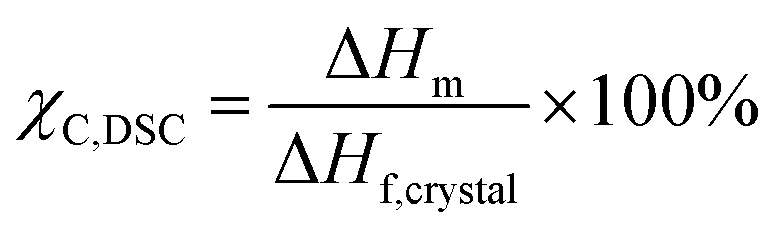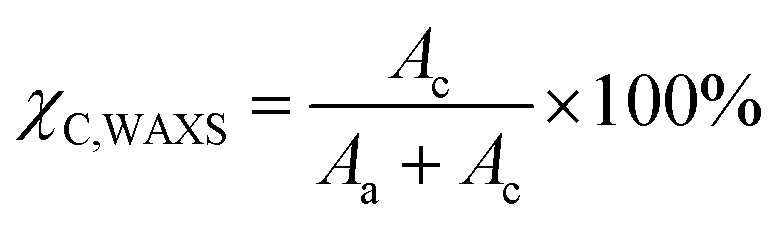Leveraging the bio-enabled muconic acid platform via phospha-Michael-addition: intrinsically flame-retardant nylon-66/DOPO copolymers†
Received
18th April 2024
, Accepted 7th August 2024
First published on 8th August 2024
Abstract
Efforts towards developing biobased chemicals primarily focus on generating molecules chemically analogous to those derived from petroleum. The compositional uniqueness of biomass can also be leveraged to reinvigorate the chemical industry with novel multifunctional molecules. We demonstrate the value and potential of these new compounds in the case of Nylon-66, a commodity polyamide that suffers from poor flame resistance. The conventional route to inhibit flammability involves blending the polymer with additives, which improves flame retardance but has mechanical property trade-offs. Herein, we address these limitations through the synthesis of a novel multifunctional comonomer derived from renewably sourced trans-3-hexenedioic acid (t3HDA). t3HDA was subjected to a one-pot isomerisation and functionalisation strategy where the alkene migrates to render this molecule active for phospha-Michael-addition (MA) with 9,10-dihydro-9-oxa-10-phosphaphenanthrene-10-oxide (DOPO), a halogen-free flame-retardant (FR). This DOPO-functional counit was polymerised into Nylon-66 copolymers and compared with physical blends of DOPO and Nylon-66 using a suite of thermomechanical techniques; analysis revealed comparable crystallinity, flame retardance, and thermomechanical properties for the DOPO-functionalised bio-advantaged polyamides. The synthesis strategy presented herein can be extended to a variety of functional groups and novel properties, a platform for creating bespoke bio-advantaged polymers.
Sustainability spotlight
In response to urgent global calls for sustainable industrial practices, this study introduces a transformative approach to synthesizing flame-retardant polyamides that significantly advances environmental sustainability. Addressing the critical issue of flammability in Nylon-66, a widely used polymer, our work circumvents the need for hazardous, leaching-prone additives by incorporating a novel, renewably sourced comonomer derived from muconic acid. This bio-based innovation mitigates health risks while preserving the mechanical integrity of the material, showcasing an exemplary case of enhancing safety without compromising performance. Aligned with the United Nations Sustainable Development Goals, particularly Goal 9 (Industry, Innovation, and Infrastructure) and Goal 12 (Responsible Consumption and Production), our research demonstrates how leveraging biobased solutions can lead to safer, more sustainable materials. The synthesis strategy presented paves the way for the development of a new class of biobased polymers with tailored properties, marking a significant step toward reducing our reliance on fossil fuels and enhancing the environmental resilience of the chemical industry.
|
Introduction
Synthetic polymers are ubiquitous to society and have served as the basis for countless advances in the fields of medicine, manufacturing, automotive, and consumer products. Most commodity polymers are derived from fossil-based carbon feedstocks. However, utilisation of biomass as a renewable alternative has been gaining prominence due to environmental concerns and growing consumer awareness. These efforts have largely focused on chemically identical replacements for petrochemicals using a bio-feedstock.1–3 However, the unique composition of biomass, for instance the high oxygen content of its building blocks and their distinct stereochemistry, offers opportunities to generate novel chemicals and materials previously unattainable through petroleum.4,5
Biobased platform intermediates that yield both direct replacements and novel molecules were recently identified through reaction network analysis and generation.6–9 The versatility of these bioprivileged compounds allows for their utilisation as a platform for the production of bio-derived commodity chemicals while derisking production of new performance-advantaged molecules. Fermentation-derived muconic acid has received increasing attention due to its bioprivileged nature.9,10 This platform molecule can be obtained from glucose and lignin derivatives using metabolically-engineered bacteria (E. coli and P. putida KT2440) or yeast (S. cerevisiae).11–17 Previous work has demonstrated the diversification of muconic acid to commodity chemicals such as adipic acid,11,13,16,18–21 hexamethylene diamine,22c-caprolactam,23,24 and terephthalic acid.25–27 Muconic acid derivatives have additionally been gaining prominence for the production of performance-advantaged fibre-reinforced polyesters28–30 and polyamides.6,26 This research fits within the broad push in recent years to increase the bio content of plastics in general, resulting in a significant amount of attention on bio-based polyamides.31–35 Two major strategies have developed out of this push. The first is bio-replacement, where chemically identical monomers derived from renewable sources are used instead of traditional petrochemical monomers. The second is bio-advantaged, where novel bio-based monomers with unique functionalities offering new properties are used in the synthesis. This work focuses on using muconic acid in a bio-advantaged strategy to introduce flame-retardance in polyamides.
The present work is based on the hypothesis that novel monomers derived from muconic acid could be molecularly engineered to become multifunctional, offering more than one target property with a single chemical. This approach could enable the chemical industry to overcome the drawbacks and tradeoffs usually associated with the utilisation of additives in polymer manufacturing. For instance, flame retardant (FR) additives are typically blended with polymers to lower their flammability36–38 and therefore broaden the range of their applications while lowering the risks of personal and property damage in the case of a fire. While vital, these FRs act as plasticisers and lower the mechanical properties of the final product compared to the parent polymer. Moreover, recent research has proposed leaching as a long-term environmental concern, leading to a desire to design polymers for their entire life cycle.39,40 Here, we explored the potential of trans-3-hexenedioic acid (t3HDA), a C6 monounsaturated diacid that is stereoselectively produced by electrochemical hydrogenation of muconic acid.41 We employed the carbon–carbon unsaturation in t3HDA as a tethering point to generate flame-inhibiting monomers that imparted improved mechanical properties in Nylon-66 (PA66). PA66 is an interesting test case owing to its widespread use as a versatile engineering thermoplastic with excellent thermal stability and hydrogen bonding enhanced mechanical properties.42,43 PA66 is found in a wide range of industries including automotives, electronics, films, and coatings. Despite its advantages, PA66 suffers from poor flame performance due to its hydrocarbon backbone, giving it a limiting oxygen index (LOI) value ranging from 21–23%44 and UL 94 rating of V-2.45 The current approach for improving flame performance includes the addition of glass and other chemical modifiers in these polyamides to achieve an improved V-0 rating.46–49 However, other polymer properties are considerably diminished due to the modifiers.
