
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceCatalytic function of ionic liquids in polyethylene terephthalate glycolysis by molecular dynamics simulations†
Mohamed Ahmed
Nosir
and
Manuel Angel
Ortuño
 *
*
Centro Singular de Investigación en Química Biolóxica e Materiais Moleculares (CIQUS), Universidade de Santiago de Compostela, 15782 Santiago de Compostela, Spain. E-mail: manuelangel.ortuno@usc.es
First published on 30th July 2024
Abstract
Chemical recycling is of paramount importance to minimise the environmental impact of plastic waste. Polyethylene terephthalate (PET) is a polar thermoplastic widely used in fibres and packaging and is amenable to chemical depolymerisation. Recent efforts are devoted to its degradation via glycolysis. Even though it requires milder conditions than hydrolysis, catalysts are still necessary. In this case, ionic liquids (ILs) come into play to catalyse the reaction. In particular, we focus on choline-based liquids due to their low toxicity and cost compared to imidazolium-based ones. However, due to the complexity of the process, detailed information on the operating mechanism is scarce, which hinders the progress towards a rational design of new and more efficient systems. Herein, we present a computational study to address the role of IL catalysts during PET glycolysis under realistic catalytic conditions, i.e., considering time, concentration, and temperature. We perform classical molecular dynamics (MD) simulations on several systems, including a complex ternary mixture formed by ethylene glycol (EG), PET oligomers, and the [Ch]3[PO4] catalyst. By means of radial/spatial distribution functions, H-bond analysis, and domain count, we present a detailed solvation scenario of the catalytic system. Our findings suggest that the IL anion (and the IL cation to a lesser extent) does participate in the nucleophilic activation of EG, while the IL cation does not play a significant role in the electrophilic activation of PET.
Sustainability spotlightPlastic waste critically damages the environment, and chemical recycling has the potential to mitigate it. This work presents a computational study at atomic level of detail regarding the depolymerisation of polyethylene terephthalate via glycolysis using ionic liquid catalysts. The study demonstrates the critical role of anions in activating the ethylene glycol nucleophile while cations do not significantly participate in the reaction. Such insight will guide the design of new ionic liquid catalysts to improve the recycling process. The project thus aligns with the objective “UN SDG 12: Ensure sustainable consumption and production patterns”. |
Introduction
Plastics have improved all aspects of our lives due to their affordability, versatility, and resistance. However, the very properties that make them useful, such as strength and durability, create irreversible damage at their end-of-life use. It is estimated that 60% of all plastic ever produced, ca. 4900 million tonnes, is dumped into landfills and oceans.1 The EU alone generates ca. 26 million tonnes of plastic waste every year.2 In this scenario, it is critical to develop strategies to handle waste and minimize its environmental impact.An important approach to address this challenge is recycling3via chemical depolymerisation to either recover the initial components (recycling to monomers) or obtain high-value derivatives (upcycling).4 Polar thermoplastics are typically suitable for this process, and herein, we focus on the popular polyethylene terephthalate, namely PET. It is widely present in synthetic fibers, packaging, and construction, accounting for ca. 10% of the global plastic production. More interestingly, it has a high recovery rate1 and advances in recycling technologies could be readily implemented. However, chemical depolymerisation is far from trivial and it usually requires harsh conditions. Ester linkages in PET can be attacked by nucleophiles such as water, amines, alcohols, or glycols. Hydrolysis would yield the initial monomers, terephthalic acid (TPA) and ethylene glycol (EG); however, the low nucleophilicity of H2O requires large energy inputs. Whereas, glycolysis requires milder conditions and the resulting product, bis(2-hydroxyethyl)terephthalate (BHET), is used as a building block for fresh PET re-polymerisation glycols (Fig. 1). Nevertheless, the kinetics of these processes is sluggish and catalysts are needed.5
 | ||
| Fig. 1 Catalytic thermal PET glycolysis to form BHET. | ||
Typical metal salts6 and N-containing bases7 face some drawbacks such as separation of products, reusability, need for organic solvents, or small contact area between the polymer and catalyst.8 To address these shortcomings, we turned to ionic liquids (ILs), which are compounds formed by cations and anions with low melting points (<100 °C). Compared to typical organic solvents, ILs offer enhanced sustainable features such as low flammability, non-volatility, and thermal stability. Despite their well-known application in catalysis,9 they are relatively new players in polymer degradation.10 The first report dates from 2007, where pyrrolidinium-based ILs converted 6-nylon to caprolactam with 4-dimethylaminopyridine in 4 h at 300 °C.11 As for PET, imidazolium-based ILs are the most commonly used media as the process can work at temperatures below 200 °C and atmospheric pressure.12–14 For instance, Wang et al. were able to degrade PET in 8 h at 180 °C, and the medium was recycled and reused six times.12 First-row-based15 and lanthanide-based16 metal-containing ILs have also shown good results in PET glycolysis.
