
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Photoresponsive hydrogel friction†
Allison L.
Chau‡
a,
Kseniia M.
Karnaukh‡
b,
Ian
Maskiewicz
b,
Javier
Read de Alaniz
 *b and
Angela A.
Pitenis
*a
*b and
Angela A.
Pitenis
*a
aMaterials Department, University of California, Santa Barbara, Santa Barbara, CA, USA. E-mail: apitenis@ucsb.edu
bDepartment of Chemistry and Biochemistry, University of California, Santa Barbara, Santa Barbara, CA, USA. E-mail: javier@chem.ucsb.edu
First published on 29th August 2024
Abstract
Photoresponsive hydrogels are an emerging class of stimuli-responsive materials that exhibit changes in physical or chemical properties in response to light. Previous investigations have leveraged photothermal mechanisms to achieve reversible changes in hydrogel friction, although few have focused on photochemical means. To date, the tribological properties of photoswitchable hydrogels (e.g., friction and lubrication) have remained underexplored. In this work, we incorporated photoresponsive methoxy-spiropyran-methacrylate monomers (methoxy-SP-MA) into a hydrogel network to form a copolymerized system of poly(N-isopropylacrylamide-co-2-acrylamido-2-methylpropane sulfonic acid-co-methoxy-spiropyran-methacrylate) (p(NIPAAm-co-AMPS-co-SP)). We demonstrated repeatable photoresponsive changes to swelling, friction, and stiffness over three light cycles. Our findings suggest that volume changes driven by the decreased hydrophilicity of the methoxy-SP-MA upon light irradiation are responsible for differences in the mechanical and tribological properties of our photoresponsive hydrogels. Our results could inform future designs of photoswitchable hydrogels for applications ranging from biomedical applications to soft robotics.
1. Introduction
Stimuli-responsive hydrogels are an important and growing class of hydrogels that change water content, structure, or mechanical and interfacial properties in response to environmental changes such as pH,1,2 temperature,3,4 solvent composition,5 electrostatics,6,7 and more recently, light.8–12 This responsive behavior makes such hydrogels attractive materials for 3D printing resins,13 biomedical applications,14–16 atmospheric water harvesting,17,18 and the rapidly emerging field of soft robotics.19–21 Light-responsive hydrogels take inspiration from biology, which utilizes light in many biological processes such as photosynthesis, phototaxis, and phototropism.22,23 Light offers bio-orthogonal advantages over other stimuli (e.g., pH, temperature, solvents, etc.) by providing non-contacting and precise spatiotemporal control with wavelength specificity. Due to this versatility, there is increasing interest in the development of photoresponsive materials.One method to form photoresponsive hydrogels is through the utilization of optoproteins, which were first reported in 197324 and are a class of light-responsive proteins that control various cell signaling pathways.25 Their discovery has formed a new field of molecular optogenetics,26 and when incorporated into biological hydrogel networks, such as collagen, the elastic modulus can be spatiotemporally controlled and increased due to light-induced dimerization and oligomerization.27–29 However, one drawback that limits the broad utilization of optoproteins is the time-consuming and low yield purification process.28 A synthetic analog to optoproteins that are commonly used to form photoresponsive polymeric systems are molecular photoswitches that change conformation and properties upon light irradiation. Photoswitches can be broadly classified by their switching mechanism, where the reversion back to its original state is driven by a thermal process (T-type) or by irradiation with another wavelength of light (P-type). Azobenzene-30,31 (either T- or P-type) and spiropyran-derived11,32,33 (T-type) photoswitches are commonly utilized and incorporated into liquid crystal elastomers34–36 and hydrogel networks resulting in light-induced actuation, bending, and locomotion.8,10,11,30,37–41
Azobenzene drives macroscopic actuation by undergoing an E/Z isomerization and elongating (trans isomer) or contracting (cis isomer) the polymer chains to which it is attached.30,31,42 Conversely, spiropyran undergoes pericyclic isomerization in response to light irradiation and exchanges between the spiropyran (SP) (ring-closed) and merocyanine (MCH+) (ring-open) conformations. The equilibrium and switching kinetics of spiropyran molecules are highly dependent on substitution pattern, solvent selection, and solution pH.32,43–45 In an acidic environment, the hydrophilic MCH+ form is often stabilized and can be converted to the hydrophobic SP upon irradiation with blue light (Fig. 1). In the absence of light, the SP form undergoes thermal relaxation to the MCH+ form. This hydrophilicity change drives hydrogel dehydration and rehydration, leading to volume changes.11,32 Due to the significant change in polarity commonly accompanied by the proton release, spiropyran/merocyanine-based photoswitches have been used as photoacids46,47 and photosurfactants.48,49
 | ||
| Fig. 1 (a) Chemical structure of methoxy-spiropyran-methacrylate (methoxy-SP-MA) monomer in acidic solution. Under 470 nm irradiation, the monomer isomerizes to the spiropyran form. In the dark, it thermally relaxes back to the merocyanine form. (b) UV-vis spectra of 0.02 mM methoxy-SP-MA monomer in acidified methanol (pH ≈ 2) at varying intervals in the dark after 1 min of 470 nm light exposure. It took approximately 2.5 h to thermally relax from SP back to MCH+ in the dark, as demonstrated by the increased absorbance at λ ≈ 450 nm. | ||
While many groups have investigated the swelling behavior and actuation of spiropyran-incorporated hydrogels,8–11,38,39 the tribological behavior (e.g., friction and lubrication) of light-responsive hydrogels remains largely unexplored. However, several studies over the past decade have investigated related phenomena, including adhesion and surface roughness changes in spiropyran-incorporated elastomers50,51 and hydrogels in response to light.52 Photocleavable groups have been attached to polymer brushes to control friction53 while others have demonstrated photoresponsive friction changes of azobenzene-derived hydrogels due to a sol–gel transition.54 The photothermal effect of thermoresponsive hydrogels has been exploited to tune friction by embedding gold nanoparticles55,56 or metals57 into poly(N-isopropylacrylamide) (pNIPAAm) networks. In these cases, the main mechanism behind the friction change is driven by the lower critical solution temperature (LCST) of pNIPAAm and the rapid phase transition and network collapse that occurs when the LCST is surpassed.
However, in our work we aimed to synthesize photoresponsive hydrogels with tunable friction coefficients that did not rely on the photothermal effect but rather utilized the photochemical effect of spiropyran photoswitches to drive chemical changes within the network that would lead to macroscopic volume changes. Methoxy-spiropyran-methacrylate (methoxy-SP-MA) was synthesized and conjugated with N-isopropylacrylamide (NIPAAm) and 2-acrylamido-2-methylpropane sulfonic acid (AMPS) and crosslinked with N,N′-methylenebisacrylamide (MBAm) to form the copolymerized hydrogel network (p(NIPAAm-co-AMPS-co-SP)). AMPS was chosen as the co-monomer for this study to increase the overall hydrophilicity of the system and, as a result, the difference in rehydration/deswelling of hydrogels in aqueous solutions. Previously, Li et al. examined different ring substituents of water-soluble spiropyrans in various solution pHs and discovered that sulfonated groups led to volume expansion upon light irradiation in acidic environments due to increased net charge.8 In addition, they demonstrated that to shift the equilibrium to the merocyanine form in the dark, acidic solutions with low pHs (pH ≈ 2) are required.8 To minimize possible hydrolysis of the spiropyran/merocyanine moiety commonly associated with the nucleophilic attack of water, we incorporated a methoxy group into the architecture, which has been previously shown to improve hydrolytic stability as well as shift the equilibrium to the merocyanine form.44,49,58
Our work was guided by the hypothesis that these hydrogels would exhibit lower friction coefficients in sliding contact with glass hemispherical probes when the methoxy-SP-MA was in the MCH+ form and higher friction in the SP form due to an increase in hydrophobicity, leading to hydrogel deswelling. To the authors’ knowledge, this is the first instance where volume changes in response to light were used to control friction of bulk hydrogels. We report herein sample volume, friction coefficient, and elastic modulus before and after irradiation. Additionally, the switching kinetics between the MCH+ and SP forms was evaluated for the monomers, polymers, and hydrogels.
