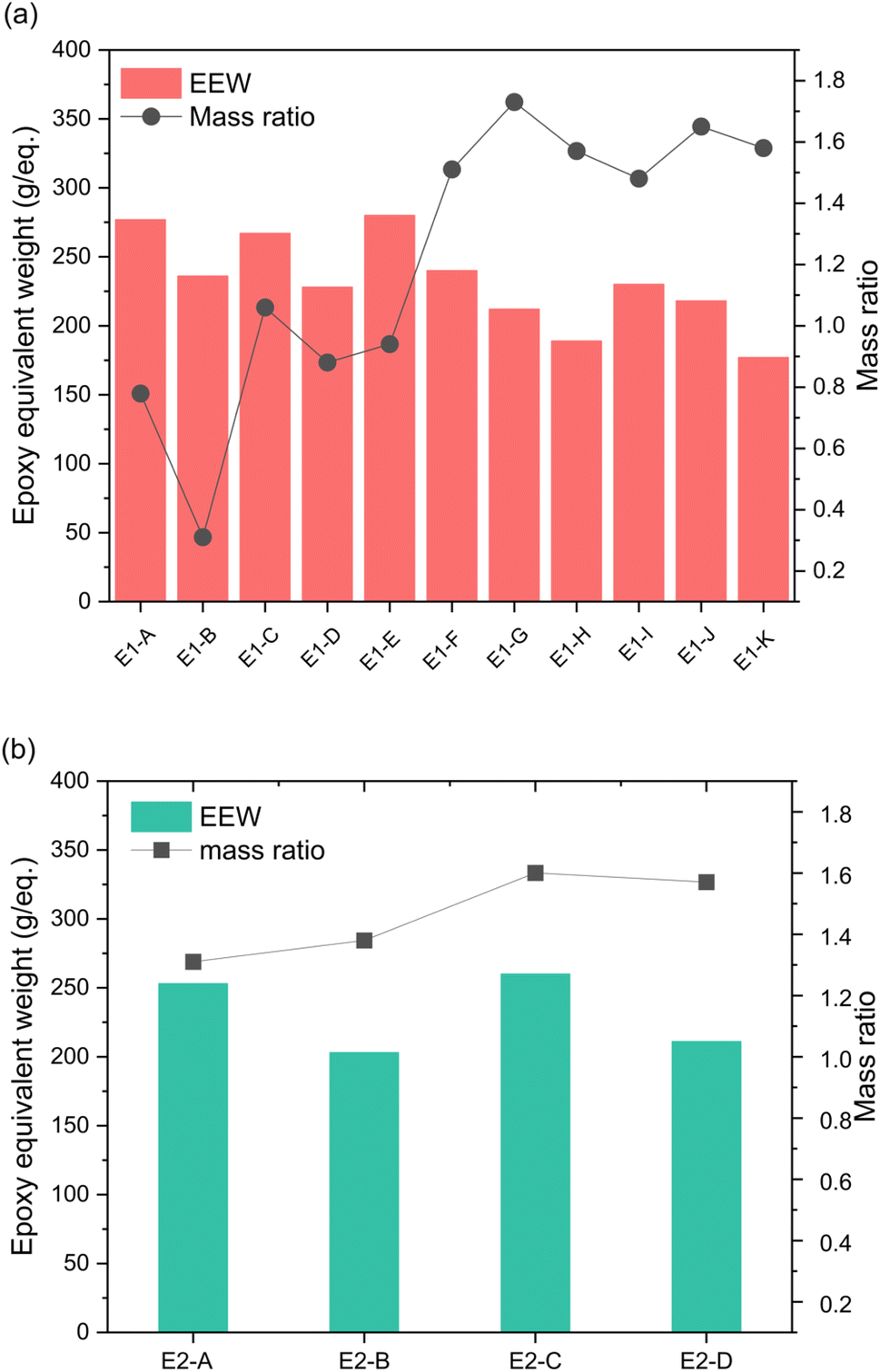
Epoxidations of 3OH and 3OH/2OH using ECH show dependence on temperature and catalyst concentration. Variation in conditions are tabulated in Table 1. Since the product using HBr to deprotect DMPP is almost exclusively DMPPO (3OH), the epoxides E1 are composed of the tri-epoxide GE-3 and its benzodioxane glycidyl ether GE-1 (Scheme 2). Molecular characterization of the epoxide products by NMR and IR are presented in the next section below. In contrast, the epoxide products from 3OH/2OH (E2 in Table 1) contain in addition the di-epoxide GE-2 and its benzodioxane glycidyl ether GE-0 (Scheme 2) as a result of MeO-DHEO being a persistent minor coproduct of the Nb2O5 catalyzed deprotection of DMPP. The mass ratio of epoxides E1A–K increases gradually as temperature increases from 40 to 100 °C (Fig. 2). The optimal molar amount of catalyst was determined based on the mass ratio and epoxy equivalent weight (EEW).
Table 1 Glycidation reaction conditions. For all entries 16 equiv. of ECH and 0.05 equiv. of TBAB were used
| Epoxide producta |
Temperature (°C) |
NaOH (M) |
EEW (g PER eq.) |
Mass ratiob |
|
All E1 epoxides are prepared from 3OH and all E2 epoxides from 3OH/2OH.