However, some commonly used ILs can be toxic and expensive, which severely limits their large-scale applications. From a sustainable point of view, there are interesting and recent efforts devoted to working with low-cost and biocompatible ILs, such as those based on cholinium cations [Ch]+.17 For instance, Sun et al. were able to dissolve and degrade PET via glycolysis using [Ch]3[PO4];18 Liu et al. obtained similar results with [Ch][AcO],19 and Marullo et al. reported PET depolymerisation under mild conditions using amino acid-based ILs, such as [Ch][Gly].20
Despite these foundations, a deep understanding of the atomic details is still missing. Experimental mechanistic studies are usually performed in conditions different from the catalytic reaction. Moreover, most techniques provide information from an average point of view, which might be limiting when certain species or features are transient in the time scale of the experiment. Instead, computational approaches allow us to look into the structure and properties of the catalytic systems at atomic resolution. Only a handful of large-scale computational studies are available in the literature: Zara et al. reported molecular dynamics (MD) simulations of the enzyme PETase in the presence of [Ch]3[PO4];21 and Sun, Zhao and co-workers conducted MD simulations of PET methanolysis22 and glycolysis23 with [Ch][AcO].
Herein, we employ classical molecular dynamics (MD) simulations to capture the dynamic behaviour of bulk liquid mixtures and picture a full solvation scenario of PET glycolysis catalysed by [Ch]3[PO4]. We further perform a deconvolution of the complex reaction media by considering the mixture of different components under similar reaction conditions (Fig. 2) and analyse possible nanosegregation patterns. With this computational approach, we aim to identify the key solvation features that promote PET glycolysis in order to lead the future design of new and more efficient IL-based catalysts.
 | ||
| Fig. 2 Representation of the components of the computational models: (a) [Ch]+ IL cation, (b) [PO4]3− IL anion, (c) EG, and (d) PET dimer. Atom legend: P (orange), O (red), N (blue), C (grey), and H (white). | ||
Results and discussion
We present classical MD simulations to provide atomistic insights into the [Ch]3[PO4]-mediated PET glycolysis with EG. In the following sections, we compute and analyse five main models to account for all possible interaction patterns: (i) pure IL, binary mixtures (ii) IL/PET, (iii) IL/EG, and (iv) PET/EG, and ternary mixture (v) IL/PET/EG. To visualise the complexity of the systems under study, Fig. 3 shows a snapshot of a typical simulation box for the ternary mixture. The relative amount of components was adapted according to available experimental data18 (see computational details). | ||
| Fig. 3 Selected snapshot of the simulation box for the ternary mixture IL/PET/EG with [Ch]+ in blue, [PO4]3− in red, PET dimers in green, and EG in pink. | ||
The manuscript is divided as follows: first, we analyse the structural features of all systems in detail; then, we evaluate the effect of temperature, H-bond lifetimes, and nanosegregation in the bulk; finally we gather all data to depict a global solvation scenario for PET glycolysis.
Structural analysis
We investigate the structural properties of five systems (IL, PET/EG, IL/EG, IL/PET, and IL/PET/EG) at 450 K by means of radial distribution functions (RDFs), namely g(r). They represent the probability of finding an atom/group/molecule at a certain distance from a reference during the simulation time.We first inspect the IL ions in pure state and in several mixtures. Fig. 4a represents the interaction between the O atoms in [PO4]3− and the H atom from the OH group in [Ch]+. There is a large peak at ca. 1.71 Å accounting for H-bonds, whose relative intensity becomes stronger between systems where the IL is more diluted. Fig. 4b represents the interaction between the O atoms in [PO4]3− and the N atom in [Ch]+. Comparatively, such electrostatic contact is much weaker than the previous H-bond.23 This analysis reveals a strong assembly of cations and anions, particularly in the ternary mixture IL/PET/EG.