The FR candidate that was chosen for this study is 9,10-dihydro-9-oxa-10-phosphaphenanthrene-10-oxide (DOPO), a phosphorus-based FR that has been extensively studied for its superior flame-inhibiting characteristics.48–50 DOPO exhibits high thermal stability and improved environmental compliance compared to traditionally used brominated FRs. For the synthesis of a DOPO functionalised diacid, dimethyl-trans-3-hexenedioate (dmt3HD) underwent a base-catalyzed one-pot isomerisation and functionalisation through Michael-addition (MA). The isomerisation of dmt3HD to dimethyl-trans-2-hexenedioate (dmt2HD) rendered the molecule active towards MA, consequently allowing DOPO to form a Michael-adduct. The resulting monomer was incorporated into the standard PA66 backbone through copolymerisation with adipic acid and hexamethylenediamine. To compare the use of the bio-derived monomer over commercially used FR-blends, a physical mixture between DOPO and PA66 was prepared and characterised. DOPO grafted to the polymer backbone showed a 20% improvement in crystallinity, comparable mechanical properties, and ∼8% improvement in flame-inhibition with respect to FR-blended polymers. Overall, this study highlights the potential of using novel bio-derived monomers for performance-advantaged polymers.
Method and materials
Materials
Dimethyl-trans-3-hexenedioate (dmt3HD), 9,10-dihydro-9-oxa-10-phosphaphenanthrene-10-oxide (DOPO), and hexamethylene diamine (HMDA) were purchased from TCI America. Dimethyl formamide (DMF), ethyl acetate, methanol, and tetrahydrofuran (THF) were supplied by Fisher Scientific. Triethylamine (TEA), lithium hydroxide (LiOH), deuterium oxide (D2O), trifluoroacetic anhydride (TFAA), and adipic acid (AA) were procured from Sigma-Aldrich.
Additionally, nitrogen gas was purchased from Airgas, deuterated chloroform (CDCl3), and dimethyl sulfoxide-d6 (DMSO-d6) from Cambridge isotopes and 1,1,1,3,3,3-hexafluoroisopropanol (HFIP) from Oakwood Chemical.
Method
Polymerisation.
Nylon-66 salt was prepared by mixing equimolar amounts of AA and HMDA in methanol solutions for 1 h at 40 °C. The precipitated salt was recovered through filtration and was dried overnight in a fume hood. DOPO-functionalised Nylon-66 was obtained following a similar procedure. Due to the low solubility of DOPO-HDA in methanol, the salt for this diacid was prepared separately using THF as solvent. Prior to polymerisation, the two salts were combined in desired ratios and mixed with 60 wt% DI water to improve mixing. Samples were polymerised in a pressurised microreactor (Parr 4900) equipped with a heating jacket. The temperature was monitored through an external thermocouple. To ensure an inert environment during polymerisation, the reactor was purged with nitrogen five times and then pressurised to 150 psig. In the initial stage of the polymerisation, the reactor was held at an external set point of 265 °C for 2 h. At the end of this stage, nitrogen and water were vented out. Subsequently, the temperature was increased to 300 °C and held there for 3 h. The mixture was stirred at 400 rpm during the entire polymerisation process.
Monomer and polymer characterisations.
1H-NMR, 13C-NMR, and 31P-NMR spectra were collected with Bruker AVIII600 and NEO 400 spectrometers. Results were analyzed with MestReNova software. About 30–50 mg of monomer and salt samples were dissolved in DMSO-d6 and D2O respectively. For polyamides, approximately 50 mg of sample were dissolved in a 3![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :2 (v/v) mixture of CDCl3 and TFAA. GC/MS was carried out on an Agilent Technologies Model 6890 equipped with a DB-1 column. 1 μL of sample solution was injected with a 3.5 min solvent delay. The temperature was held at 50 °C for 0.2 min, ramped at 12.5 °C min−1 to 250 °C, and finally ramped at 25 °C min−1 to 340 °C. Analysis of the MS results was done through AMDIS software (NIST).
:2 (v/v) mixture of CDCl3 and TFAA. GC/MS was carried out on an Agilent Technologies Model 6890 equipped with a DB-1 column. 1 μL of sample solution was injected with a 3.5 min solvent delay. The temperature was held at 50 °C for 0.2 min, ramped at 12.5 °C min−1 to 250 °C, and finally ramped at 25 °C min−1 to 340 °C. Analysis of the MS results was done through AMDIS software (NIST).
Gel permeation chromatography (GPC).
EcoSEC gel permeation chromatography (GPC) system was used to measure polymer molecular weight distributions. The GPC system was equipped with UV and refractive index detectors and consisted of a Tosoh TSKgel SuperH6000 column in series with two Agilent PL HFIP gel columns. Approximately 5 mg polyamide samples were dissolved in 1,1,1,3,3,3-hexafluoroisopropanol (HFIP) and filtered through a 0.45 μm PTFE filter. Sample aggregation and ionic interactions were suppressed by the addition of sodium trifluoroacetate at a concentration of 0.02 mol L−1 HFIP (1.7 g kg−1 HFIP). A 10 μL sample volume was injected and analyzed at 45 °C under a 0.3 mL min−1 flow rate. Mn and Mw of samples were measured using polymethyl methacrylate (PMMA) standards.
Dynamic scanning calorimetry (DSC).
Thermal characterisations were conducted using a TA DSC Q2500 instrument. Specimens measuring under 5 mg were sealed in hermetic aluminum pans and were cycled thrice under nitrogen over a range of 30 °C to 300 °C with a ramp rate of 10 °C min−1. Results were calculated using Trios software from the third cycle to minimise the influence of the polymer's thermal history. ΔHm was calculated by integrating the melting peak while Tm and Tc were calculated at the peak of the melting and crystallisation events respectively. The degree of crystallinity χC, DSC was determined using the following equation:| | 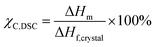 | (1) |
where ΔHf,crystal = 57.8 kJ mol−1 and refers to the enthalpy of fusion of a single crystal of Nylon-66.
Thermogravimetric analysis (TGA)
TGA measurements were performed in triplicate on a Netzsch STA449 F1 with Proteus software for data analysis. Specimens measuring 5–10 mg were placed in alumina pans. A 5 minutes isothermal stage was followed by a 10 °C min−1 ramp to 800 °C under 20 mL min−1 nitrogen flow. The decomposition temperature Td,10 was calculated at 10% mass loss and the residual char was measured at 500 °C. Differential thermogravimetric (DTG) curves were calculated using the first derivative of the TGA data and were plotted as the rate of mass loss versus temperature.