Mass ratio = 3OH or 3OH/2OH input (g)/isolated epoxide (g).
|
|
E1-A
|
40 |
1 |
277 |
0.78 |
|
E1-B
|
40 |
2 |
236 |
0.31 |
|
E1-C
|
60 |
0.5 |
267 |
1.06 |
|
E1-D
|
60 |
1 |
228 |
0.88 |
|
E1-E
|
60 |
2 |
280 |
0.94 |
|
E1-F
|
80 |
0.5 |
240 |
1.51 |
|
E1-G
|
80 |
1 |
212 |
1.73 |
|
E1-H
|
80 |
2 |
189 |
1.57 |
|
E1-I
|
100 |
0.5 |
230 |
1.48 |
|
E1-J
|
100 |
1 |
218 |
1.65 |
|
E1-K
|
100 |
2 |
177 |
1.58 |
|
E2-A
|
80 |
1 |
253 |
1.31 |
|
E2-B
|
80 |
2 |
203 |
1.38 |
|
E2-C
|
100 |
1 |
260 |
1.6 |
|
E2-D
|
100 |
2 |
211 |
1.57 |
 |
| | Scheme 2 (a) Reaction of DMPPO with ECH to form glycidyl ether GE-3 and the benzodioxane glycidyl ether GE-1. (b) Reaction of MeO-DHEO with ECH to form glycidyl ether GE-2 and the benzodioxane glycidyl ether GE-0. Numbering of GE products reflects the number of epoxide groups in the molecule. | |
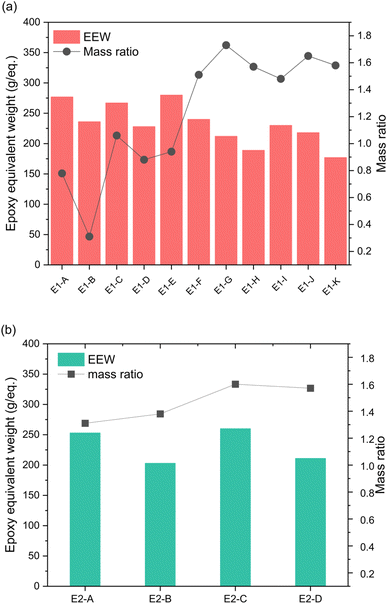 |
| | Fig. 2 Effect of temperature and NaOH concentration on EEW and the mass ratio of (a) 3OH and (b) 3OH/2OH epoxidation products. Mass ratio is the conversion of phenols to epoxides and is obtained by measuring the epoxides' weight. The x-axis represents the different experimental conditions shown in Table 1. | |
For epoxide synthesis starting with 3OH, the NaOH catalyst to phenolic hydroxyl molar ratio was varied from 0.5–2.0. At NaOH catalyst to phenolic hydroxyl molar ratio of 0.5 the lowest mass ratio and highest EEW were observed. Therefore, E1-G, E1-H, E1-J and E1-K were chosen as the optimal epoxides to cure and to study their properties.
Epoxides formed from E2-A through E2-D are GE-0 through GE-4, so temperature and catalyst effects were also studied. Since higher temperature (80–100 °C) and molar concentration of NaOH (1 M and 2 M) were favored for 3OH epoxies, these conditions were used for the 3OH/2OH studies. Similar to 3OH epoxies, mass ratio and epoxy content did not vary much between 80 and 100 °C with the EEW slightly higher at 1.0 M NaOH. Zhao et al., showed DHEO favored 1.0 M NaOH and a lower temperature of 60 °C.24 Due to the slightly lower EEW values, E2-B and E2-D were chosen as optimal expoxy samples for curing into thermosets.
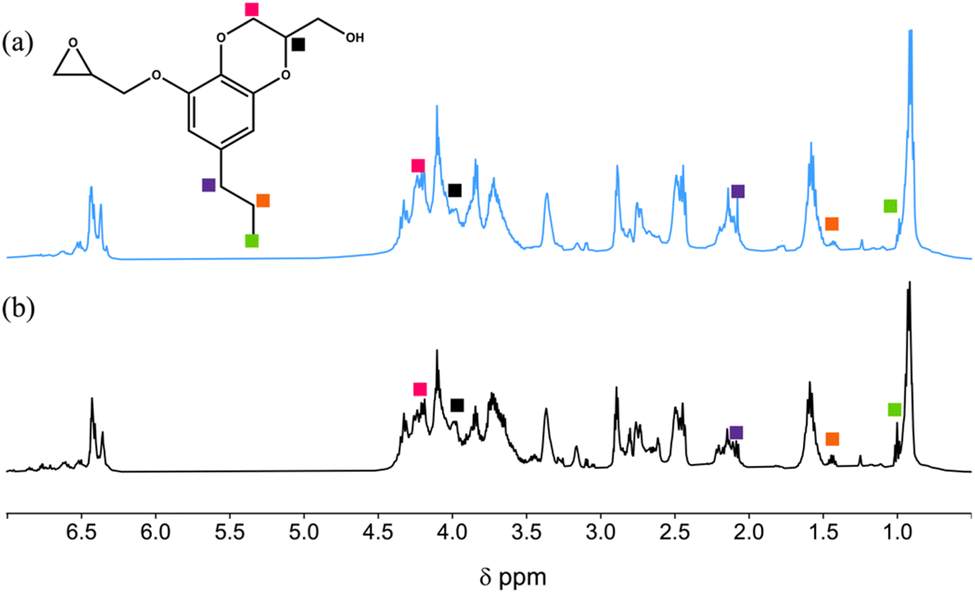
Characterization of epoxides E1 and E2
The resulting epoxides were characterized by 1H NMR. Fig. 3 displays representative 1H spectra for E1-H and E2-B. In both spectra, it is difficult to assign each peak because the epoxide products are mixtures of GE-0 through GE-3 as illustrated in Scheme 2 above. For example, in E2-B, the benzdioxane product GE-0 gives rise to a complex pattern that is difficult to assign definitively. Nevertheless, in both spectra, the benzodioxane coproducts GE-0 and GE-1 are observed at δ 1.00, 1.43, and 2.09 ppm (propyl chain), and peaks in the δ 4.11–4.35 ppm region due to the oxirane rings.