 | ||
| Fig. 4 Radial distribution functions (RDFs) between IL anion and (a) OH and (b) N in IL cation for different systems at 450 K. | ||
Next, we evaluate the involvement of EG in the media. Fig. 5a shows the interaction between the H from the OH group in EG and the O atoms in [PO4]3−. They are expected to be important as EG and [Ch]-based IL can form deep-eutectic solvents.24 There is a sharp peak at ca. 1.74 Å, indicative of H-bonds between these two species. According to the intensity, the probability of forming these H-bonds is higher in the ternary system. The mechanistic implication of this feature is that the O–H bond from EG would be greatly polarized by the IL anion, and therefore the O atom from EG will hold more partial negative charge. These findings align with the proposed mechanism where the nucleophilicity of EG is enhanced by the IL anion, thus favouring the later nucleophilic attack to the carbonyl group of PET. Fig. 5b shows the interaction between the H from the OH group in EG and the O atom in [Ch]+, displaying H-bond interactions at ca. 1.79 Å. This suggests that the protic part of the IL cation may also contribute to the above-mentioned activation of EG. Fig. 5c displays the self-interaction between EG molecules, with an H-bond peak at also 1.79 Å of similar intensity to that for [Ch]+. Thus, these two interactions likely compete in the solution. Finally, Fig. 5d plots the interaction between the H from the OH group in EG and the O atom in the carbonyl groups of PET. There is a peak located at ca. 1.84 Å but it has low intensity (<1). This points to H-bonds that are weaker and more labile, as we discuss in a later section. It seems that EG does not necessarily enhance the electrophilicity of the carbonyl group in PET.
 | ||
| Fig. 5 Radial distribution functions (RDFs) between EG and (a) O in IL anion, (b) O in IL cation, (c) O in EG, and (d) O in PET for different systems at 450 K. | ||
In view of the little interaction between EG and PET, we analyse the solvation patterns around the carbonyl groups of PET for the IL and Fig. 6a and b show their interaction with the N atom in [Ch]+ and the H atom from the OH group in [Ch]+, respectively. In the former, a peak at ca. 5 Å is clearly present in the binary mixture IL/PET, but its intensity decreases in the ternary system. In the latter case, no H-bonds are detected, even in the absence of EG. These results suggest that PET may eventually interact with the cationic N moiety of [Ch]+ through electrostatics, but the protic part has no influence. It is possible that, under catalytic conditions, EG displaces IL when interacting with PET.
 | ||
| Fig. 6 Radial distribution functions (RDFs) between OC PET and (a) N and (b) OH in IL cation for different systems at 450 K. | ||
Further, we observe that PET dimers tend to agglomerate (Fig. S2†), likely due to their aliphatic nature, which points to a heterogenization of the liquid as discussed later on. This behaviour could explain previous results, as PET self-interaction may hinder the approach of other species such as EG or [Ch]+ towards the carbonyl group.