Wide-angle X-ray scattering (WAXS)
WAXS patterns were collected for annealed samples using a XENOCS Xeuss 2.0 SWAXS system equipped with Dectris Pilatus3 R 1M detector calibrated using a silver behenate standard. The WAXS measurements were performed under vacuum with the samples fixed directly on the holder. Monochromatised light (λ = 1.54 Å) from Cu Kα radiation was used over an acquisition period of 600 s. Results were analysed with Foxtrot 3.3.4 (SOLEIL Synchrotron France) for absolute intensity correction of the background signal. The degree of crystallinity (χC,WAXS) was calculated through the following equation based on the ratio of the area of the crystalline fraction (Ac) to the total area (Aa + Ac) under the WAXS curve:| | 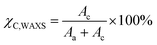 | (2) |
Microscale combustion calorimetry (MCC)
A micro-combustion calorimeter (MCC) was used to measure the heat of combustion of gases evolved during the controlled pyrolysis of the polyamides. The samples were analysed under nitrogen using a 1 °C s−1 heating rate from 200 to 700 °C using method A of ASTM D7309 (pyrolysis under nitrogen). The measurements were performed in triplicate to evaluate their reproducibility. The MCC provided the heat release rate as a function of temperature where total heat released (THR) was calculated as the area under the curve. Char yield was obtained by measuring the difference in sample mass before and after pyrolysis. Fire growth capacity (FGC) was calculated using ASTM D7309 to provide insights into the potential for the material to ignite and grow in fire intensity once ignited.
Dynamic mechanical analysis (DMA)
Polyamide samples in the ISO527-2-1BB geometry were prepared by injection moulding under an argon atmosphere using a HAAKE MiniJet Pro. Samples were subsequently annealed under inert conditions at 150 °C for 24 h. Torsional DMA was carried out for annealed samples using a TA Ares G2 instrument. Specimens were heated using a 5 °C min−1 ramp in the linear viscoelastic regime from 0 °C to 100 °C with an angular frequency of ω = 10 rad s−1. Analysis was performed using Trios software. The storage (E′) and loss (E′′) moduli were measured at 0 °C. The tan δ curve was calculated as a ratio between E′ and E′′. Tg values are reported at the peak of the tan δ.
Tensile tests
Tensile properties were measured using an Instron Universal Testing Machine (3369 series) with a 10 mm min−1 extension rate. Triplicates were tested with ISO527-2-1BB geometry and averages were reported.
Leaching test
DOPO leaching was measured by placing 20 mg of 10F and 10B in 6 mL of chloroform at room temperature for 24 hours and 3 mL of water at 80 °C for 72 hours. The solids were filtered then 1H-NMR was run on the remaining solution.
Results and discussion
Synthesis of FR-grafted monomer
A one-pot base-catalyzed isomerisation and Michael-addition (MA) was carried out to graft flame-retardant (DOPO) onto dmt3HD (Scheme 1). Equal volumes of TEA and DMF were used to shift the double bond in dmt3HD from the β-γ to α-β position (Fig. S1†), rendering it active for MA. Subsequently, DOPO underwent a nucleophilic 1,4-phospha-MA onto dmt2HD. The addition of the β position over the α position was confirmed using 2D HMBC proton-phosphorous NMR (Fig. S2†). This synthesis was performed on the diester (dmt3HD) over the diacid (t3HDA) since the methyl groups prevented ring-closing and formation of lactone by-products under basic conditions. Additionally, t3HDA exhibited limited reactivity towards MA compared to dmt3HD. The selective formation of the desired phospha-MA product was confirmed through GC/MS (Fig. S3†) and 1H-NMR (Fig. 1) which showed DOPO-HDA as the sole product. Peaks corresponding to DOPO were observed in the aromatic range of 7–8.5 ppm whereas the alkene peak of dmt3HD (5.6 ppm) disappeared completely. Recovery of DOPO-HDA was achieved through base-catalyzed hydrolysis (overall yield: 79.9%) and characterised by 1H-NMR (Fig. 1). DOPO-HDA was further utilised for copolymerisation and insertion in the Nylon-66 backbone.
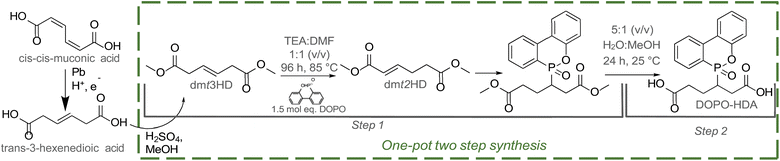 |
| | Scheme 1 Synthesis of DOPO-grafted diacid (DOPO-HDA) through a one-pot isomerisation and Michael addition (MA) reaction of dimethyl-trans-3-hexenedioate (dmt3HD). | |
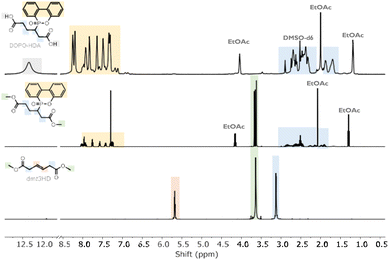 |
| | Fig. 1
1H NMR spectra of the (a) starting diester (dmt3HD) (b) DOPO-grafted diester in CDCl3 (c) and desired product (DOPO-HDA) in DMSO-d6. | |
Polymerisation and characterisation of monomer incorporation
Conventional Nylon-66 salt was prepared by precipitating HMDA and AA in MeOH. Next, DOPO-nylon blends were prepared by polymerising the Nylon-66 salt in the presence of a desired quantity of DOPO (5 or 10 wt%). This range was selected recognising the potential adverse impact of incorporating large quantities of a bulky pendant group on the resulting mechanical properties of the polymer. Post-polymerisation, these FR-blended polyamides were referred to as PA66-5B and PA66-10B for the samples containing 5 and 10 wt% DOPO, respectively (Scheme 2). Similarly, a zwitterionic salt was prepared by independently precipitating HMDA and DOPO-HDA in THF (Scheme 2 and Fig. S4†). The novel FR-salt was then mixed with the standard Nylon 66 salt in the desired ratios to emulate the DOPO loading in the blends. The polyamides wherein DOPO was functionalised to the backbone are referred to as PA66-5F and PA66-10F.
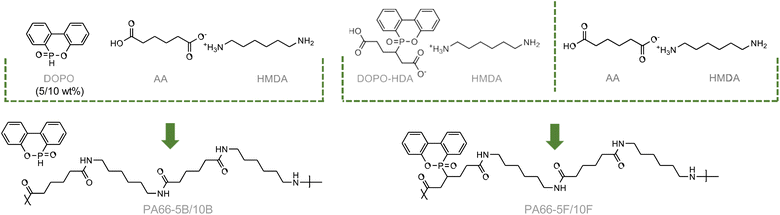 |
| | Scheme 2 Polymerisation of 5 and 10 wt% DOPO physically blended with PA66 (left) versus equivalent. | |
Table 1 compares the theoretical and actual incorporation of DOPO in the polyamide as determined by 1H-NMR (Fig. S5†). Actual DOPO incorporation was calculated by integrating the aromatic peaks belonging to DOPO with respect to the HMDA peak at 3.7 ppm. The calculated values confirmed that the concentration of DOPO in the blends and functionalised samples closely followed the expected values (Table 1) and therefore the flame-retardant properties of both sample series can be directly compared (vide infra). Additional confirmation for the survival of the grafted DOPO post-polymerisation was provided by 31P NMR (Fig. S6†) and the 1H–31P heteronuclear multiple bond correlation (HMBC) spectra presented in Fig. S7 and S8.† PA66-10B exhibited one phosphorus peak showing cross-peaks only in the aromatic region which substantiated that DOPO remains as a free molecule in the polymer matrix. In contrast, two phosphorus peaks were observed for PA66-10F (Fig. S7b†). The sharper peak (44 ppm) was associated with the functionalised DOPO established by cross-peaks appearing in both the aromatic and aliphatic regions. The ratio of the two phosphorus peak integrations showed that ∼25% DOPO detached from the polymeric backbone and, therefore, is present as a free molecule, such as with the blends. However, as a majority of the DOPO remained attached to the polyamide backbone differences in properties between blended and functionalised samples are still driven by the functionalised groups.