2. Materials and methods
2.1 Chemicals
Hydrogel samples were prepared using a combination of commercially available and synthesized constituents. N-Isopropylacrylamide 97% (NIPAAm), ammonium persulfate >98% (APS), and N,N,N′,N′-tetramethylethylenediamine 99% (TEMED) were purchased from Sigma Aldrich. N,N′-methylenebisacrylamide 99+% (MBAm) and 1,4-dioxane 99+% were purchased from ThermoFisher Scientific. 2-Acrylamido-2-methylpropane sulfonic acid >98% (AMPS) was purchased from Tokyo Chemical Industry Co., Ltd. All reagents were used as received. 2-(5′-methoxy-3′,3′-dimethylspiro[chromene-2,2′-indolin]-1′-yl) ethyl methacrylate (methoxy-SP-MA) (molecular weight: 405.494 g mol−1) was synthesized and characterized as detailed in ESI† Section S1 (Fig. S1–S7). Stock solutions of APS (10 wt%) were prepared in ultrapure Milli-Q water (18.2 MΩ cm). Phosphate buffer solution (PBS) (pH ≈ 2) was prepared by adding 1 M HCl to 100 mM NaH2PO4 solution. The pH of the final solution was measured using the Thermo Scientific Orion StarTM A111 Benchtop pH meter.2.2 Light source
Throughout polymerization, swelling experiments, and mechanical testing, the irradiation source was 470 nm collimated LED lights (Thorlabs, Inc. M470L5-C1). Light intensity was measured using a power meter (Thorlabs PM100D) with a standard photodiode power sensor (Thorlabs S121C, 10 mm diameter, 400–1100 nm, 500 nW–500 mW). A light intensity of I = 35 ± 1 mW cm−2 was obtained at 1 A current and maximum power while the blue light source was 35 mm away from the sensor. Similar light intensities and experimental conditions were used for all photoswitching measurements in this study.2.3 Hydrogel synthesis
Copolymerized p(NIPAAm-co-AMPS-co-SP) hydrogel disks were prepared by dissolving 140 mg NIPAAm, 1.4 mg AMPS (0.5 mol% relative to total polymer concentration), 13 mg methoxy-SP-MA (2.5 mol%), and 5.1 mg of MBAm (0.5 mol%) in a solution of 800 μL dioxane, 100 μL DI water, and 7.4 μL TEMED. The precursor solution was bubbled with nitrogen for 30 min. 100 μL of 10 wt% APS was added (forming a 4![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 vol/vol solution of 1,4-dioxane:DI water) to initiate polymerization, and the precursor was polymerized between two glass slides with 1.7 mm thick VitonTM spacers in a nitrogen-rich environment for 1 h. During polymerization, the samples were irradiated with 470 nm light to drive the monomer equilibrium to the SP form to facilitate more efficient incorporation into the network. Due to the difference in reactivity rate between acrylamide and methoxy-spiropyran-methacrylate, it is important to conduct the polymerization of hydrogels in an oxygen-free atmosphere to ensure complete incorporation of methoxy-SP-MA.59,60 After polymerization, the gels were sectioned with an 18 mm circular punch (pre-swollen thickness, t = 1.6 mm) and swollen in 100 mM phosphate buffer solution (pH ≈ 2) in the dark for at least 36 h prior to testing to ensure the gels reached equilibrium swelling in the MCH+ conformation. All mechanical tests and volume measurements were completed two days after synthesis. Control hydrogels of p(NIPAAm-co-AMPS) were polymerized in a similar manner as the p(NIPAAm-co-AMPS-co-SP) hydrogels utilizing the same solvent system of 4:1 v/v 1,4-dioxane:DI water, except 144 mg NIPAAm was added to keep the polymer concentration consistent.
:1 vol/vol solution of 1,4-dioxane:DI water) to initiate polymerization, and the precursor was polymerized between two glass slides with 1.7 mm thick VitonTM spacers in a nitrogen-rich environment for 1 h. During polymerization, the samples were irradiated with 470 nm light to drive the monomer equilibrium to the SP form to facilitate more efficient incorporation into the network. Due to the difference in reactivity rate between acrylamide and methoxy-spiropyran-methacrylate, it is important to conduct the polymerization of hydrogels in an oxygen-free atmosphere to ensure complete incorporation of methoxy-SP-MA.59,60 After polymerization, the gels were sectioned with an 18 mm circular punch (pre-swollen thickness, t = 1.6 mm) and swollen in 100 mM phosphate buffer solution (pH ≈ 2) in the dark for at least 36 h prior to testing to ensure the gels reached equilibrium swelling in the MCH+ conformation. All mechanical tests and volume measurements were completed two days after synthesis. Control hydrogels of p(NIPAAm-co-AMPS) were polymerized in a similar manner as the p(NIPAAm-co-AMPS-co-SP) hydrogels utilizing the same solvent system of 4:1 v/v 1,4-dioxane:DI water, except 144 mg NIPAAm was added to keep the polymer concentration consistent.
2.4 UV-vis spectroscopy
UV-vis absorption spectra were measured on Shimadzu UV 3600 UV-vis-NIR Spectrometer using an Absolute Absorbance stage with a working range of 180 to 3600 nm. The photoinduced optical absorption kinetics was measured on a home-built pump–probe setup, as previously reported by our group.61 For more details, see ESI† Section S2.1. UV-vis absorption spectra and kinetics were obtained for methoxy-SP-MA monomer in acidified MeOH (Fig. S8, ESI†) and for linear p(NIPAAm-co-AMPS-co-SP) (Fig. S9, ESI†) and p(NIPAAm-co-AMPS-co-SP) hydrogels (Fig. S10, ESI†) in 100 mM phosphate buffer solution (pH ≈ 2). For more details about the preparation of the UV-vis samples and detailed characterization of materials, see ESI† Section S2.2–S2.5.2.5 Hydrogel characterization
 and in the light after at least 5 h of blue light irradiation (λ = 470 nm, I = 35 ± 1 mW cm−2) to determine
and in the light after at least 5 h of blue light irradiation (λ = 470 nm, I = 35 ± 1 mW cm−2) to determine  . Three dark–light cycles were conducted, and gels equilibrated in the dark at least 16 h before further characterization the following day. Representative indentation curves are displayed in Fig. 2a and Fig. S11 (ESI†), and the reported E* is the average and standard deviation across three separate hydrogels (n = 3 samples, 45 total indentations (Fig. S12, ESI†).