To better visualise the microenvironments within the ternary mixture IL/PET/EG, we present spatial distribution functions (SDFs), which are a 3D extension of the above-mentioned RDFs. Fig. 7 displays the closest contacts in [Ch]+ and EG. We observe a remarkable concentration of O from [PO4]3− (blue isosurface) near OH from both [Ch]+ and EG. Additionally, the carbonyl group from PET (green isosurface) may interact with OH from EG but in a weakly fashion, since the isovalue had to be enhanced for proper visualization.
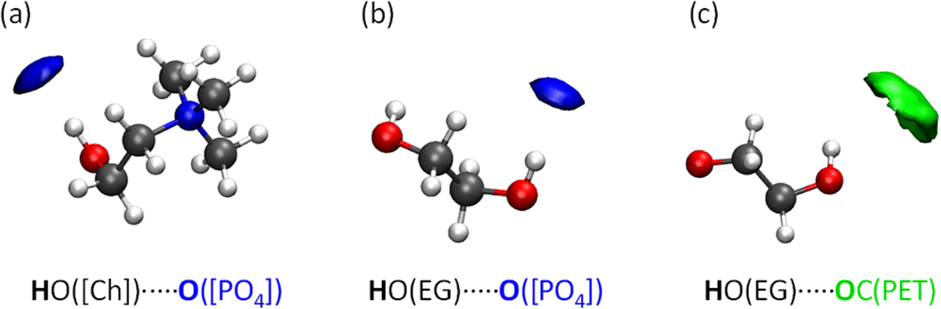 | ||
| Fig. 7 Isosurfaces of the spatial distribution functions (SDFs) between (a) [Ch]+ and [PO4]3− (isovalue = RDF max.), (b) EG and [PO4]3− (isovalue = RDF max.), and (c) EG and PET (isovalue = ∼10 × RDF max.) for the IL/PET/EG system at 450 K. Atom legend: O (red), N (blue), C (grey), and H (white). | ||
Effect of temperature
Since in the previous section, we identified some weak interactions regarding PET, we next evaluated the impact of temperature on those for the ternary mixture at 300, 350, 400, and 450 K. In Fig. 8a, the intensity of the H-bond between the carbonyl in PET and the OH in EG does not significantly decrease with T, which means that such contact is not thermally dependent. In Fig. 8b, there is a very small H-bond interaction between the carbonyl in PET and the OH in [Ch]+ as previously reported;23 however, it is only found at 300 K and quickly vanishes at higher T. This suggests that the IL cation does not significantly interact with PET via H-bonds at the catalytic operating conditions (450 K). | ||
| Fig. 8 Radial distribution functions (RDFs) between OC PET and (a) OH in EG and (b) OH in IL cation for the ternary system at different temperatures. | ||
H-bond lifetimes
Previous sections have highlighted the importance of H-bonds (or the absence of them) in the solution. To delve deeper into this feature, we estimate the H-bond lifetimes along the simulation for the IL/PET/EG system. We consider an intermolecular H-bond when the maximum distance between the H-bond donor and acceptor is limited to 3.50 Å, the maximum distance between the H atom and the acceptor atom is set to 2.45 Å, and the angle between the donor/acceptor and donor/H is less than 30°.The computed H-bond lifetimes are shown in Table 1. According to these results, the sturdiest H-bonds correspond to those involving [PO4]3− with average values of 24–27 ps. They are closely followed by the H-bonds of EG and [Ch]+, with similar values of 18–20 ps. Finally, the H-bonds concerning PET are very labile and transient in time, with values of barely 2 ps.
| H-bond | τ lf/ps |
|---|---|
| O([PO4])⋯HO([Ch]) | 23.6 |
| O([PO4])⋯HO(EG) | 26.9 |
| O([Ch])⋯HO(EG) | 19.4 |
| O(EG)⋯HO(EG) | 18.6 |
| OC(PET)⋯HO(EG) | 1.6 |
| OC(PET)⋯HO([Ch])) | 2.0 |
From a mechanistic perspective, the longer lifetimes between EG and H-bond acceptors suggest that EG will be activated more frequently. Statistically, EG will be more accessible to enter the catalytic cycle, which could mean a potential increase in reaction rates. Contrarily, the shorter lifetimes between EG and PET are indicative of a transient interaction which is expected to have little impact on catalysis. Overall, these data further support the trend in H-bond relevance suggested by RDFs.