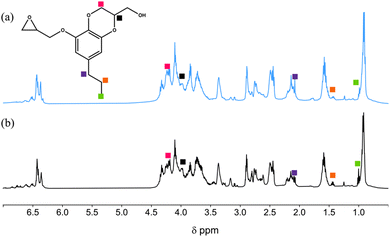 |
| | Fig. 3
1H NMR spectra of (a) E1-H and (b) E2-B epoxides. | |
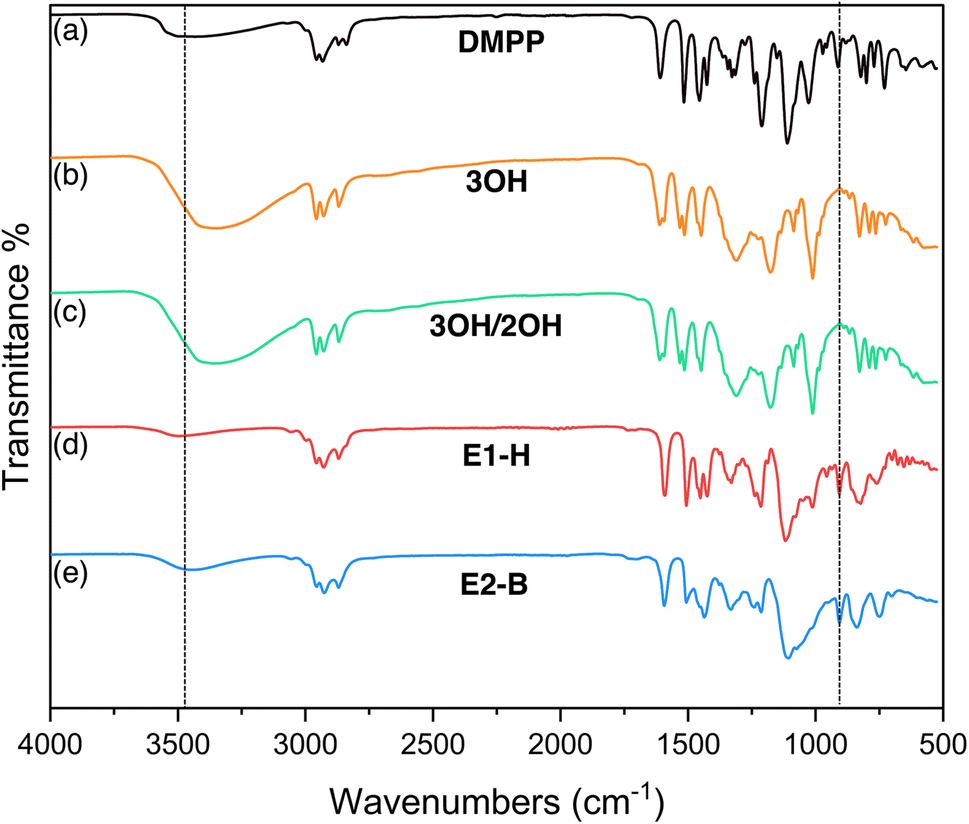
The epoxides were also characterized by FTIR. The O-demethylation of DMPP and formation of 3OH and 3OH/2OH show an increase in O–H stretching around 3331 cm−1. After epoxidation, bands at 927 cm−1 are characteristic of an epoxide ring and at 1015 cm−1 are characteristic of a C–O–C ether linkage are observed. The O–H bands significantly decrease as a result of epoxidation, indicating the hydroxyl groups are consumed but with possible benzodioxane side products GE-0 and GE-1 present at 3300 cm−1 (Fig. 4).