. Three dark–light cycles were conducted, and gels equilibrated in the dark at least 16 h before further characterization the following day. Representative indentation curves are displayed in Fig. 2a and Fig. S11 (ESI†), and the reported E* is the average and standard deviation across three separate hydrogels (n = 3 samples, 45 total indentations (Fig. S12, ESI†).
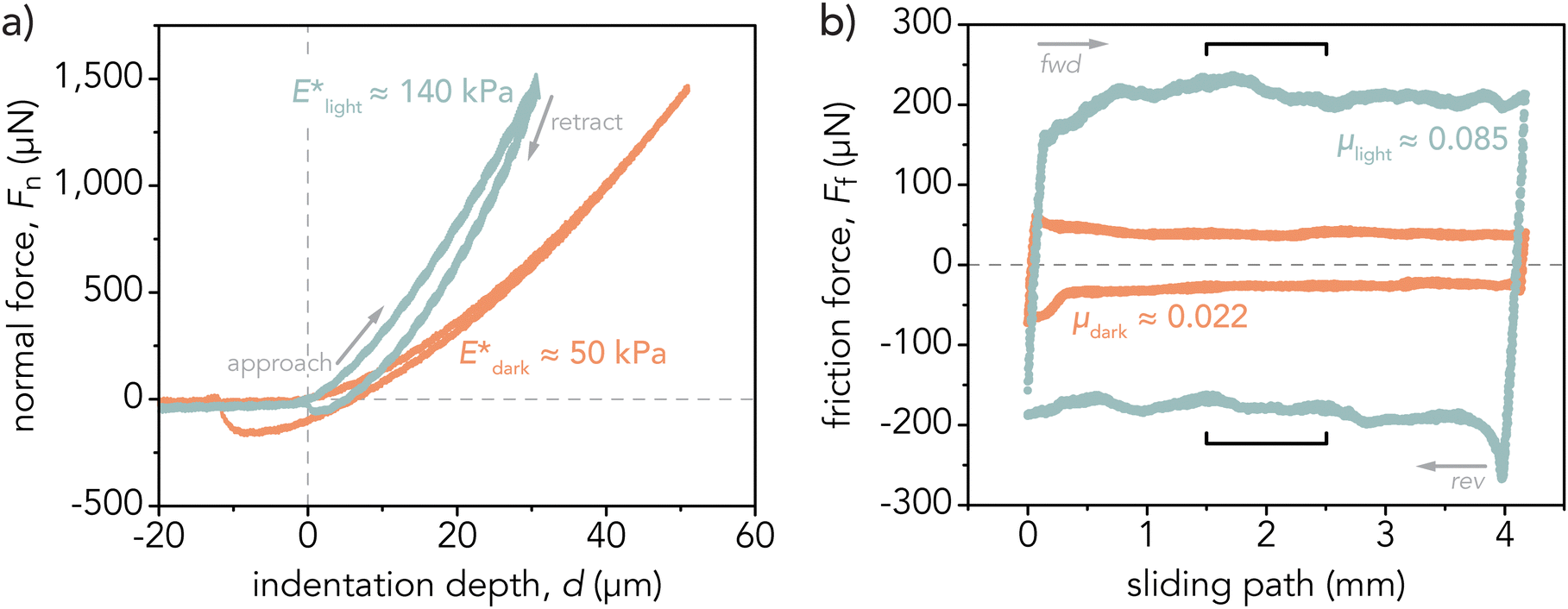 | ||
| Fig. 2 Representative (a) indentation curves and (b) friction force loops for p(NIPAAm-co-AMPS-co-SP) hydrogels in the dark (orange) and after light irradiation (blue). The reduced elastic modulus and friction force both increased in the SP state. Indentations were conducted at an applied force of Fn = 1.5 mN and indentation velocity of 10 μm s−1. The labeled E* values are the approximate average elastic moduli across three samples (n = 3 hydrogels, 45 indents) for one cycle in the dark or light respectively. Friction coefficients were measured at an applied force of Fn = 2 mN and sliding velocity of v = 0.1 mm s−1 by analyzing the free sliding regime in the middle 25% of the sliding path (indicated by brackets). The labeled μ values are the approximate average friction coefficients across three samples (n = 3 hydrogels). | ||
The friction ratio between the light and dark state was calculated as fc = μlight,c/μdark,c. A friction ratio fc > 1 indicates that the gel exhibited increased friction coefficients within the light state.
3. Results and discussion
3.1 Switching and swelling kinetics
UV-vis spectroscopy data demonstrated that methoxy-spiropyran-methacrylate monomers photoisomerized from MCH+ to SP, within a minute under the application of 470 nm light (I = 25 ± 1 mW cm−2) and thermally relaxed in the dark to MCH+ within 4 h (Fig. 1b and Fig. S8, ESI†). Due to the poor solubility of the methoxy-SP-MA in the phosphate buffer solution (pH ≈ 2), the UV-vis spectroscopy experiments were performed in an acidified MeOH solution to mimic the conditions of the phosphate buffer (pH ≈ 2) (ESI† Sections S2.2 and S2.3). To determine the photoswitching properties of a spiropyran/merocyanine molecule when incorporated into a network, we synthesized linear and crosslinked p(NIPAAm-co-AMPS-co-SP) (ESI† Sections S2.4 and S2.5). Due to the improved solubility, the UV-vis spectroscopy characterization was performed in 100 mM phosphate buffer (pH ≈ 2). The photoswitching behavior of the linear p(NIPAAm-co-AMPS-co-SP) in PBS (pH ≈ 2) (Fig. 3) was similar to the methoxy-SP-MA monomer (Fig. 1b) and linear p(NIPAAm-co-AMPS-co-SP) in acidified methanol (Fig. S9b, ESI†). Both systems can be irradiated for several cycles without a loss of photoswitching properties. Characterization of the crosslinked p(NIPAAm-co-AMPS-co-SP) hydrogel network (Fig. S10, ESI†) was challenging due to absorption oversaturation, even when the concentration of methoxy-SP-MA was decreased to 0.5 mol% for UV-vis spectroscopy experiments compared to the 2.5 mol% methoxy-SP-MA used for mechanical testing. Despite that, we were able to observe similar photoswitching properties to the linear p(NIPAAm-co-AMPS-co-SP). | ||
| Fig. 3 (a) Chemical structure of linear p(NIPAAm-co-AMPS-co-SP) in the merocyanine and spiropyran forms. (b) UV-vis spectrum of 0.150 mg mL−1 linear p(NIPAAm-co-AMPS-co-SP) in 100 mM phosphate buffer (pH ≈ 2) with 5 mol% AMPS. The absorbance at λ ≈ 450 nm increased over time in the dark as the methoxy-SP-MA thermally relaxed from the SP conformation back to the MCH+ conformation upon removal of 470 nm light. (c) Pump–probe kinetics measurements upon irradiation with 470 nm light for approximately 1 min (Thorlabs, Inc. M470F3, I = 25 ± 1 mW cm−2), followed by the thermal relaxation of the molecule over 4 h in the dark, monitored at the λ ≈ 450 nm absorbance peak. | ||
To demonstrate photoswitchability, p(NIPAAm-co-AMPS-co-SP) hydrogels were equilibrated in 100 mM PBS (pH ≈ 2) for at least 36 h and then irradiated from above with 470 nm light at an intensity of 35 mW cm−2. In addition to hydrophilicity changes upon light exposure, methoxy-SP-MA molecules are photochromic, possessing a red color in the ring-open MCH+ state and becoming pale-yellow/colorless in the ring-closed SP state.47,65–67 Within the hydrogel network, this color change was perceived as transitioning from red to light yellow (Fig. 4) with the MCH+ quickly converting to SP within seconds of light irradiation (Video S1, ESI†). Over 10 min, light penetrated deeper into the bulk of the hydrogel network due to increased transparency at the surface, creating a photobleaching front that propagated through the thickness of the hydrogel. Eventually, the entire hydrogel changed color (Fig. 4b), supporting conversion throughout the bulk of the hydrogel.