At lower temperatures, the lifetime trends are the same but the values increase up to one order of magnitude. These data can be consulted in Table S5.†
Nanosegregation analysis
To obtain further insight into the behaviour of bulk mixtures, we evaluate the nanosegregation of species via domain analysis.25Table 2 shows the number of domains for each species (subunit) of the IL/PET/EG system, where 1.0 means that the subunits are connected while larger values mean that the subunits are homogeneously distributed. The first interesting result is that PET dimers form a domain with themselves (1.6) and with EG (1.0). They also present partial aggregation with IL cations (8.1) but no aggregation at all with IL anions (99.4). Secondly, it is known that EG and choline-based ILs are nanostructured,26,27 and we do observe that behaviour in the IL/EG system (1.0). However, in the presence of PET, such domain breaks down into several ones (ca. 7), although some heterogeneity remains. Notably, this behaviour only happens at high temperatures, since decreasing T favours a single domain (Tables S2–4†). Finally, the IL ions do not present aggregation, neither in IL/PET/EG nor IL/EG systems, likely due to their low concentration compared to EG.| PET | EG | IL cation | IL anion | |
|---|---|---|---|---|
| a The table is symmetric, so values are displayed only once. | ||||
| PET | 1.6 | 1.0 | 8.1 | 99.4 |
| EG | 6.9 (1.0) | 6.8 (1.0) | 6.9 (1.0) | |
| IL cation | 92.7 (62.5) | 42.4 (21.6) | ||
| IL anion | 100 (100) | |||
Alternatively, we also study the heterogenization of the mixture through cluster analysis. The results are in line with previous RDFs and can be consulted in Fig. S5.†
Solvation scenario
Based on the analysis of the above-mentioned MD simulations of complex mixtures via RDFs, H-bond lifetimes, and domain count, Fig. 9 presents a final picture of the solvation of key components of PET glycolysis under catalytic conditions. EG is the nucleophile of the reaction, and its activity is clearly enhanced by the IL anion,28 while [Ch]+ and other EG molecules can also participate to a lesser extent (Fig. 9a). PET is the electrophile of the reaction, but it does not form strong H-bond interactions with EG or IL and it tends to self-aggregate. Occasionally, the carbonyl group could be surrounded by the positively-charged N group of [Ch]+ (Fig. 9b). But under typical reaction conditions (high T), the role of the OH of [Ch]+ in stabilising a developing negative charge is not apparent from our MD simulations, which is in line with very recent DFT studies.29Fig. 9c shows an exemplary snapshot highlighting this network of interactions.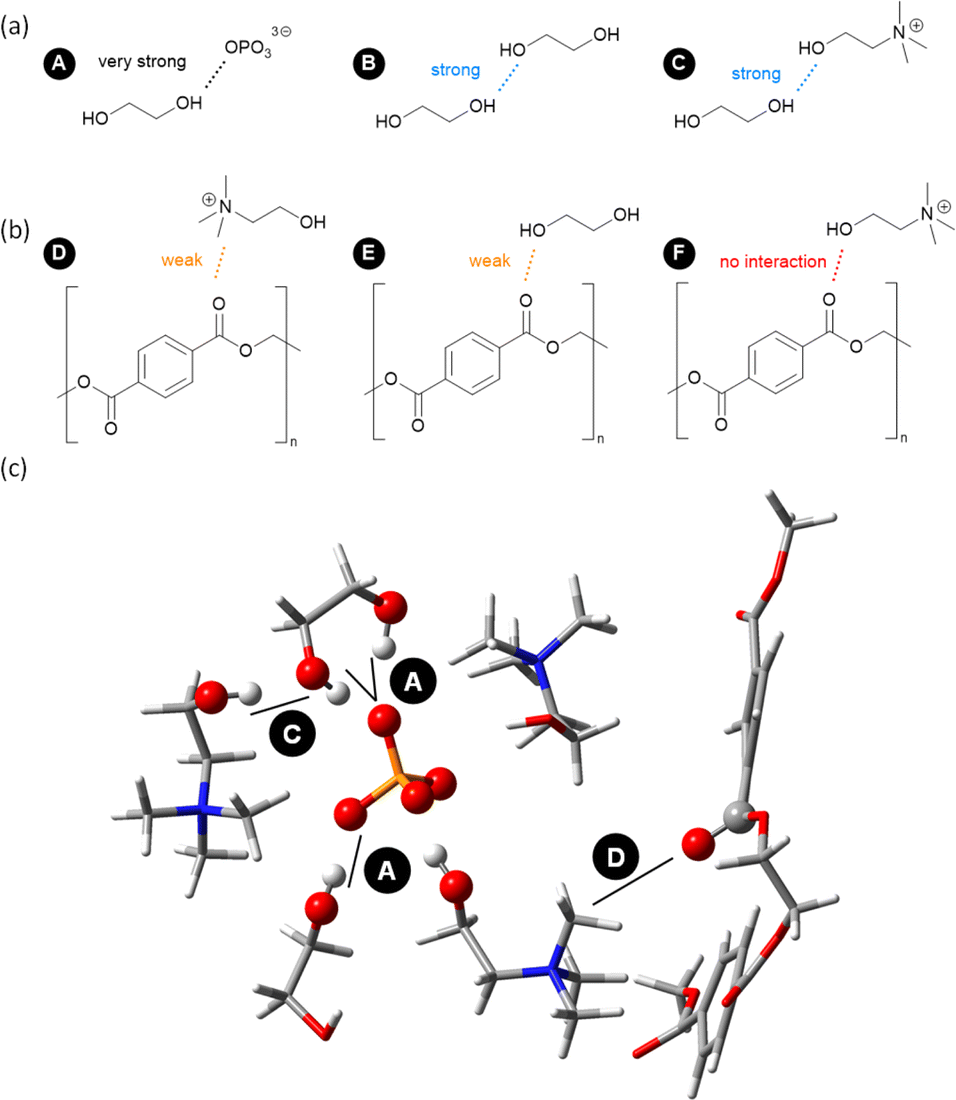 | ||
| Fig. 9 Main solvation patterns of IL-catalysed PET glycolysis to (a) enhance the nucleophilicity of EG and (b) enhance electrophilicity of PET, as well as (c) a 3D model of a selected snapshot. Atom legend: P (orange), O (red), N (blue), C (grey), and H (white). | ||
To draw further conclusions about the mechanism, more sophisticated ab initio MD simulations would be required to sample rare events,30,31 such as bond-breaking and bond-forming processes. Due to the large size of the systems under study, this is out of the scope of the present contribution.