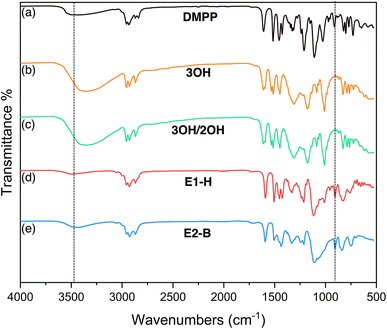 |
| | Fig. 4 FTIR spectra of (a) DMPP (b) 3OH (c) 3OH/2OH (d) E1-H (e) E2-B. The appearance and disappearance of the –OH band is highlighted at 3331 cm−1 and the appearance of the epoxy band around 927 cm−1 is highlighted. | |
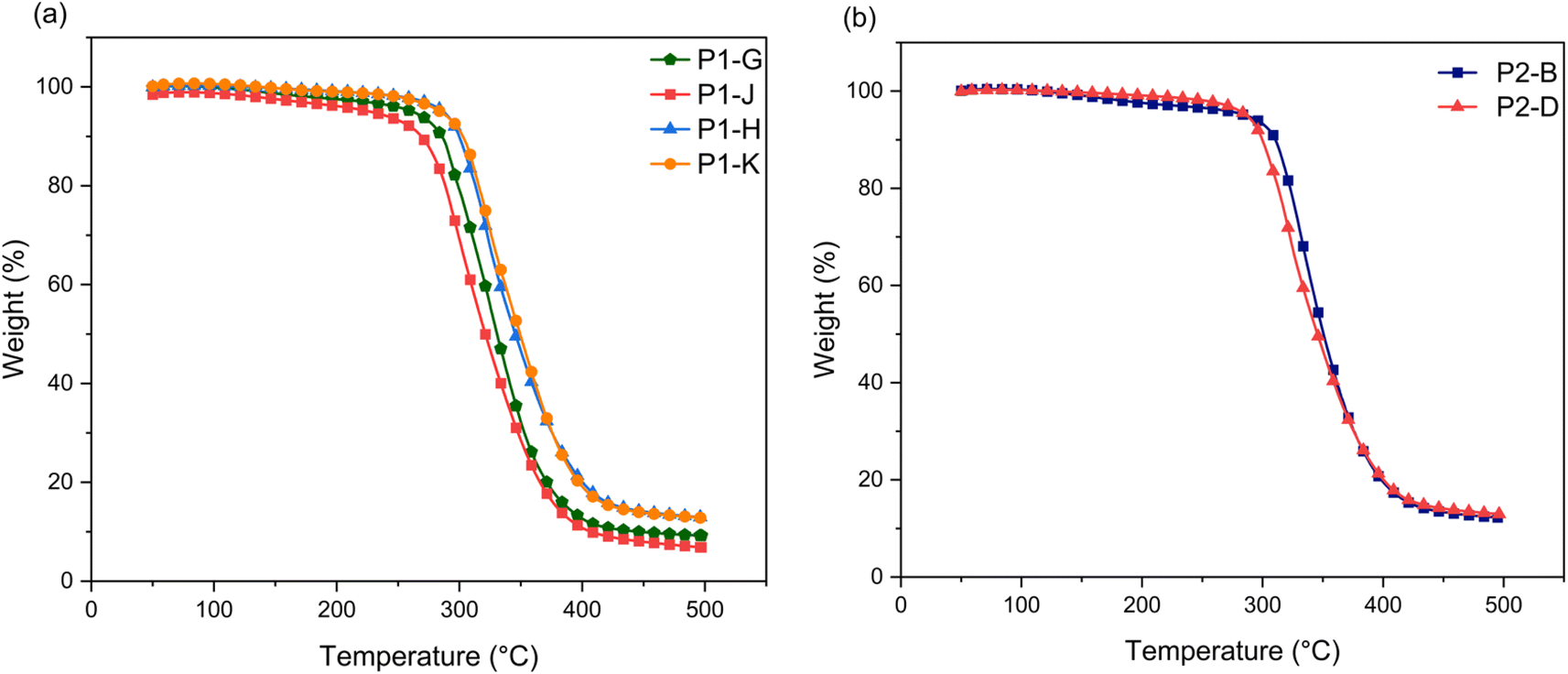
Thermal stability of epoxy thermoset networks
Epoxides of 3OH and 3OH/2OH with lower EEW and higher mass ratio were cured with a 1:1 stoichiometric ratio of DETA to form polymer networks (P). Thermogravimetric analysis was performed to evaluate the thermal stability of the epoxy thermoset polymers. The characteristic values from these curves are given in Table 2 including onset temperature degradation (Td5), statistic heat-resistant index temperature, and char%. Fig. 5 shows a one-step degradation curve for all epoxy thermoset polymers, which is attributed to the decomposition of the cross-linked polymer network.30P1-G and P1-J show lower degradation temperatures of up to 30 °C compared to the other epoxy thermosets, presumably due to reduced cross linking and greater presence of the benzodioxane GE-1 side product, leading to more free space and mobility.6 Thermal stability of cured networks is calculated through the statistic heat-resistant index temperature (Ts), which is determined by the 5% and 30% weight loss of the sample as shown in eqn (1).31| | | Ts = 0.49 [Td5 + 0.6 (Td30 − Td5)] | (1) |
Table 2 Thermogravimetric data of weight loss at 5%, 30% and 50%, statistic heat-resistance index temperature (Ts), and char residue at 500 °C of epoxy networks. Epoxy networks (P) are labeled after epoxidation reaction conditions listed in Table 1. For example, P1-G is formed using epoxy E1-G and DETA with a 1:1 stoichiometric ratio of epoxy to –NH
| Epoxy network |
T
d5 (°C) |
T
d30 (°C) |
T
d50 (°C) |
T
s (°C) |
Char500 (%) |
|
P1-G
|
263 |
310 |
331 |
142 |
9.2 |
|
P1-J
|
248 |
301 |
323 |
137 |
6.7 |
|
P1-H
|
288 |
335 |