 | ||
| Fig. 4 (a) Chemical structure of p(NIPAAm-co-AMPS-co-SP) hydrogel. (b) Top- and side-view of hydrogel demonstrating the photobleaching front propagating through the thickness of the hydrogel over 10 min with image of hydrogel before and after photobleaching. Black arrows and white lines are used to guide the eye to indicate the photobleaching front. | ||
3.2 Volume change
The p(NIPAAm-co-AMPS-co-SP) hydrogels demonstrated repeatable photoswitchable contraction and swelling, as displayed in Fig. 5.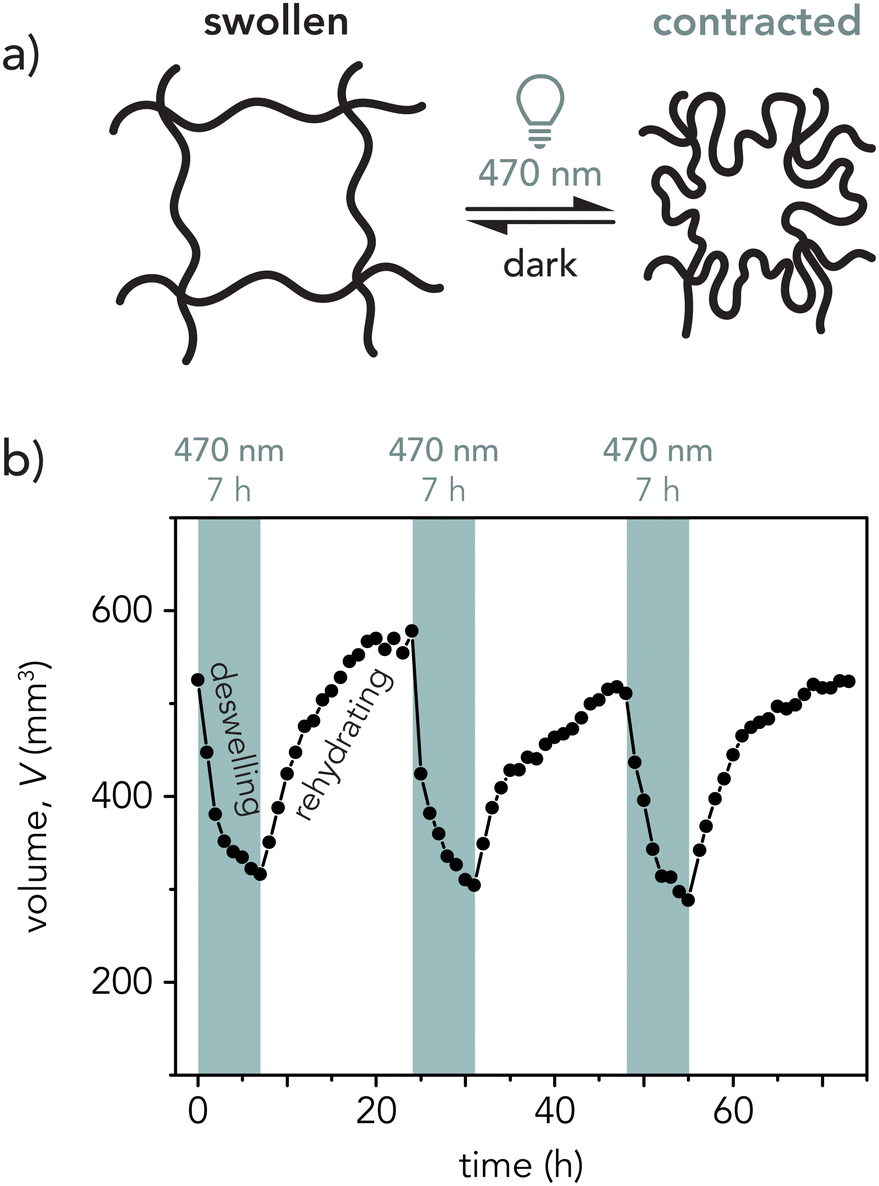 | ||
| Fig. 5 (a) Hydrogel schematic depicting the possible microstructural change within the hydrogel mesh upon light irradiation. (b) Representative plot of volume as a function of time for three light–dark cycles (7 h blue light on, 17 h off). | ||
As MCH+ converts to SP upon light irradiation, not only is there a color change but a hydrophilicity change as the molecule becomes more hydrophobic due to the loss of the positively charged nitrogen upon ring-closing, leading to deswelling of the hydrogel mesh (Fig. 5a). Within the first 3 h, sample volume decreased quickly (−38 ± 12%, n = 14 hydrogels, see Fig. S13, ESI†) then slowed over the following 2–4 h (−14 ± 5%, n = 10 hydrogels). Once the light was turned off, the hydrogel slowly rehydrated over 17 h as the SP started to thermally equilibrate back to the MCH+ form. Occasionally, as demonstrated in Fig. 5b, the hydrogel reached its initial volume after the third light cycle. However, most gels in this study only rehydrated to about 80% of the initial sample volume. Similar declines in rehydration percentage have been observed in other photoresponsive hydrogel systems,39,68 while others have seen more consistent rehydration.9,30 This may be caused by inherent sample-to-sample variation compounded by inherent error in volume measurements. Additionally, there may be incomplete conversion between the MCH+ and SP states between light cycles. However, the underlying mechanism responsible for this effect is not well understood and warrants further study.