Experimental
Computational details
Classical molecular dynamics (MD) simulations were carried out with the open-source Large-scale Atomic/Molecular Massively Parallel Simulator (LAMMPS) package.32 All molecules were described with nonpolarisable force fields including bonded (bonds, angles, dihedrals, and impropers) and non-bonded (van der Waals and electrostatic interactions) parameters. Such nonpolarisable force fields predict correct geometries and provide qualitative dynamics at lower computational costs compared to polarisable ones.33 For the IL, the cation [Ch]+ was described with OPLS-AA as proposed by Canongia Lopes et al.34 and the anion [PO4]3− with parameters from Demichelis et al.35 Charges of ionic species were not scaled. PET oligomers were described with CHARMM-GUI36 and EG molecules with parameters from Kaiser et al.37To obtain a comprehensive scenario of the glycolysis of PET in ILs, we investigated different systems: pure liquids, binary mixtures, and ternary mixtures. Each system contains a constant number of molecules for each of its components, according to the conditions from the following ternary mixture. We aimed to build a simulation box with 100 IL units (100 [PO4]3− and 300 [Ch]+) to have enough statistical data about the catalyst. To estimate an appropriate number of PET monomeric units and EG molecules, we turned to experiments where a typical catalytic run contained 0.05 g of IL, 0.5 g of PET, and 2.0 g of EG;18 or ca. 0.1 mmol of IL, 2.6 mmol of PET monomeric units, and 32.2 mmol of EG. Since using the actual molar ratio would have greatly increased the size and cost of the simulations, requiring >30![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 EG molecules, we roughly decreased the amount of EG by one order of magnitude as a feasible approximation. The resulting ternary mixture was then formed by 100 IL units (100 [PO4]3− and 300 [Ch]+), 500 PET dimeric units, and 4000 EG molecules, all randomly distributed inside the simulation box.
000 EG molecules, we roughly decreased the amount of EG by one order of magnitude as a feasible approximation. The resulting ternary mixture was then formed by 100 IL units (100 [PO4]3− and 300 [Ch]+), 500 PET dimeric units, and 4000 EG molecules, all randomly distributed inside the simulation box.
Periodic boundary conditions were applied in the three directions. The initial spatial configurations of the simulation box were generated with PACKMOL38 using the above-mentioned number of molecules. A sufficient cut-off radius of 10 Å was set for non-bonded Lennard-Jones and van der Waals interactions. The long-range electrostatic interactions were taken into account by implementing the particle–particle–particle–mesh (P3M) solver39 with a relative accuracy of 10−6. The steepest descent minimization technique was employed to initially relax the system for each initial configuration. The propagation was performed using the Verlet integrator with a time step of 0.5 fs. The systems underwent equilibration for 4 ns in the NPT ensemble (300, 350, 400, and 450 K; 1 atm) using the Nosé–Hoover thermostat and barostat with coupling times for T and P as 0.1 and 0.5 ps, respectively. After that, the systems underwent production for 10 ns in the NVT ensemble (300, 350, 400, and 450 K). NVT MD trajectories with a duration of 500 ps were generated for data analysis, storing the trajectory every 5 ps. H-bond lifetimes were performed following the methodology by Gehrke and Kirchner.40 Cluster analyses were performed with the hierarchical clustering algorithm by Frömbgen et al.41,42 Analysis and visualization of MD trajectories were performed with TRAVIS43 and VMD44 software packages, respectively.
We validated the above-mentioned force fields and the computational protocol correctly provided the expected experimental densities for our models (Table S1†). We further tested different lengths of PET oligomers (10, 25, and 100 units),23 and they behave similarly to PET dimers (Fig. S3 and S4†).
Conclusions
In this study, we present all-atom classical molecular dynamics simulations to shed light on the catalytic role of the ionic liquid [Ch]3[PO4] in PET glycolysis under realistic experimental conditions. We thoroughly analyse the structural features of the ternary mixture IL/PET/EG via radial and spatial distribution functions, supplemented by H-bond lifetimes and domain analysis, and we compared them with binary mixtures to identify key interactions. On the one hand, the nucleophilicity of the OH groups in EG can greatly increase via the formation of strong H-bonds with the IL anion. The OH groups of the IL cation as well as other EG molecules can also contribute to such activation. On the other hand, the electrophilicity of the carbonyl groups in PET could slightly increase via electrostatic interactions with the IL cation, but its OH group does not provide any apparent benefit towards catalytic activity, particularly at operating high temperatures. The domain analysis indicates that PET oligomers are nanostructured and can partially disturb the known nanosegregation of IL/EG under catalytic conditions.Future endeavours will entail (i) computational screening of anions, which have a strong impact on EG activity, as well as (ii) tuning of cations to promote electrostatic interactions with PET while minimising toxicity and cost.