360 |
155 |
13.1 |
|
P1-K
|
285 |
327 |
350 |
152 |
12.8 |
|
P2-B
|
287 |
335 |
352 |
155 |
12.3 |
|
P2-D
|
288 |
324 |
327 |
152 |
12.9 |
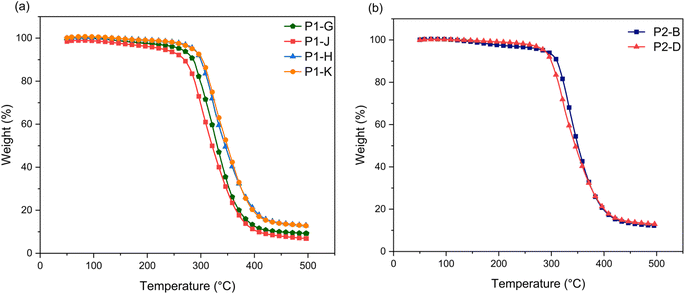 |
| | Fig. 5 Thermogravimetric analysis (TGA) thermograms of (a) P1 and (b) P2 epoxy thermoset networks. | |
For thermosets prepared from 2.0 M NaOH epoxides, the thermal stability temperature is similar (152–155 °C), which show the crosslinked networks don't allow for mobility of the polymer chains and creates resistance to increases of free space.32 The higher char% yields also show increased thermal stability at elevated temperatures.3,33
Mechanical properties of epoxy thermoset networks
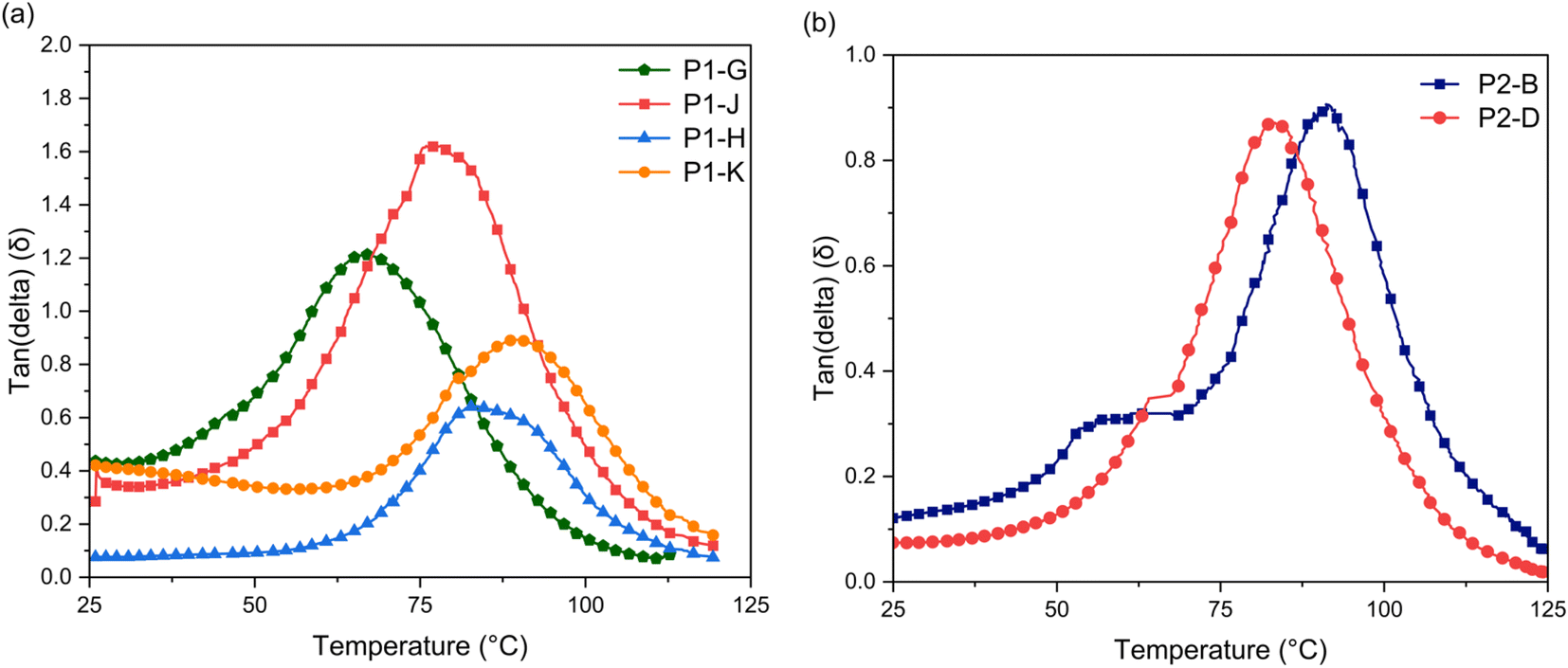
To investigate the thermo-mechanical properties of the epoxy networks, glass-transition temperature (Tg) and crosslinking density (ρ) of cured epoxy resins were calculated by using dynamic mechanical analysis (DMA) (Fig. 6). Table 3 shows the glass transition temperatures corresponding to the peak of maximum tanδ (Tα), indicating that the phase of the sample changes. A higher Tg can reflect the higher cross-linking density of polymer networks.34 In P1 epoxy networks, Tg increases as molar ratio increases to 2 (P1-H and P1-K), which shows a decrease of polymer segments movement and increase of covalent bonds.25,32,34
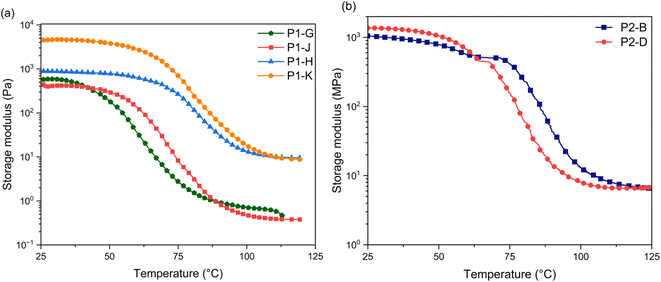 |
| | Fig. 6 Storage modulus values of (a) P1 and (b) P2 thermosets as a function of temperature. | |
Table 3 Dynamic mechanical properties and crosslink density of epoxy networks. Tα is α-relaxation temperature and 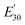 is the storage modulus at 30 °C
is the storage modulus at 30 °C
| Epoxy network |
T
α (°C) |
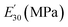
|
E′ at Tα + 30 °C (MPa) |
ρ (mol dm−3) |
|
P1-G
|
67 |
586 |
0.753 |
0.238 |
|
P1-J
|
73 |
420 |
0.403 |
0.129 |
|
P1-H
|
85 |
880 |
9.72 |
3.01 |
|
P1-K
|
90 |
4591 |
8.59 |
3.05 |
|
P2-B