3.3 Photoresponsive lubricity
The friction coefficients of p(NIPAAm-co-AMPS-SP) hydrogels were measured before and after illumination with 470 nm light. Hydrogels were equilibrated in 100 mM phosphate buffer solution (pH ≈ 2) for at least 36 h before mechanical characterization. Fig. 6a and c demonstrate a representative friction experiment: friction coefficients and hydrogel volumes were obtained while the gels were in the dark and after direct irradiation (λ = 470 nm, I = 35 ± 1 mW cm−2) for 3 h. Hydrogel volumes were measured again after mechanical testing, and gels were left to equilibrate in the dark for at least 16 h before the next cycle began to account for the slow rehydration times. Fig. 6a demonstrates that friction repeatedly increased at least 5 times the initial value after light irradiation for a representative gel. Fig. 6b shows the average friction coefficients and standard deviations in the dark and light state across three hydrogel samples for three dark–light cycles. For friction coefficient values for each hydrogel, see Fig. S14 and S15 (ESI†). The average initial friction coefficient in the dark was μdark,1 = 0.022 ± 0.002. After direct light irradiation for 3 h, the friction coefficient almost quadrupled to μlight,1 = 0.085 ± 0.039 leading to a friction ratio (μlight,1/μdark,1) of f1 = 3.9. However, the friction ratio increased with each dark–light cycle (μdark,2 = 0.023 ± 0.003, μlight,2 = 0.170 ± 0.010, μdark,3 = 0.019 ± 0.003, μlight,3 = 0.160 ± 0.030), with f2 = 7.4 and f3 = 8.5 for the second and third light cycles, respectively. This indicates that the friction increase with light irradiation is repeatable. The friction ratios reported herein surpass other pNIPAAm-based systems that depend on the combined effects of temperature and light for photoresponsive hydrogel friction.56,57 | ||
| Fig. 6 Representative (a) friction coefficient and (c) normalized volume ((V/Vdark,1) × 100%) as a function of time for a p(NIPAAm-co-AMPS-co-SP) hydrogel sample. Friction and volume measurements were recorded after the gel underwent 3 dark–light cycles, during which gels equilibrated in the dark for about 16 h and were irradiated with 470 nm light for 3 h. Upon irradiation, friction increased 5 times while the volume of the gel decreased 60% due to increased hydrophobicity upon conversion to the SP state. Multiple dark–light cycles demonstrate the repeatability of the friction change between the MCH+ and SP states. (b) Bar plot demonstrating the average friction coefficient and standard deviation for 3 individual hydrogels in the dark and under 470 nm light with the friction ratio (f = μlight/μdark) labeled for each light cycle. (d) Bar plot of average normalized volume ((V/Vdark,1) × 100%) across 3 hydrogels for each dark–light cycle. Gels generally recovered about 75% of their initial volume after the first light cycle. | ||
Representative hydrogel swelling behavior corresponding with friction measurements for a single hydrogel sample is depicted in Fig. 6c, which shows normalized volume ((V/Vdark,1) × 100%) over time. For this experiment, hydrogel volume decreased after the first light cycle and reswelled to approximately 50% of the initial volume overnight. The sample deswelled and rehydrated to similar volumes during the second and third light cycles.
Fig. 6d demonstrates the average normalized volume ((V/Vdark,1) × 100%) for three hydrogels across the dark–light cycles. On average, gels only reached about 75% of their initial volume after the first cycle but consistently deswelled to 50% of their initial volume (Vdark,1) after 3 h of irradiation for all the light cycles. This may indicate that not all the methoxy-SP-MA fully converted back to the MCH+ form after the first light cycle. This is consistent with previous results, where poly(N-isopropylacrylamide-co-acrylic acid) hydrogels also did not reach full volume recovery after the first light cycle but were able to achieve consistent recovery in subsequent cycles.39
In addition to friction and volume changes, the elastic modulus also changed with light. 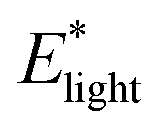 was consistently 2 to 3 times greater than
was consistently 2 to 3 times greater than  for each dark–light cycle and across three hydrogel samples (Fig. S12, ESI†). According to rubber elasticity theory, modulus is dependent on the density of elastically active polymer chains (ρel) within a network, with greater density leading to higher moduli.69–71 For hydrogels, mesh size (ξ), which can be defined as the average spacing between polymer chains in a network, is inversely proportional to ρel.72 Therefore, a higher density of chains often indicates a smaller mesh. And an increase in elastic modulus due to hydrogel deswelling, as was observed in our photoresponsive gels, may indicate decreases in ξ. Based on de Gennes’ elastic modulus scaling relationship of E ∝ ξ−3,70 if Elight ≈ 2Edark, then ξlight ≈ 0.79 ξdark. Therefore, mesh size may decrease roughly 20 to 30% depending on the elastic modulus increase with light. This estimation is also supported by the volume change that occurs upon light irradiation.
for each dark–light cycle and across three hydrogel samples (Fig. S12, ESI†). According to rubber elasticity theory, modulus is dependent on the density of elastically active polymer chains (ρel) within a network, with greater density leading to higher moduli.69–71 For hydrogels, mesh size (ξ), which can be defined as the average spacing between polymer chains in a network, is inversely proportional to ρel.72 Therefore, a higher density of chains often indicates a smaller mesh. And an increase in elastic modulus due to hydrogel deswelling, as was observed in our photoresponsive gels, may indicate decreases in ξ. Based on de Gennes’ elastic modulus scaling relationship of E ∝ ξ−3,70 if Elight ≈ 2Edark, then ξlight ≈ 0.79 ξdark. Therefore, mesh size may decrease roughly 20 to 30% depending on the elastic modulus increase with light. This estimation is also supported by the volume change that occurs upon light irradiation.
While de Gennes’ scaling concepts link the elastic modulus with hydrogel mesh size,70 friction coefficients have also been shown to scale with mesh size for Gemini hydrogel (gel-on-gel) sliding configurations with larger mesh sizes leading to lower friction coefficients.73 Similarly, others have demonstrated that mesh size increases with water content.74,75 Therefore, we postulate that friction increased upon light irradiation due to decreasing mesh size – indicated by hydrogel deswelling and increased elastic modulus – associated with the increased hydrophobicity of the methoxy-SP-MA molecules.
3.4 Challenges and opportunities for photoresponsive hydrogels
This study presents, to the authors’ knowledge, the first instance that photoswitchable friction relying on a photochemical effect rather than photothermal56,57 or a sol–gel transition54 has been demonstrated in bulk hydrogel materials. Zhu et al. demonstrated friction changes for gold nanoparticle-incorporated pNIPAAm hydrogels about 1 mm thick. A green light with 3.5 W cm−2 intensity, which is an order of magnitude greater than what was used here (I = 35 ± 1 mW cm−2), was able to locally increase the temperature within the pNIPAAm network and surpass the LCST, leading to a rapid collapse of the gel. They reported that the friction coefficient increased from approximately μ ≈ 0.25 to μ ≈ 0.7 (f = 2.8) within 10 s and decreased within 30 s.56 Similarly, Wu et al. demonstrated photothermal friction changes for MXene-p(NIPAAm-co-AMPS) hydrogels irradiated with near-infrared light (I = 1 W cm−2). In their study, they observed that friction increased from μ ≈ 0.02 to μ ≈ 0.13 (f = 6.5) within 5 min and decreased within 1 min.57 However, both hydrogel systems relied on the LCST behavior of pNIPAAm to drive the change in friction behavior. Here, we demonstrated photoresponsive friction due to the hydrophilic transition of SP-MA which led to light-induced deswelling and repeatably higher friction ratios.It is worth noting that NIPAAm, which is thermoresponsive, was required for our studies due to its solubility in both water and dioxane. This solvent mixture was necessary to dissolve monomers and the photochromic material for these studies. Additionally, AMPS was chosen as the comonomer to increase swelling within aqueous solutions and to add a source of internal protons.39 It has been demonstrated that the incorporation of AMPS with NIPAAm leads to an increased lower critical solution temperature (LCST), but homogeneous pNIPAAm hydrogels can shrink under irradiation due to a slight temperature increase even several degrees below the LCST,39 which was something also observed herein (Fig. S13 and S16, ESI†). Even though there was a small volume change (−8 ± 7%, n = 6 hydrogels), there was no significant change in friction coefficient after 3 h of 470 nm light irradiation (I = 35 ± 1 mW cm−2) (Fig. S16, ESI†), indicating that the driving force behind the friction change for our p(NIPAAm-co-AMPS-co-SP) hydrogels is most likely the methoxy-SP-MA incorporated monomers.