Table 1 Comparison of theoretical and actual DOPO incorporation into polyamide through 1H-NMR and molecular weights of polymer series
| Sample |
Expected mol% incorporation |
Actual mol% incorporationa |
M
n
(kDa) |
Đ
|
|
Incorporation of DOPO was calculated through 1H NMR analysis of the polymers.
M
n: Number-average molecular weight of polymers were calculated with respect to PMMA standards.
|
| PA66 |
— |
— |
65.6 |
1.78 |
| PA66-5B |
4.8 |
4.9 |
78.1 |
1.79 |
| PA66-5F |
4.8 |
5.0 |
83.6 |
1.65 |
| PA66-10B |
9.6 |
8.7 |
62.3 |
1.72 |
| PA66-10F |
9.6 |
11.0 |
62.6 |
1.68 |
Molecular weight of polymer series
The GPC chromatograms for the polyamide series are shown in Fig. S9.† The corresponding molecular weights calculated with respect to PMMA standards are displayed in Table 1. The extent of DOPO loading (5 wt% versus 10 wt%) had minimal influence on Mn as PA66-5B/10B showed comparable weights intermediate to the PA66 control.
Thermal and crystal properties
The effect of blended and functionalised DOPO on thermal properties and crystallinity was investigated through DSC, WAXS, and TGA (Fig. 2, 3, S10 S11,† and Table 2). PA66 exhibited the highest melting temperature (Tm = 260.5 °C) and degree of crystallinity (χC,DSC = 26%) based on DSC (Fig. 2). DOPO incorporation, whether as a blend or a pendant group, inhibited crystal formation and led to a decrease in Tm and χC,DSC. While PA66-5B/5F exhibited similar ΔHm and Tm, a prominent difference was observed with the higher DOPO-loaded samples. PA66-10B showed the lowest Tm (239.7 °C) and χC,DSC (15%). In contrast, the tethering of DOPO to the PA66 backbone in the 10F sample showed superior crystallinity of over 18%. To evaluate the crystalline structure, room temperature WAXS was performed on polymer samples annealed for 24 h (Fig. S11†). The semi-crystalline nature of these polyamides was established by the sharp peaks representing crystalline segments and the characteristic broad halo corresponding to the amorphous regions. The percent crystallinity (χC,WAXS) is reported as the ratio of the areas under the peaks normalised over the total area.
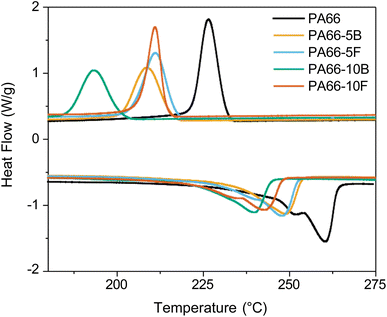 |
| | Fig. 2 DSC thermograms for the flame-retardant polyamide series with respect to PA66 showing melting/crystallisation event. | |
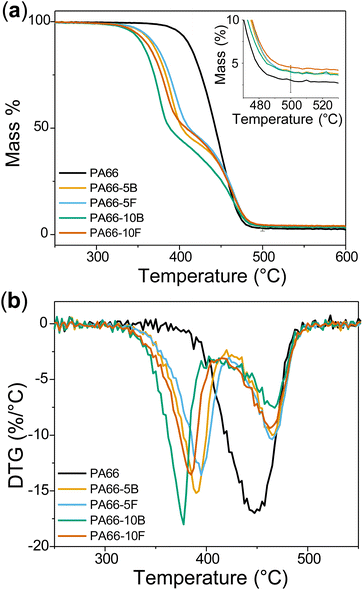 |
| | Fig. 3 (a) TGA thermograms for neat PA66 and flame-retardant polyamide series under nitrogen atmosphere. (b) Differential thermogravimetry (DTG) shows the temperature when maximum thermal decomposition occurs. | |
Table 2 Thermal properties of the polyamides with different loadings of DOPO as blends or functionalised into the backbone
| Sample |
DSC analysis |
WAXS |
TGA analysis |
|
T
m (°C)a |
ΔHm (J g−1) |
χ
c, DSC (%) |
T
c (°C) |
χ
c, WAXSb (%) |
Char500°Cc (%) |
T
d10 (°C) |
DTG peak 1d (°C) |
DTG peak 2d (°C) |
|
T
m: melting temperature; ΔHm: enthalpy of melting; Tc: crystallisation temperature; χc,DSC : percent crystallinity (DSC).
χ
c,WAXS: percent crystallinity from WAXS (annealed).
Char500 °C : residual mass at 500 °C; Td10: decomposition temperature at 10% mass loss under N2 from TGA.
DTG peaks: calculated from the derivative of TGA curves.
|
| PA66 |
260.5 |
66.4 |
26.0 |
226.7 |
42.2 |
3.04 ± 1.4 |
487.2 ± 0.3 |
448.4 ± 3.6 |
— |
| PA66-5B |
248.8 |
50.8 |
19.9 |
208.8 |
35.0 |
4.05 ± 0.6 |
330.7 ± 4.0 |
391.2 ± 0.2 |
466.2 ± 1.6 |
| PA66-5F |
247.9 |
49.5 |
19.4 |
211.2 |
38.8 |
4.04 ± 0.3 |
325.0 ± 3.5 |
393.5 ± 1.9 |
462.3 ± 3.1 |
| PA66-10B |
239.7 |
38.2 |
15.0 |
193.3 |
31.8 |
3.96 ± 0.8 |
327.5 ± 5.2 |
377.2 ± 0.2 |
458.3 ± 7.8 |
| PA66-10F |
242.9 |
46.8 |
18.3 |
211.0 |
38.2 |
4.57 ± 2.2 |
302.7 ± 2.3 |
384.4 ± 1.3 |
463.5 ± 0.3 |
PA66 exhibited two characteristic peaks at approximately 20° and 24° corresponding to the (100) and (010)/(110) doublet of the α-phase. PA66 possesses a triclinic structure in which the (100) peak represents intrasheet scattering caused by adjacent polymer chains in a sheet and the (010)/(110) peak corresponds to the intersheet scattering between polymer sheets. Following the results from DSC, it was expected that both modes of DOPO incorporation would retard crystallisation. While that effect was observed, the difference in % crystallinity between blends and functionalised samples was amplified post-polymer annealing. In the case of the blended specimens at 5 and 10 wt% loadings (PA66-5/10B), the polymers showed a decrease in crystallinity to 35.0% and 31.8%, respectively, compared to 42.2% in neat PA66. The functionalised counterparts, PA66-5/10F, exhibited higher crystallinity values of 37.2% and 33.0% respectively. These results confirmed that the attachment of DOPO to the polymer backbone improved the overall crystallinity compared to the blended systems. Thermogravimetric analysis (TGA) was performed on PA66 and FR polyamides to investigate the influence of DOPO on thermal decomposition and flame inhibition (Fig. 3a).36 The DTG curve (1st derivate of the TGA curve) shows thermal decomposition events more prominently (Fig. 3b). PA66 showed one decomposition peak consistent with the literature, whereas polymers containing DOPO exhibited two decomposition events. The first stage corresponds to the evolution of DOPO out of the polymer matrix, which is corroborated by the boiling point of DOPO reported at 380 °C. Tethering DOPO to the polymer backbone allowed it to evolve marginally later compared to the DOPO blended samples. This is demonstrated in Table 2 where PA66-10B showed its 1st decomposition peak 8 °C lower than its functionalised counterpart (PA66-10F). From Fig. 3a and Table 2, char yield for all samples containing DOPO increased compared to PA66. Phosphorus is known to promote char formation through phosphoric acid derivatives, which in turn may act as cross-linkers to decomposed polymeric byproducts.42 The char layer subsequently acts as a mass and heat transfer barrier to slow flame spread.42 Char yield was seen to be the highest for the 10F sample, which could be the result of tethering DOPO to the polyamide backbone and allowing it to interact with the solid phase. Therefore, tethering FR to a polymeric backbone reduced the volatilisation of the FR and enabled it to increase interaction with the condensed phase.