|
91 |
1009 |
6.74 |
2.06 |
|
P2-D
|
75 |
1317 |
6.59 |
2.09 |
Cross linking density is an indicator of the materials performance.35 To determine cross-linking density, it is calculated using the equilibrium storage modulus in the rubbery region over the α-relaxation temperature (Tα) according to the rubber elasticity theory as stated in eqn (2).36,37
E′ is the storage modulus at
Tα + 30 °C,
ϕ is the front factor (approximated to 1 in the Flory theory),
R the gas constant, and
T the absolute temperature. Here, the
P1-H and
P1-K epoxy networks are calculated to have a crosslinking density of 3 mol dm
−3, which is about 15 to 20 times higher than
P1-G and
P1-J (
Fig. 7a). The lower thermal stability temperature and low crosslinking density of
P1-G and
P1-J show there is less functionality and covalent bonding occurring. The height of
P1-G and
P1-J is at
δ > 1, showing the material is reaching elastomeric networks rather than a glassy and rubbery state indicating more free volume in the system, allowing for the network to deform easier (
Fig. 7a).
38,39 Increasing the molar ratio of NaOH allows for the completion of the epoxy reaction and reduces the formation of benzodioxane co-product.
40 In
P2 thermosets, the
Tg is slightly different specifically in the
P1-D epoxy. The presence of
GE-2 and
GE-0 has created a change in the chemical structure, allowing for a less compact material. The less compact material permits more movement as the temperature increases. Although there is more movement of the polymer chains of
P2-D, the cross-linking density of the two is similar. The decrease in crosslinking density of 3.0 mol dm
−3 to 2.0 mol dm
−3 can be attributed to the overall increase in EEW, which demonstrates an increase in benzodioxane product either from
GE-0 or
GE-1 (
Fig. 7b). The
MeO-DHEO epoxidation (
Scheme 2b) may favor lower temperatures and less NaOH concentration similar to what was observed previously for DHEO epoxy.