One limitation in our experiments is that the deswelling and rehydration of the hydrogels are diffusion-controlled, which limits the rate at which friction can change due to diffusion of solution in and out of the network. One way to potentially accelerate this process is by reducing the dimensions of the hydrogel, which is why smaller and thinner (typically < 0.5 mm) hydrogels are often used, leading to deswelling and swelling times on the order of minutes rather than hours.10 Intentionally synthesizing porous hydrogels can also decrease swelling times from hours to seconds but may sacrifice strength.76 In contrast, the gels herein are approximately 2 mm thick and 20 mm in diameter when fully swollen. These thicknesses are required for friction and indentations using our existing microtribometer configuration to ensure that we remain within the small strain limit (d/t ≤ 0.1) and to avoid any substrate effects. Additionally, thicker substrates help reduce bending or wrinkling effects during light penetration that often occur due to gradients in light intensity and strain.
The photoresponsive behavior demonstrated herein is not limited to bulk hydrogels but can be applied to thin (<1 mm) hydrogel coatings as well. Thin layers of p(NIPAAm-co-AMPS-co-SP) were covalently attached to glass (see Section S8 for synthesis details, ESI†) to constrain the gels from swelling in the radial direction. Despite this restriction, there were still noticeable volume changes that occurred upon irradiation due to deswelling in the vertical direction, and both the friction coefficient and elastic modulus increased (Fig. S17, ESI†). These results indicate that bulk volume changes are not necessary to increase friction. However, to fully explore whether volume change is needed to drive photoresponsive friction changes or if the hydrophilicity change of the methoxy-SP-MA is enough, polymer brushes of p(NIPAAm-co-AMPS-co-SP) could be synthesized to eliminate any volume changes due to hydrogel swelling. This interesting topic will be left to future studies.
The rich and vast design space of our photoswitchable hydrogel platform offers opportunities to achieve more controlled friction behavior by tuning parameters such as light intensity, methoxy-SP-MA concentration, AMPS concentration, polymer concentration, and solution pH. This work demonstrates a new route to control friction coefficients of hydrogels using light, which could lead to the design of new photoresponsive materials and coatings for soft robotics and biomedical applications to control underwater locomotion or object manipulation.
4. Conclusion
While the tribological behavior of hydrogels responsive to other stimuli such as temperature,77,78 solvent,79,80 and pH81,82 have been explored, there are fewer studies on light-induced interfacial changes. Our findings demonstrate that light-tunable friction repeatably was achieved with p(NIPAAm-co-AMPS-co-SP-MA)) hydrogels across three light cycles. Hydrogels with low friction (μdark ≈ 0.02) were formed when the gels were in the dark (primarily MCH+) while high friction (μlight > 0.08) was achieved in the light (primarily SP), indicating that the friction coefficient increased at least 4× upon light irradiation. Elastic moduli ( ) and volume changes (Vlight ≈ 0.5Vdark) were also observed upon light irradiation. The underpinning mechanism may be linked to photo-induced reduction in mesh size due to decreased hydrophilicity of methoxy-SP-MA monomers. Photoswitchable gels offer an exciting new arena to investigate the fundamental mechanisms of hydrogel surface physics and their programmable and reversible tribological properties and could have far-reaching impacts from biomedicine to soft robotics.
) and volume changes (Vlight ≈ 0.5Vdark) were also observed upon light irradiation. The underpinning mechanism may be linked to photo-induced reduction in mesh size due to decreased hydrophilicity of methoxy-SP-MA monomers. Photoswitchable gels offer an exciting new arena to investigate the fundamental mechanisms of hydrogel surface physics and their programmable and reversible tribological properties and could have far-reaching impacts from biomedicine to soft robotics.
Data availability
The original data for all the figures in the main text and Supplementary Information are available at Data Dryad, a free and searchable digital repository (https://doi.org/10.5061/dryad.280gb5mzv).Conflicts of interest
There are no conflicts to declare.Acknowledgements
Thanks to Dr Sophia Bailey, Conor Pugsley, and Madeleine Miyamoto for their help with initial experiments, and to Lisa Månsson for helpful discussions. Special thanks to Dr Alexander A. Mikhailovsky, the Optical Characterization Lab Manager at UC Santa Barbara, for assistance in conducting pump–probe spectroscopy experiments. K. M. K. thanks Dr Amanda Strom, TEMPO Facilities Manager at the Materials Research Laboratory (MRL) at UC Santa Barbara, for training on the Shimadzu UV 3600 UV-vis-NIR Spectrometer. This work was supported by the National Science Foundation (NSF) Materials Research Science and Engineering Center (MRSEC) at UC Santa Barbara through DMR-2308708 (IRG-2). The research reported here made use of the shared facilities of the Materials Research Science and Engineering Center (MRSEC) at UC Santa Barbara: NSF DMR–2308708. The UC Santa Barbara MRSEC is a member of the Materials Research Facilities Network (https://www.mrfn.org). A. L. C. acknowledges support from the UC President's Dissertation Year Fellowship. I. M. gratefully acknowledges the MRL Research Internships in Science and Engineering (RISE) program at UC Santa Barbara. A. A. P. acknowledges funding support from the NSF CAREER Award (CMMI-CAREER-2048043).References
- E. Prouvé, B. Drouin, P. Chevallier, M. Rémy, M. C. Durrieu and G. Laroche, Macromol. Biosci., 2021, 21, 1–12 CrossRef PubMed.
- T. Tanaka, D. Fillmore, S.-T. Sun, I. Nishio, G. Swislow and A. Shah, Phys. Rev. Lett., 1980, 45, 1636–1639 CrossRef CAS.
- R. Fei, J. T. George, J. Park, A. K. Means and M. A. Grunlan, Soft Matter, 2013, 9, 2912–2919 RSC.
- W. Liu, R. Xie, J. Zhu, J. Wu, J. Hui, X. Zheng, F. Huo and D. Fan, npj Flexible Electron., 2022, 6, 1–10 CrossRef.
- M. V. Martinez, M. Molina and C. A. Barbero, J. Phys. Chem. B, 2018, 122, 9038–9048 CrossRef CAS PubMed.
- Z. Wang, J. Li, Y. Liu and J. Luo, Mater. Des., 2020, 188, 108441 CrossRef CAS.
- S. Xiao, X. He, Z. Zhao, G. Huang, Z. Yan, Z. He, Z. Zhao, F. Chen and J. Yang, Eur. Polym. J., 2021, 148, 110350 CrossRef CAS.
- C. Li, A. Iscen, L. C. Palmer, G. C. Schatz and S. I. Stupp, J. Am. Chem. Soc., 2020, 142, 8447–8453 CrossRef CAS PubMed.