Flame-inhibition analysis
The industry standard of flame-retardancy testing is the LOI and UL-94 tests. LOI measures the amount of oxygen required to sustain combustion, while UL-94 evaluates the material's self-extinguishing capability when exposed to an open flame. These tests give a practical and detailed analysis of how materials behave in flame-rich environments. However, they remain industry standards outside the practical limits of laboratory research due to the relatively large amounts of material needed to perform accurate testing. Producing novel materials for heuristic screenings is fundamentally a material-limited endeavour. As a result, MCC has become the de facto test for small-scale material analysis.51–54 Should this work attract enough industry interest to perform scale-up, these tests would become critical to perform.
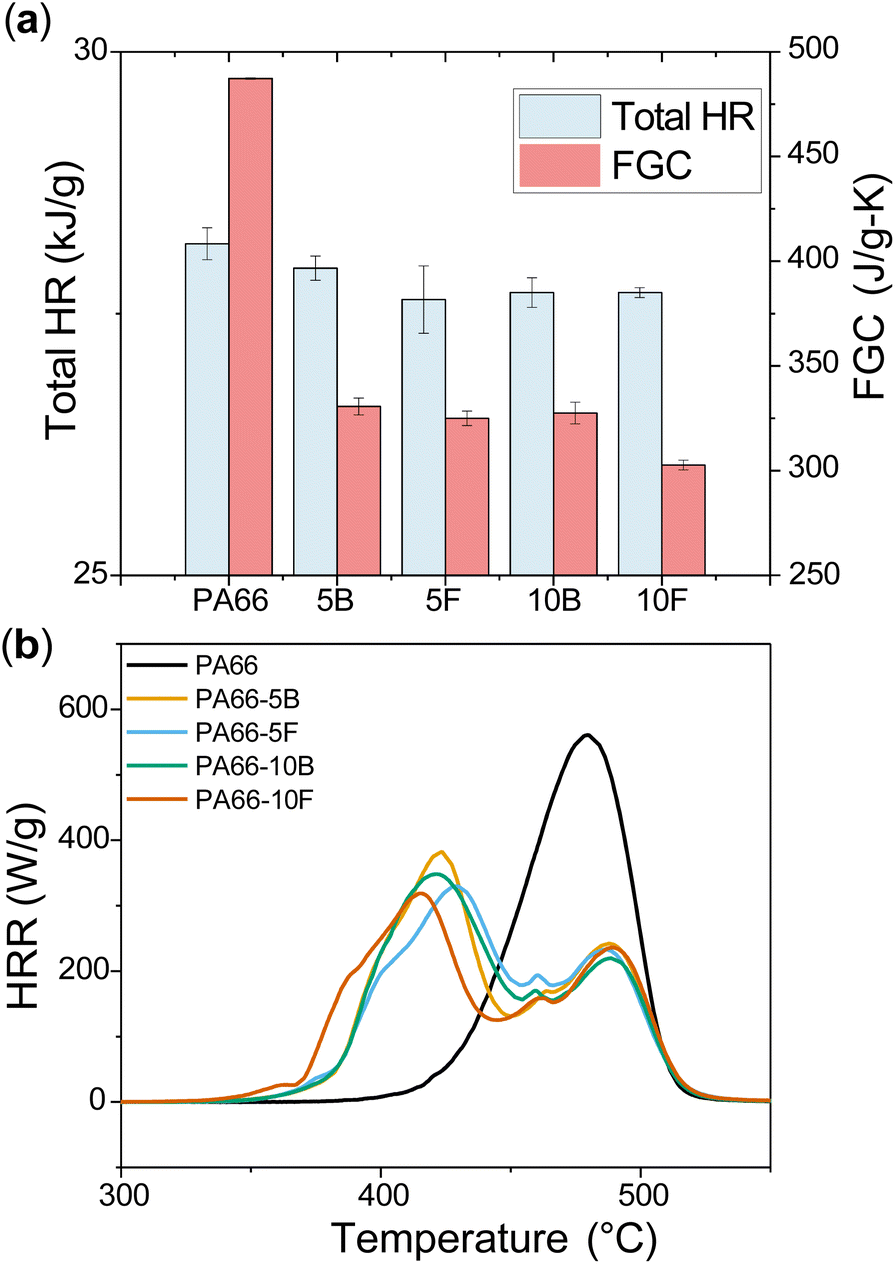
MCC was used to measure the heat release rate (HRR) versus temperature to gauge flame performance for the polyamide series (Fig. 4). Compared to the PA66 control sample, all DOPO-containing materials showed improvements in terms of heat release and char yield (Table S1 and Fig. S12†). In contrast to PA66, the FR-samples showed two peaks of heat release, indicating that DOPO chemistry is changing the thermal decomposition characteristics of the polyamides. Fire growth capacity (FGC) is a parameter calculated through ASTM D7309, which is dependent on chemical structure and gives insights into ignition and flame growth once ignited.43 The FGC of PA66 decreased from 500 J g−1 K−1 to 327.5 J g−1 K−1 for PA66-10B and 302.6 J g−1 K−1 for PA66-10F. Additionally, a large reduction in peak HRR was observed for the PA66-10B and PA66-10F samples, showing that higher loadings of DOPO enhance flame inhibition (Table S1†). Overall, it can be seen that DOPO enhanced flame retardance compared to neat PA66; however, its mode of incorporation affected polymer performance minimally. A more drastic difference might be expected between blends and functionalised FRs if lower boiling point modifying chemicals are used.