Data availability
The data supporting this article have been included as part of the ESI.†Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors acknowledge MICIU/AEI/10.13039/501100011033 and European Union NextGenerationEU/PRTR for funding the project TED2021-131460A-I00, as well as Xunta de Galicia (Xunta Distinguished Researcher program ED431H 2020/21, Grupo de Referencia Competitivo ED431C 2020/22, Centro singular de investigación de Galicia accreditation 2019–2022, ED431G 2019/03) and European Union (European Regional Development Fund─ERDF) for additional support. The authors acknowledge CESGA (“Centro de Supercomputación de Galicia”) for providing generous computational resources.Notes and references
- R. Geyer, J. R. Jambeck and K. L. Law, Sci. Adv., 2017, 3, e1700782 CrossRef PubMed.
- European Commission, Energy, Climate Change, Environment, https://environment.ec.europa.eu/topics/plastics_en, accessed 20th May, 2024 Search PubMed.
- J. M. García and M. L. Robertson, Science, 2017, 358, 870 CrossRef PubMed.
- G. W. Coates and Y. D. Y. L. Getzler, Nat. Rev. Mater., 2020, 5, 501 CrossRef CAS.
- M. Chu, Y. Liu, X. Lou, Q. Zhang and J. Chen, ACS Catal., 2022, 12, 4659 CrossRef CAS.
- R. López-Fonseca, I. Duque-Ingunza, B. de Rivas, S. Arnaiz and J. I. Gutiérrez-Ortiz, Polym. Degrad. Stab., 2010, 95, 1022 CrossRef.
- C. Jehanno, M. M. Pérez-Madrigal, J. Demarteau, H. Sardon and A. P. Dove, Polym. Chem., 2019, 10, 172 RSC.
- I. Vollmer, M. J. F. Jenks, M. C. P. Roelands, R. J. White, T. van Harmelen, P. de Wild, G. P. van der Laan, F. Meirer, J. T. F. Keurentjes and B. M. Weckhuysen, Angew. Chem., Int. Ed., 2020, 59, 15402 CrossRef CAS PubMed.
- (a) J. P. Hallett and T. Welton, Chem. Rev., 2011, 111, 3508 CrossRef CAS PubMed; (b) P. McNeice, P. C. Marr and A. C. Marr, Catal. Sci. Technol., 2021, 11, 726 RSC.
- (a) T. Christoff-Tempesta and T. H. Epps III, ACS Macro Lett., 2023, 12, 1058 CrossRef CAS PubMed; (b) M. Zunita, H. P. Winoto, M. F. K. Fauzan and R. Haikal, Polym. Degrad. Stab., 2023, 211, 110320 CrossRef CAS.
- A. Kamimura and S. Yamamoto, Org. Lett., 2007, 9, 2533 CrossRef CAS PubMed.
- H. Wang, Z. Li, Y. Liu, X. Zhang and S. Zhang, Green Chem., 2009, 11, 1568 RSC.
- H. Wang, Y. Liu, Z. Li, X. Zhang, S. Zhang and Y. Zhang, Eur. Polym. J., 2009, 45, 1535 CrossRef CAS.
- Q. Yue, C. Wang, L. Zhang, Y. Ni and Y. Jin, Polym. Degrad. Stab., 2011, 96, 399 CrossRef CAS.
- Q. Wang, Y. Geng, X. Lu and S. Zhang, ACS Sustainable Chem. Eng., 2015, 3, 340 CrossRef CAS.
- N. G. Bush, C. H. Dinh, C. L. Catterton and M. E. Fieser, RSC Sustainability, 2023, 1, 938 RSC.
- B. L. Gadilohar and G. S. Shankarling, J. Mol. Liq., 2017, 227, 234 CrossRef CAS.
- J. Sun, D. Liu, R. P. Young, A. G. Cruz, N. G. Isern, T. Schuerg, J. R. Cort, B. A. Simmons and S. Singh, ChemSusChem, 2018, 11, 781 CrossRef CAS.
- Y. Liu, X. Yao, H. Yao, Q. Zhou, J. Xin, X. Lu and S. Zhang, Green Chem., 2020, 22, 3122 RSC.
- S. Marullo, C. Rizzo, N. T. Dintcheva and F. D'Anna, ACS Sustainable Chem. Eng., 2021, 9, 15157 CrossRef CAS.
- Z. Zara, D. Mishra, S. K. Pandey, E. Csefalvay, F. Fadaei, B. Minofar and D. Řeha, Molecules, 2022, 27, 119 CrossRef CAS PubMed.
- C. Liu, Y. Ling, Z. Wang, W. Zheng, W. Sun and L. Zhao, Chem. Eng. Sci., 2022, 247, 117024 CrossRef CAS.