- A. Aggarwal, C. Li, S. I. Stupp and M. Olvera de la Cruz, Soft Matter, 2022, 18, 2193–2202 RSC.
- W. Francis, A. Dunne, C. Delaney, L. Florea and D. Diamond, Sens. Actuators, B, 2017, 250, 608–616 CrossRef CAS.
- T. Satoh, K. Sumaru, T. Takagi and T. Kanamori, Soft Matter, 2011, 7, 8030–8034 RSC.
- J. C. Roback, M. B. Minnis, K. Ghebreyessus and R. C. Hayward, ACS Appl. Polym. Mater., 2024, 6(11), 6627–6634 CrossRef CAS.
- D. Podstawczyk, M. Nizioł, P. Szymczyk, P. Wiśniewski and A. Guiseppi-Elie, Addit. Manuf., 2020, 34, 101275 CAS.
- Z. Li, Y. Zhou, T. Li, J. Zhang and H. Tian, View, 2022, 3, 1–26 Search PubMed.
- Y. Yu, M. Brió Pérez, C. Cao and S. de Beer, Eur. Polym. J., 2021, 147, 110298 CrossRef CAS.
- F. Andrade, M. M. Roca-Melendres, E. F. Durán-Lara, D. Rafael and S. Schwartz, Cancers, 2021, 13, 1–17 CrossRef PubMed.
- A. Feng, C. Onggowarsito, S. Mao, G. G. Qiao and Q. Fu, ChemSusChem, 2023, 16, e202300137 CrossRef CAS PubMed.
- G. Chen, Phys. Chem. Chem. Phys., 2022, 24, 12329–12345 RSC.
- B. Ying and X. Liu, iScience, 2021, 24, 103174 CrossRef PubMed.
- A. López-Díaz, A. Martín-Pacheco, A. Naranjo, C. Martín, M. A. Herrero, E. Vázquez and A. S. Vázquez, in 3rd IEEE International Conference on Soft Robotics (RoboSoft), 2020, pp. 13–18.
- Y. Lee, W. J. Song and J.-Y. Sun, Mater. Today Phys., 2020, 15, 100258 CrossRef.
- K. Thimann and G. M. Curry, Comparative Biochemistry V1: A Comprehensive Treatise, Academic Press, Inc., 1960, pp. 243–309 Search PubMed.
- P. G. Falkowski and J. A. Raven, Aquatic Photosynthesis, Princeton University Press, 2nd edn, 2007 Search PubMed.
- D. Oesterhelt and W. Stoeckenius, Proc. Natl. Acad. Sci. U. S. A., 1973, 70, 2853–2857 CrossRef CAS PubMed.
- C. P. O’Banion and D. S. Lawrence, ChemBioChem, 2018, 19, 1201–1216 CrossRef PubMed.
- A. Armbruster, A. M. Mohamed, H. T. Phan and W. Weber, Curr. Opin. Biotechnol, 2024, 87, 103126 CrossRef CAS PubMed.
- L. K. Månsson, A. A. Pitenis and M. Z. Wilson, Front. Bioeng. Biotechnol., 2022, 10, 1–13 Search PubMed.
- M. Hörner, J. Becker, R. Bohnert, M. Baños, C. Jerez-Longres, V. Mühlhäuser, D. Härrer, T. W. Wong, M. Meier and W. Weber, Adv. Mater. Technol., 2023, 8, 1–10 Search PubMed.
- E. Hopkins, E. Valois, A. Stull, K. Le, A. A. Pitenis and M. Z. Wilson, ACS Biomater. Sci. Eng., 2021, 7, 408–414 CrossRef CAS PubMed.
- C. Li, M. Kazem-Rostami, J. S. W. Seale, S. Zhou and S. I. Stupp, Chem. Mater., 2023, 35, 3923–3930 CrossRef CAS.
- S. Tamesue, Y. Takashima, H. Yamaguchi, S. Shinkai and A. Harada, Angew. Chem., Int. Ed., 2010, 49, 7461–7464 CrossRef CAS PubMed.
- T. Satoh, K. Sumaru, T. Takagi, K. Takai and T. Kanamori, Phys. Chem. Chem. Phys., 2011, 13, 7322–7329 RSC.
- D. Moldenhauer and F. Gröhn, Chem. – Eur. J., 2017, 23, 3966–3978 CrossRef CAS PubMed.
- S. Fredrich, T. Engels and A. P. H. J. Schenning, ACS Appl. Polym. Mater., 2022, 4, 7751–7758 CrossRef CAS.
- L. B. Braun, T. Hessberger, E. Pütz, C. Müller, F. Giesselmann, C. A. Serra and R. Zentel, J. Mater. Chem. C, 2018, 6, 9093–9101 RSC.
- D. Liu and D. J. Broer, Angew. Chem., Int. Ed., 2014, 53, 4542–4546 CrossRef CAS PubMed.
- A. Aggarwal, C. Li, S. I. Stupp and M. Olvera de la Cruz, Soft Matter, 2022, 18, 2193–2202 RSC.
- C. Li, Y. Xue, M. Han, L. C. Palmer, J. A. Rogers, Y. Huang and S. I. Stupp, Matter, 2021, 4, 1377–1390 CrossRef CAS.
- B. Ziółkowski, L. Florea, J. Theobald, F. Benito-Lopez and D. Diamond, Soft Matter, 2013, 9, 8754–8760 RSC.
- S. Long, J. Huang, J. Xiong, C. Liu, F. Chen, J. Shen, Y. Huang and X. Li, Polymers, 2023, 15, 786 CrossRef CAS PubMed.
- W. Qiu, J. M. P. Scofield, P. A. Gurr and G. G. Qiao, Macromol. Rapid Commun., 2022, 43, 1–35 CrossRef PubMed.
- H. M. D. Bandara and S. C. Burdette, Chem. Soc. Rev., 2012, 41, 1809–1825 RSC.
- E. Inoue, H. Kokado, I. Shimizu, H. Kobayashi and Y. Takahashi, Bull. Chem. Soc. Jpn., 1972, 45, 1951–1956 CrossRef CAS.
- L. Wimberger, S. K. K. Prasad, M. D. Peeks, J. Andreásson, T. W. Schmidt and J. E. Beves, J. Am. Chem. Soc., 2021, 143, 20758–20768 CrossRef CAS PubMed.
- V. K. Johns, Z. Wang, X. Li and Y. Liao, J. Phys. Chem. A, 2013, 117, 13101–13104 CrossRef CAS PubMed.
- L. Wimberger, J. Andréasson and J. E. Beves, Chem. Commun., 2022, 58, 5610–5613 RSC.
- M. S. Zayas, N. D. Dolinski, J. L. Self, A. Abdilla, C. J. Hawker, C. M. Bates and J. Read de Alaniz, ChemPhotoChem, 2019, 3, 467–472 CrossRef CAS.
- L. Zhao, S. Seshadri, X. Liang, S. J. Bailey, M. Haggmark, M. Gordon, M. E. Helgeson, J. Read de Alaniz, P. Luzzatto-Fegiz and Y. Zhu, ACS Cent. Sci., 2022, 8, 235–245 CrossRef CAS PubMed.