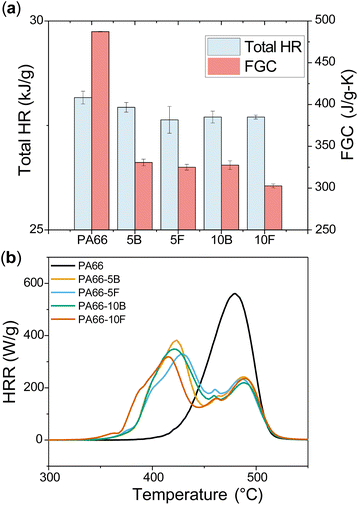 |
| | Fig. 4 (a) Total heat release of pure PA66 as compared to both blended and functionalised samples. The presence of DOPO decreased the total HR significantly. The FGC is the fire growth capacity, which gives insight into the ignition and subsequent flame growth. (b) MCC results showing heat release rate (HRR) curves for neat PA66 and flame-retardant polyamide series. | |
Leaching analysis
The use of additive packages into commercial resins has become ubiquitous due to the excellent thermal and mechanical property modification advantages offered. Recent research has raised environmental concerns due to the release of these additive packages as the polymer breaks down in the environment,39,40 leading to a desire to design polymers for their entire life-cycle, including their eventual waste management. DOPO is a popular flame-retardant additive due to its lower health risk as compared to other FRs, such as bromine-based flame inhibitors. The health effects of the DOPO molecule are less than the halogen based FRs, but are unlikely to be zero. Therefore, designing covalently-bonded FR molecules lowers the risk of bioaccumulation and should be pursued. The amount of leaching between the blend and the functionalized sample was tested using two chemical washes. Both 10F and 10B powders were subjected to a chloroform bath at room temperature for 24 hours. The water bath was heated to 80°, above the Tg, and held for 72 hours. The powders were then filtered, and 1H-NMR was performed on the filtered solution. In both cases, the resulting 1H-NMR demonstrated no NMR-detectable quantities of DOPO had been removed from the polymer during their chemical washes. This implies the PA66/DOPO interactions are sufficiently strong enough to prevent significant leaching even in artificially contrived high surface area states with a favourable solvent over the time scales explored. These results indicate that further investigations into quantifying the leaching rates of common nylon additives to screen for high-impact functionalisation candidates are needed. If the appropriate additive can be established, this work would provide a template for the creation of nylon with significant differences in the leaching rate between a physical blend and the new functionalised polyamide.
Rheology and mechanical properties
Torsional dynamic mechanical analysis (DMA) was conducted to understand the viscoelastic response of the annealed polyamides to a known deformation as a function of temperature (Fig. 5). Values for the storage modulus (E′), loss modulus (E′′), and tan δ are summarised in Table 3. E′ and E′′ were reported at the glassy plateau of 0 °C. Tan δ is calculated through the ratio between E′ and E′′ where δ is the phase angle between the stress and the strain. In this study, the glass-transition temperature of the polymers (Tg) was located using the maxima of tan δ. Tg is a key polymer property and is characterised as the temperature at which long-range motion in the amorphous domains ceases as a polymer melt is cooled. Over this transition, dramatic changes in properties are observed as the polymer shifts from a rubbery to a glassy state. PA66 showed the lowest E′ followed by PA66-5B/F. A slight reduction in E′ was also observed for the 10F sample relative to 10B, potentially due to their reduced Mn. However, a significant increase in Tg (∼10 °C) was observed between PA66-10B and 10F. In the blends, free DOPO molecules act as plasticizers in the amorphous domains of the polymer. This enables chain mobility at lower temperatures, which is revealed by the suppressed Tg exhibited by the blends. In contrast, when DOPO is functionalised to the polyamide backbone, chain mobility and diffusivity are reduced due to the anchored pendant group. This effect is observed through the consistently increased Tg in both functionalised samples (5F/10F) compared to their corresponding blends.
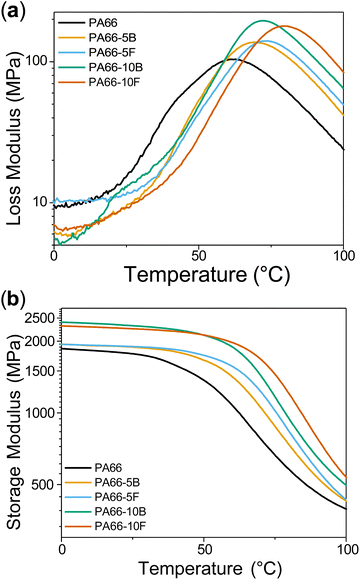 |
| | Fig. 5 DMA results showing (a) loss modulus (E′′) and the (b) storage modulus (E′) curves for neat PA66 and flame-retardant polyamide series. | |
Table 3 Viscoelastic properties of polyamides measured through torsional DMA
| Sample |
E′ @ 0 °C (GPa) |
E′′@ 0 °C (MPa) |
T
g
(°C) |
|
T
g values are reported at the peak of the tan δ curve.
|
| PA66 |
1.86 |
9.5 |
71.6 |
| PA66-5B |
1.94 |
6.2 |
79.9 |
| PA66-5F |
1.94 |
9.9 |
83.1 |
| PA66-10B |
2.40 |
3.9 |
80.8 |
| PA66-10F |
2.32 |
6.9 |
90.3 |
Mechanical properties of the samples were assessed using tensile tests that were run in triplicate (Table 4). Mechanical properties of polymers are related to the crystallinity and molecular weights of the samples. At 5% loading, the functionalised sample (5F) showed a significantly higher UTS and Young's modulus compared to both neat PA66 and PA66-5B. This could be explained in part due to the suppressed crystallisation in the 5B sample compared to the 5F sample. This trend did not extend to the 10 wt% DOPO samples, however. The 10F samples were significantly more brittle than others, which is reflected in the lower tensile toughness values and the large variance in the UTS value. This increase in brittleness is due to the unique processing conditions necessary for 10F. As the quantity of DOPO-functionalised monomer increased, the temperature gap between Tm and degradation narrowed. At 10% incorporation, the gap was small enough to cause considerable difficulties in processing. As a result, the 10F samples were not injection moulded but solvent cast in n-cresol. While this allowed for analysis of mechanical properties, the unique process embrittled the material. The increase in mechanical properties in the sample 5F combined with the challenge to process 10F suggests a practical upper limit to the addition of the functionalised monomer. We note the increase in Tg observed in the 10F continued into the Tm, with melting temperature approaching the degradation temperature. This made thermal processing unfeasible; solvent casting was thus necessary and led to a more complicated product. Future experimentation and scaling should, therefore, focus on 5F. This material requires less functionalised monomer, a costly addition, has nearly equal flame-retardance, and superior mechanical performance, making it the prime candidate for future research. Additionally, we note that these samples were tested bone dry and without added plasticizer. Atmospheric moisture is well known to plasticize PA66, significantly reducing its brittleness.55 In the future, retesting with larger tensile specimens conditioned in a humidified environment would provide a complementary picture of how these materials could be expected to perform under everyday use conditions.