- W. Zheng, C. Liu, X. Wei, W. Sun and L. Zhao, Chem. Eng. Sci., 2023, 267, 118329 CrossRef CAS.
- (a) E. L. Smith, A. P. Abbott and K. S. Ryder, Chem. Rev., 2014, 114, 11060 CrossRef CAS PubMed; (b) T. Chowdhury, S. Chatterjee, S. H. Deshmukh and S. Bagchi, J. Phys. Chem. B, 2023, 127, 7299 CrossRef CAS.
- M. Brehm, H. Weber, M. Thomas, O. Hollóczki and B. Kirchner, ChemPhysChem, 2015, 16, 3271 CrossRef CAS PubMed.
- V. Alizadeh, D. Geller, F. Malberg, P. B. Sánchez, A. A. H. Padua and B. Kirchner, ChemPhysChem, 2019, 20, 1789 Search PubMed.
- (a) V. Alizadeh, F. Malberg, A. A. H. Padua and B. Kirchner, J. Phys. Chem. B, 2020, 124, 7433 CrossRef CAS PubMed; (b) S. Rozas, M. Atilhan and S. Aparicio, J. Chem. Phys., 2022, 156, 204506 CrossRef CAS PubMed.
- Z. Ju, L. Zhou, X. Lu, Y. Li, X. Yao, S. Cheng, G. Chen and C. Ge, Phys. Chem. Chem. Phys., 2021, 23, 18659 RSC.
- D. Bura, L. Pedrini, C. Trujillo and S. J. Connon, RSC Sustainability, 2023, 1, 2197 RSC.
- G. Bussi and A. Laio, Nat. Rev. Phys., 2020, 2, 200 CrossRef.
- M. Heshmat, M. Leven, O. Linker, M. Sebastian, C. Gürtler and M. R. Machat, Phys. Chem. Chem. Phys., 2023, 25, 20485 RSC.
- (a) S. Plimpton, J. Comput. Phys., 1995, 117, 1 CrossRef CAS; (b) LAMMPS Site. https://lammps.sandia.gov/, accessed 20th May, 2024 Search PubMed.
- V. Lesch, H. Montes-Campos, T. Méndez-Morales, L. J. Gallego, A. Heuer, C. Schröder and L. M. Varela, J. Chem. Phys., 2016, 145, 204507 CrossRef PubMed.
- J. N. Canongia Lopes, J. Deschamps and A. A. H. Pádua, J. Phys. Chem. B, 2004, 108, 2038 CrossRef.
- R. Demichelis, N. A. Garcia, P. Raiteri, R. I. Malini, C. L. Freeman, J. H. Harding and J. D. Gale, J. Phys. Chem. B, 2018, 122, 1471 CrossRef CAS PubMed.
- S. Jo, T. Kim, V. G. Iyer and W. Im, J. Comput. Chem., 2008, 29, 1859 CrossRef CAS PubMed.
- A. Kaiser, O. Ismailova, A. Koskela, S. E. Huber, M. Ritter, B. Cosenza, W. Benger, R. Nazmutdinov and M. Probst, J. Mol. Liq., 2014, 189, 20 CrossRef CAS PubMed.
- L. Martínez, R. Andrade, E. G. Birgin and J. M. Martínez, J. Comput. Chem., 2009, 30, 2157 CrossRef PubMed.
- B. A. Luty and W. F. van Gunsteren, J. Phys. Chem., 1996, 100, 2581 CrossRef CAS.
- S. Gehrke and B. Kirchner, J. Chem. Eng. Data, 2020, 65, 1146 CrossRef CAS.
- T. Frömbgen, J. Blasius, V. Alizadeh, A. Chaumont, M. Brehm and B. Kirchner, J. Chem. Inf. Model., 2022, 62, 5634 CrossRef PubMed.
- T. Frömbgen, J. N. Canongia Lopes, B. Kirchner and K. Shimizu, J. Phys. Chem. B, 2024, 128, 3937 CrossRef PubMed.
- (a) M. Brehm and B. Kirchner, J. Chem. Inf. Model., 2011, 51, 2007 CrossRef CAS; (b) M. Brehm, M. Thomas, S. Gehrke and B. Kirchner, J. Chem. Phys., 2020, 152, 164105 CrossRef CAS PubMed.
- W. Humphrey, A. Dalke and K. Schulten, J. Mol. Graphics, 1996, 14, 33 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Additional computational details and radial distribution functions. See DOI: https://doi.org/10.1039/d4su00251b |
| This journal is © The Royal Society of Chemistry 2024 |