24
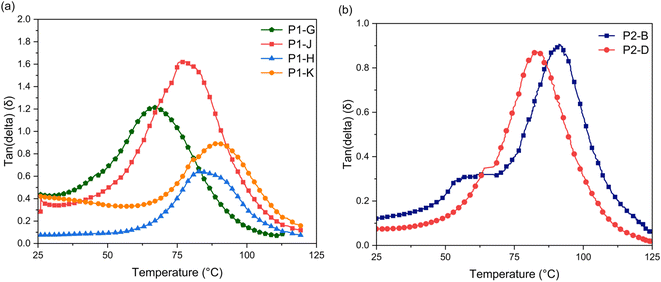 |
| | Fig. 7 Tan(delta) of (a) P1 and (b) P2 thermosets as a function of temperature. The ratio of the loss modulus (E′′) to the storage modulus (E′) is defined as tan(delta). Tg is the peak temperature in the tan(delta). | |
Conclusion
Two synthesis routes and the characterizations of a novel lignin-based tri-epoxide systems have been shown. One pathway through the use of HBr affords exclusively the pyrogallol 3OH while the synthesis over Nb2O5 catalyst in water yields a mixture of 3OH/2OH, possibly due to the lower acidity of the niobia catalyst. Despite the slight difference in the purity of the monomers, the properties (curing characteristics and thermal stability) of the resulting thermoset polymers are comparable. The performance of renewable lignin-based DMPPO epoxy thermosets in terms of thermal and mechanical properties opens avenues for the direct utilization of biorefinery products in the fabrication of bio-based polymers and offers a sustainable alternative to petroleum-based polymer materials. The prepared P1 epoxy networks exhibit higher cross-linking densities of 3 mol dm−3 compared to their P2 counterparts of 2 mol dm−3. This difference in performance can be attributed to the resulting products E1 and E2 from the epoxidation synthesis based on the phenol starting mixtures. Specifically, the presence of GE-2 and GE-0 in P2 epoxides alters the compactness of the material, allowing for more movement of polymer chains with increasing temperature. In contrast, Zhao et al. describe lignin-based DHEO (di-epoxy) thermosets exhibiting a crosslinking of 0.39 mol dm−3. A dimer DHEO (tetra-epoxy) increased the crosslinking to a comparable 3.28 mol dm−3.23 The DHEO dimer enhances mechanical properties but requires an additional synthesis step, whereas P1 and P2 achieve higher densities without extra modifications of the DMPPO monomer. Also, mixtures containing lignin-derived multi-functional phenols (P2) can be utilized without the need of thorough purification of the individual monomers, albeit with a potential slight decrease in crosslinking. Although, the decrease in cross-linking density isn't inherently negative; DGEBA-based materials like adhesives and coatings often share similar properties. The desired application significantly determines its optimal properties. In short, the dynamic mechanical analysis (Fig. 6 and 7) of P2 can exhibit storage modulus (E′) and glass transition temperature (Tg) comparable to those of P1 and while P2 has unexpected lower crosslinking density, both P1 and P2 show promising thermal and mechanical properties and demonstrate viability of biorefinery products to replace petroleum-based polymer materials.
Data availability
The data supporting the outcomes of this study have been incorporated within the manuscript.
Author contributions
Melissa Sanchez: formal analysis, investigation, methodology, visualization, writing – original draft. Peter C. Ford: conceptualization, methodology, project administration, supervision, visualization, writing – review & editing. Mahdi M. Abu-Omar: conceptualization, funding acquisition, methodology, project administration, supervision, visualization, writing – review & editing.
Conflicts of interest
MMAO is founder and part owner of Spero Renewables, LLC, a technology company making biomass-based renewable alternative to petrochemicals.
Acknowledgements
This work was supported by the US Department of Energy, Office of Science, Basic Energy Science, and the Mellichamp Sustainability Initiative, UCSB.
References
- J. Huang, P. Fu, W. Li, L. Xiao, J. Chen and X. Nie, RSC Adv., 2022, 12, 23048–23056 RSC.
- S. Benyahya, C. Aouf, S. Caillol, B. Boutevin, J. P. Pascault and H. Fulcrand, Ind. Crops Prod., 2014, 53, 296–307 CrossRef CAS.
- H. Nouailhas, C. Aouf, C. Le Guerneve, S. Caillol, B. Boutevin and H. Fulcrand, J. Polym. Sci., Part A: Polym. Chem., 2011, 49, 2261–2270 CrossRef CAS.
- Q. Ruiz, S. Pourchet, V. Placet, L. Plasseraud and G. Boni, Polymers, 2020, 12, 229 CrossRef CAS PubMed.
- M. Cabo, M. N. Prabhakar and J. Song, Sci. Rep., 2021, 11, 24140 CrossRef PubMed.
- X. Pan, P. Sengupta and D. C. Webster, Biomacromolecules, 2011, 12, 2416–2428 CrossRef CAS PubMed.
- Y. Jiang, D. Ding, S. Zhao, H. Zhu, H. I. Kenttämaa and M. M. Abu-Omar, Green Chem., 2018, 20, 1131–1138 RSC.
- R. Ménard, S. Caillol and F. Allais, Ind. Crops Prod., 2017, 95, 83–95 CrossRef.
- E. Manarin, F. Da Via, B. Rigatelli, S. Turri and G. Griffini, ACS Appl. Polym. Mater., 2023, 5, 828–838 CrossRef CAS PubMed.
- Y. Jiang, J. Yun and X. Pan, ACS Sustain. Chem. Eng., 2022, 10, 16555–16562 CrossRef CAS.
- C. Gioia, M. Colonna, A. Tagami, L. Medina, O. Sevastyanova, L. A. Berglund and M. Lawoko, Biomacromolecules, 2020, 21, 1920–1928 CrossRef CAS PubMed.
- F. Hernández-Ramos, M. G. Alriols, T. Calvo-Correas, J. Labidi and X. Erdocia, ACS Sustain. Chem. Eng., 2021, 9, 3608–3615 CrossRef.
- E. Melro, A. Filipe, D. Sousa, B. Medronho and A. Romano, New J. Chem., 2021, 45, 6986–7013 RSC.