Table 4 Mechanical properties of polymer specimens (triplicates) as determined through tensile testing
| Sample |
UTS (MPa) |
Toughness (MPa) |
Young's modulus (GPa) |
Elongation at break (%) |
| PA66 |
84.2 ± 10.6 |
3.69 ± 1.03 |
1.18 ± 0.00 |
8.03 ± 1.20 |
| PA66-5B |
89.0 ± 13.0 |
3.50 ± 1.10 |
1.47 ± 0.00 |
7.25 ± 1.20 |
| PA66-5F |
129 ± 6.40 |
9.97 ± 3.90 |
1.47 ± 0.01 |
12.7 ± 2.80 |
| PA66-10B |
90.4 ± 4.50 |
3.39 ± 0.42 |
1.56 ± 0.01 |
7.02 ± 0.50 |
| PA66-10F |
68.7 ± 24.9 |
2.15 ± 1.17 |
1.46 ± 0.22 |
5.50 ± 1.30 |
Conclusion
Utilisation of bio-based molecules can be leveraged to give access to novel multifunctional monomers that are key towards producing performance-advantaged polymers. Demonstrated in this study is the potential of a multifunctional monomer produced using fermentation-derived t3HDA for flame-retardant applications. Currently, flame retardance in polymers is primarily achieved by physically blending flame-retardant molecules. However, this approach can lead to leaching of the FRs that ultimately diminishes flame-inhibiting performance and also detrimentally affects mechanical properties in the polymer caused by the disruption in crystal formation. To address these drawbacks, a novel diacid was prepared by tethering FR molecule DOPO onto the unsaturation offered by t3HDA and copolymerised into a Nylon-66 backbone. For equivalent comparison, PA66 was physically blended with DOPO and the impact of the two modes of DOPO incorporation on polymer properties was investigated. Tethering the FR group achieved improved flame retardance at higher loadings compared to FR blends. However, significant improvements in crystallinity were offered by DOPO-functionalised polyamides over the blends. χc was minimally affected compared to neat PA66, and its effect was observed through improved mechanical properties when molecular weights were comparable. As the DOPO segments and free molecules were relegated to the amorphous regions, properties observed via DMA seemed to be affected compared to neat PA66. Tg and E′ were increased due to the rigidity offered by the aromatic rings in DOPO. This study leverages bio-advantaged and bio-privileged concepts and uses t3HDA as a vehicle for inserting flame retardance while minimally compromising other key polymer properties. Extension of this chemistry would create new opportunities to investigate halogen-free nitrogen and phosphorus-based FRs that were previously not considered due to their volatility as free molecules.56 Furthermore, the synthesis framework can be used to accelerate the development of biopolymers by allowing selective alteration of polymer properties.
Author contributions
Prerana Carter, PhD (data curation: equal; formal analysis: equal; investigation: lead; methodology: equal; visualisation: equal; writing – original draft: lead); Peter M. Meyer (data curation: equal; formal analysis: equal; investigation: equal; visualisation: equal; writing – review & editing: lead); Michael J. Forrester, PhD (data curation: supporting; formal analysis: equal; investigation: supporting; methodology: supporting; supervision: supporting; writing – review & editing: equal); Dhananjay Dileep (data curation: supporting; formal analysis: supporting; methodology: supporting); Ting-Han Lee, PhD (data curation: supporting; investigation: supporting; methodology: supporting); Sohaima Noreen (data curation: supporting); Nickolas L. Chalgren (data curation: supporting); Brent Shanks, PhD (conceptualisation: equal; project administration: lead; resources: lead; supervision: equal; writing – review & editing: supporting); Jean-Philippe Tessonnier, PhD (conceptualisation: equal; supervision: equal; writing – review & editing: equal); Eric W. Cochran, PhD (conceptualisation: equal; data curation: supporting; formal analysis: supporting; funding acquisition: supporting; investigation: supporting; methodology: supporting; writing – original draft: supporting; writing – review & editing: equal).
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
This material is based upon work supported by the U.S. Department of Energy's Office of Energy Efficiency and Renewable Energy (EERE) under the Bioenergy Technologies Office Award Number DE-EE0008492. We also acknowledge the support of the National Science Foundation through NSF-2113695. We thank Dr Sarah Cady (ISU Chemical Instrumentation Facility) for training and assistance pertaining to the NMR results included in this publication.
Notes and references
- E. S. Lipinsky, Science, 1981, 212, 1465–1471 CrossRef CAS PubMed.
- S. Choi, C. W. Song, J. H. Shin and S. Y. Lee, Metab. Eng., 2015, 28, 223–239 CrossRef CAS PubMed.
- S. Takkellapati, T. Li and M. A. Gonzalez, Clean Technol. Environ. Policy, 2018, 20, 1615–1630 CrossRef CAS PubMed.
-
N. Fitzgerald and A. Bailey, in Moving beyond Drop-In Replacements: Performance-Advantaged Biobased Chemicals, 2018 Search PubMed.
- R. M. Cywar, N. A. Rorrer, C. B. Hoyt, G. T. Beckham and E. Y.-X. Chen, Nat. Rev. Mater., 2022, 7, 83–103 CrossRef CAS.
- J. Huo and B. H. Shanks, Annu. Rev. Chem. Biomol. Eng., 2020, 11, 63–85 CrossRef PubMed.
- X. Zhou, Z. J. Brentzel, G. A. Kraus, P. L. Keeling, J. A. Dumesic, B. H. Shanks and L. J. Broadbelt, ACS Sustainable Chem. Eng., 2019, 7, 2414–2428 CrossRef CAS.
- B. H. Shanks and P. L. Keeling, Green Chem., 2017, 19, 3177–3185 RSC.
- B. H. Shanks and L. J. Broadbelt, ChemSusChem, 2019, 201900323 Search PubMed.
- I. Khalil, G. Quintens, T. Junkers and M. Dusselier, Green Chem., 2020, 22, 1517–1541 RSC.
-
N. Saito and H. Nakajima, U.S. Pat., US5487987A, 1996 Search PubMed.
- K. A. Curran, J. M. Leavitt, A. S. Karim and H. S. Alper, Metab. Eng., 2013, 15, 55–66 CrossRef CAS PubMed.
- D. R. Vardon, M. A. Franden, C. W. Johnson, E. M. Karp, M. T. Guarnieri, J. G. Linger, M. J. Salm, T. J. Strathmann and G. T. Beckham, Energy Environ. Sci., 2015, 8, 617–628 RSC.
- M. Suástegui and Z. Shao, J. Ind. Microbiol. Biotechnol., 2016, 43, 1611–1624 CrossRef PubMed.
- M. Suastegui, J. E. Matthiesen, J. M. Carraher, N. Hernandez, N. Rodriguez Quiroz, A. Okerlund, E. W. Cochran, Z. Shao and J.-P. Tessonnier, Angew. Chem., Int. Ed., 2016, 55, 2368–2373 CrossRef CAS PubMed.
-
J. M. Carraher, J. M. Carraher, T. Pfennig, R. G. Rao, B. H. Shanks and J.-P. Tessonnier, U.S. Pat., US8592189B2, 2013 Search PubMed.