- X. Liang, K. M. Karnaukh, L. Zhao, S. Seshadri, A. J. DuBose, S. J. Bailey, Q. Cao, M. Cooper, H. Xu, M. Haggmark, M. E. Helgeson, M. Gordon, P. Luzzatto-Fegiz, J. Read de Alaniz and Y. Zhu, ACS Cent. Sci., 2024, 10, 684–694 CrossRef CAS PubMed.
- P. Tannouri, K. M. Arafeh, J. M. Krahn, S. L. Beaupré, C. Menon and N. R. Branda, Chem. Mater., 2014, 26, 4330–4333 CrossRef CAS.
- T. J. Gately, W. Li, S. H. Mostafavi and C. J. Bardeen, Macromolecules, 2021, 54, 9319–9326 CrossRef CAS.
- J. E. Stumpel, B. Ziółkowski, L. Florea, D. Diamond, D. J. Broer and A. P. H. J. Schenning, ACS Appl. Mater. Interfaces, 2014, 6, 7268–7274 CrossRef CAS PubMed.
- J. Erath, J. Cui, J. Schmid, M. Kappl, A. Del Campo and A. Fery, Langmuir, 2013, 29, 12138–12144 CrossRef CAS PubMed.
- J. Wang, X. Zhang, S. Zhang, J. Kang, Z. Guo, B. Feng, H. Zhao, Z. Luo, J. Yu, W. Song and S. Wang, Matter, 2021, 4, 675–687 CrossRef CAS.
- I. Rehor, C. Maslen, P. G. Moerman, B. G. P. Van Ravensteijn, R. Van Alst, J. Groenewold, H. B. Eral and W. K. Kegel, Soft Robot., 2021, 8, 10–18 CrossRef PubMed.
- Q. L. Zhu, C. Du, Y. Dai, M. Daab, M. Matejdes, J. Breu, W. Hong, Q. Zheng and Z. L. Wu, Nat. Commun., 2020, 11, 1–11 CrossRef PubMed.
- P. Wu, C. Zeng, J. Guo, G. Liu, F. Zhou and W. Liu, Friction, 2024, 12, 39–51 CrossRef CAS.
- C. Berton, D. M. Busiello, S. Zamuner, R. Scopelliti, F. Fadaei-Tirani, K. Severin and C. Pezzato, Angew. Chem., Int. Ed., 2021, 60, 21737–21740 CrossRef CAS PubMed.
- T. Pirman, M. Ocepek and B. Likozar, Ind. Eng. Chem. Res., 2021, 60, 9347–9367 CrossRef CAS.
- E. Takács and L. Wojnárovits, Radiat. Phys. Chem., 1995, 46, 1007–1010 CrossRef.
- J. R. Hemmer, S. O. Poelma, N. Treat, Z. A. Page, N. D. Dolinski, Y. J. Diaz, W. Tomlinson, K. D. Clark, J. P. Hooper, C. Hawker and J. Read de Alaniz, J. Am. Chem. Soc., 2016, 138, 13960–13966 CrossRef CAS PubMed.
- J. Schindelin, I. Arganda-Carreras, E. Frise, V. Kaynig, M. Longair, T. Pietzsch, S. Preibisch, C. Rueden, S. Saalfeld, B. Schmid, J. Y. Tinevez, D. J. White, V. Hartenstein, K. Eliceiri, P. Tomancak and A. Cardona, Nat. Methods, 2012, 9, 676–682 CrossRef CAS PubMed.
- A. L. Chau, C. E. R. Edwards, M. E. Helgeson and A. A. Pitenis, ACS Appl. Mater. Interfaces, 2023, 15, 43075–43086 CrossRef CAS PubMed.
- A. L. Chau, C. D. Pugsley, M. E. Miyamoto, Y. Tang, C. D. Eisenbach, T. E. Mates, C. J. Hawker, M. T. Valentine and A. A. Pitenis, Tribol. Lett., 2023, 71, 1–12 CrossRef.
- H. Komber, S. Müllers, F. Lombeck, A. Held, M. Walter and M. Sommer, Polym. Chem., 2014, 5, 443–453 RSC.
- V. K. Seiler, K. Callebaut, K. Robeyns, N. Tumanov, J. Wouters, B. Champagne and T. Leyssens, CrystEngComm, 2018, 20, 3318–3327 RSC.
- C. Berton, D. M. Busiello, S. Zamuner, E. Solari, R. Scopelliti, F. Fadaei-Tirani, K. Severin and C. Pezzato, Chem. Sci., 2020, 11, 8457–8468 RSC.
- G. Liman, E. Mutluturk and G. Demirel, ACS Mater. Au, 2024, 4(4), 385–392 CrossRef CAS PubMed.
- L. R. G. Treloar, The Physics of Rubber Elasticity, Oxford University Press, 3rd edn, 1975 Search PubMed.
- P. G. de Gennes, Scaling Concepts in Polymer Physics, Cornell University Press, Ithaca, 1979 Search PubMed.
- P. J. Flory, Principles of Polymer Chemistry, Cornell University Press, Ithaca, 1953 Search PubMed.
- Y. Tsuji, X. Li and M. Shibayama, Gels, 2018, 4, 1–12 CrossRef PubMed.
- J. M. Urueña, A. A. Pitenis, R. M. Nixon, K. D. Schulze, T. E. Angelini and W. G. Sawyer, Biotribology, 2015, 1–2, 24–29 CrossRef.
- A. A. Pitenis and W. G. Sawyer, Tribol. Lett., 2018, 66, 1–7 CrossRef CAS.
- T. Canal and N. A. Peppas, J. Biomed. Mater. Res., 1989, 23, 1183–1193 CrossRef CAS PubMed.
- H. Choudhary and S. R. Raghavan, ACS Appl. Mater. Interfaces, 2022, 14, 13733–13742 CrossRef CAS PubMed.
- D. P. Chang, J. E. Dolbow and S. Zauscher, Langmuir, 2007, 23, 250–257 CrossRef CAS PubMed.
- A. A. Pitenis, K. D. Schulze, R. M. Nixon, A. C. Dunn, B. A. Krick, W. G. Sawyer and T. E. Angelini, Soft Matter, 2014, 10, 8955–8962 RSC.
- Y. Yu, M. Cirelli, B. D. Kieviet, E. S. Kooij, G. J. Vancso and S. de Beer, Polymer, 2016, 102, 372–378 CrossRef CAS.
- E. O. McGhee, A. L. Chau, M. C. Cavanaugh, J. G. Rosa, C. L. G. Davidson, J. Kim, J. M. Urueña, B. S. Sumerlin, A. A. Pitenis and W. G. Sawyer, Biotribology, 2021, 26, 100170 CrossRef.
- S. Ma, M. Scaraggi, P. Lin, B. Yu, D. Wang, D. Dini and F. Zhou, J. Phys. Chem. C, 2017, 121, 8452–8463 CrossRef CAS.
- A. L. Chau, P. T. Getty, A. R. Rhode, C. M. Bates, C. J. Hawker and A. A. Pitenis, Front. Chem., 2022, 10, 1–12 Search PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4sm00677a |
| ‡ Equal contribution. |
| This journal is © The Royal Society of Chemistry 2024 |