- D. Watkins, M. Nuruddin, M. Hosur, A. Tcherbi-Narteh and S. Jeelani, J. Mater. Res. Technol., 2015, 4, 26–32 CrossRef CAS.
- J. Zakzeski, P. C. A. Bruijnincx, A. L. Jongerius and B. M. Weckhuysen, Chem. Rev., 2010, 110, 3552–3599 CrossRef CAS PubMed.
- L. Shuai, M. T. Amiri, Y. M. Questell-Santiago, F. Héroguel, Y. Li, H. Kim, R. Meilan, C. Chapple, J. Ralph and J. S. Luterbacher, Science, 2016, 354, 329–333 CrossRef CAS PubMed.
- X. Liu, F. P. Bouxin, J. Fan, V. L. Budarin, C. Hu and J. H. Clark, ChemSusChem, 2020, 13, 4296–4317 CrossRef CAS PubMed.
- R. Dinu, C. Cantarutti and A. Mija, ACS Sustain. Chem. Eng., 2020, 8, 6844–6852 CrossRef CAS.
- P. Ortiz, R. Vendamme and W. Eevers, Molecules, 2020, 25, 1158 CrossRef CAS PubMed.
- J. Podschun, B. Saake and R. Lehnen, Eur. Polym. J., 2015, 67, 1–11 CrossRef CAS.
- S. Zhao and M. M. Abu-Omar, Macromolecules, 2017, 50, 3573–3581 CrossRef CAS.
- S. Zhao, X. Huang, A. J. Whelton and M. M. Abu-Omar, ACS Sustain. Chem. Eng., 2018, 6, 10628–10636 CrossRef CAS.
- S. Zhao and M. M. Abu-Omar, ACS Sustain. Chem. Eng., 2016, 4, 6082–6089 CrossRef CAS.
- S. Zhao and M. M. Abu-Omar, Biomacromolecules, 2015, 16, 2025–2031 CrossRef CAS PubMed.
- Y. Ecochard, M. Decostanzi, C. Negrell, R. Sonnier and S. Caillol, Molecules, 2019, 24, 1818 CrossRef CAS PubMed.
- B. Liu, M. Sanchez, J. Truong, P. C. Ford and M. M. Abu-Omar, Green Chem., 2022, 24, 4958–4968 RSC.
- S. Li, B. Liu, J. Truong, Z. Luo, P. C. Ford and M. M. Abu-Omar, Green Chem., 2020, 22, 7406–7416 RSC.
- F. Ng, G. Couture, C. Philippe, B. Boutevin and S. Caillol, Molecules, 2017, 22, 149 CrossRef PubMed.
- S. Li, W. Guan, Z. Wu, J. Lu and J. Guo, Polym.-Plast. Technol. Eng., 2007, 46, 901–903 CrossRef CAS.
- D. Puglia, L. B. Manfredi, A. Vazquez and J. M. Kenny, Polym. Degrad. Stab., 2001, 73, 521–527 CrossRef CAS.
- Z. Cheng, M. Fang, X. Chen, Y. Zhang, Y. Wang, H. Li and J. Qian, ACS Omega, 2020, 5, 4200–4212 CrossRef CAS PubMed.
- A. Bandyopadhyay, P. K. Valavala, T. C. Clancy, K. E. Wise and G. M. Odegard, Polymer, 2011, 52, 2445–2452 CrossRef CAS.
- Y. Chiu, H. Tsai and T. Imae, J. Appl. Polym. Sci., 2012, 124, 1234–1240 CrossRef CAS.
- J. P. Patel, Z. G. Xiang, S. L. Hsu, A. B. Schoch, S. A. Carleen and D. Matsumoto, Int. J. Adhes. Adhes., 2017, 78, 256–262 CrossRef CAS.
- Y. Wang, S. Wang, C. Bian, Y. Zhong and X. Jing, Polym. Degrad. Stab., 2015, 111, 239–246 CrossRef CAS.
- R. Everaers, A. Y. Grosberg, M. Rubinstein and A. Rosa, Soft Matter, 2017, 13, 1223–1234 RSC.
- P. J. Flory, Polym. J., 1985, 17, 1–12 CrossRef CAS.
- C. W. H. Rajawasam, O. J. Dodo, M. A. S. N. Weerasinghe, I. O. Raji, S. V. Wanasinghe, D. Konkolewicz and N. De Alwis Watuthanthrige, Polym. Chem., 2024, 15, 219–247 RSC.
- L. M. De Espinosa, J. C. Ronda, M. Galià and V. Cádiz, J. Polym. Sci., Part A: Polym. Chem., 2009, 47, 1159–1167 CrossRef.
-
H. Q. Pham and M. J. Marks, in Ullmann's Encyclopedia of Industrial Chemistry, Wiley-VCH, Wiley, 1st edn, 2005 Search PubMed.
|
| This journal is © The Royal Society of Chemistry 2024 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence a and
Mahdi M.
Abu-Omar
a and
Mahdi M.
Abu-Omar


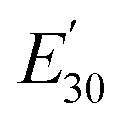 is the storage modulus at 30 °C
is the storage modulus at 30 °C