- H. Almqvist, H. Veras, K. Li, J. G. Hidalgo, C. Hulteberg, M. Gorwa-Grauslund, N. S. Parachin and M. Carlquist, ACS Sustainable Chem. Eng., 2021, 9(24), 8097–8106 CrossRef CAS.
- D. R. Vardon, N. A. Rorrer, D. Salvachúa, A. E. Settle, C. W. Johnson, M. J. Menart, N. S. Cleveland, P. N. Ciesielski, K. X. Steirer, J. R. Dorgan and G. T. Beckham, Green Chem., 2016, 18, 3397–3413 RSC.
- S. Capelli, D. Motta, C. Evangelisti, N. Dimitratos, L. Prati, C. Pirola and A. Villa, ChemCatChem, 2019, 11, 3075–3084 CrossRef CAS.
- J. E. Matthiesen, J. M. Carraher, M. Vasiliu, D. A. Dixon and J.-P. Tessonnier, ACS Sustainable Chem. Eng., 2016, 4, 3575–3585 CrossRef CAS.
- W. Niu, K. M. Draths and J. W. Frost, Biotechnol. Prog., 2002, 18, 201–211 CrossRef CAS PubMed.
-
M. Lu, F. Lu, J. Chen, W. Yu, Q. Huang, J. Zhang and J. Xu, WO Patent, WO2015086819A1, 2015 Search PubMed.
-
R. Beerthuis, G. Rothenberg and N. R. Shiju, U.S. Pat., US20130085255, 2013 Search PubMed.
- R. Beerthuis, G. Rothenberg and N. R. Shiju, Green Chem., 2015, 17, 1341–1361 RSC.
- D. I. Collias, A. M. Harris, V. Nagpal, I. W. Cottrell and M. W. Schultheis, Ind. Biotechnol., 2014, 10, 91–105 CrossRef CAS.
- J. M. Carraher, T. Pfennig, R. G. Rao, B. H. Shanks and J.-P. Tessonnier, Green Chem., 2017, 19, 3042–3050 RSC.
- R. Lu, F. Lu, J. Chen, W. Yu, Q. Huang, J. Zhang and J. Xu, Angew. Chem., Int. Ed., 2016, 55, 249–253 CrossRef CAS PubMed.
- N. A. Rorrer, S. Nicholson, A. Carpenter, M. J. Biddy, N. J. Grundl and G. T. Beckham, Joule, 2019, 3, 1006–1027 CrossRef CAS.
- N. A. Rorrer, J. R. Dorgan, D. R. Vardon, E. J. Gjersing and G. T. Beckham, ACS Sustainable Chem. Eng., 2016, 4, 6867–6876 CrossRef CAS.
- N. A. Rorrer, D. R. Vardon, J. R. Dorgan, E. J. Gjersing and G. T. Beckham, Green Chem., 2017, 19, 2812–2825 RSC.
- A. Patti and D. Acierno, Polymers, 2022, 14, 692 CrossRef CAS PubMed.
- M. Galià, L. M. de Espinosa, J. C. Ronda, G. Lligadas and V. Cádiz, Eur. J. Lipid Sci. Technol., 2010, 112, 87–96 CrossRef.
- P. Radzik, A. Leszczyńska and K. Pielichowski, Polym. Bull., 2020, 77, 501–528 CrossRef CAS.
- M. M. Kleybolte, L. Zainer, J. Y. Liu, P. N. Stockmann and M. Winnacker, Macromol. Rapid Commun., 2022, 43, 2200185 CrossRef CAS PubMed.
- S. Huf, S. Krügener, T. Hirth, S. Rupp and S. Zibek, Eur. J. Lipid Sci. Technol., 2011, 113, 548–561 CrossRef CAS.
- B. A. Howell, K. L. Oberdorfer and E. A. Ostrander, Int. J. Polym. Sci., 2018, 2018, e7237236 Search PubMed.
- B. A. Howell, K. E. Carter and H. Dangalle, Renewable and Sustainable Polymers, Am. Chem. Soc., 2011, 1063, 133–152 CAS.
- B. A. Howell and W. Sun, Polym. Degrad. Stab., 2018, 157, 199–211 CrossRef CAS.
- A. Blum and B. N. Ames, Science, 1977, 195, 17–23 CrossRef CAS PubMed.
- A. B. Morgan, Polym. Rev., 2019, 59, 25–54 CrossRef CAS.
- M. N. Dell'Anna, M. Laureano, H. Bateni, J. E. Matthiesen, L. Zaza, M. P. Zembrzuski, T. J. Paskach and J.-P. Tessonnier, Green Chem., 2021, 23, 6456–6468 RSC.
-
M. I. Kohan, Nylon Plastics Handbook, Hanser Publishers, Munich; New York: Cincinnati, 1995 Search PubMed.
- M. Winnacker and B. Rieger, Macromol. Rapid Commun., 2016, 37, 1391–1413 CrossRef CAS PubMed.
- D. W. van Krevelen, Polymer, 1975, 16, 615–620 CrossRef CAS.
- E. Braun and B. C. Levin, Fire Mater., 1987, 11, 71–88 CrossRef CAS.
- U. Braun, B. Schartel, M. A. Fichera and C. Jäger, Polym. Degrad. Stab., 2007, 92, 1528–1545 CrossRef CAS.
- M. S. Subbulakshmi, N. Kasturiya, P. B. Hansraj, P. Bajaj and A. K. Agarwal, J. Macromol. Sci., Part C: Polym. Rev., 2000, 40, 85–104 CrossRef.
- K. A. Salmeia and S. Gaan, Polym. Degrad. Stab., 2015, 113, 119–134 CrossRef CAS.
- X. Wang, Y. Hu, L. Song, W. Xing, H. Lu, P. Lv and G. Jie, Polymer, 2010, 51, 2435–2445 CrossRef CAS.
- J. Sag, P. Kukla, D. Goedderz, H. Roch, S. Kabasci, M. Döring and F. Schönberger, Polymers, 2020, 12, 778 CrossRef CAS PubMed.
- Q. Xu, R. A. Mensah, C. Jin and L. Jiang, J. Therm. Anal. Calorim., 2022, 147, 6001–6013 CrossRef CAS.
- Q. Xu, L. Jiang, A. Majlingova, N. Ulbrikova, R. A. Mensah, O. Das and F. Berto, Polymers, 2022, 14, 45 CrossRef CAS PubMed.
- R. E. Lyon, R. Walters and S. Stoliarov, Polym. Eng. Sci., 2007, 47, 1501–1510 CrossRef CAS.
- C. Q. Yang, Q. He, R. E. Lyon and Y. Hu, Polym. Degrad. Stab., 2010, 95, 108–115 CrossRef CAS.
- J. Sag, D. Goedderz, P. Kukla, L. Greiner, F. Schönberger and M. Döring, Molecules, 2019, 24, 3746 CrossRef CAS PubMed.
- Y. Cao, X.-L. Wang, W.-Q. Zhang, X.-W. Yin, Y.-Q. Shi and Y.-Z. Wang, Ind. Eng. Chem. Res., 2017, 56, 5913–5924 CrossRef CAS.
|
| This journal is © The Royal Society of Chemistry 2024 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence ab,
Peter M.
Meyer‡
ab,
Peter M.
Meyer‡