
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceImpact of a poly(ethylene glycol) corona block on drug encapsulation during polymerization induced self-assembly†
Guanrui
Li
 ,
Cassie
Duclos
and
Ralm G.
Ricarte
*
,
Cassie
Duclos
and
Ralm G.
Ricarte
*
Department of Chemical and Biomedical Engineering, FAMU-FSU College of Engineering, Tallahassee, FL 32310, USA. E-mail: rricarte@eng.famu.fsu.edu
First published on 3rd September 2024
Abstract
Polymerization induced self-assembly (PISA) provides a facile platform for encapsulating therapeutics within block copolymer nanoparticles. Performing PISA in the presence of a hydrophobic drug alters both the nanoparticle shape and encapsulation efficiency. While previous studies primarily examined the interactions between the drug and hydrophobic core block, this work explores the impact of the hydrophilic corona block on encapsulation. Poly(ethylene glycol) (PEG) and poly(2-hydroxypropyl methacrylate) (PHPMA) are used as the model corona and core blocks, respectively, and phenylacetic acid (PA) is employed as the model drug. Attachment of a dithiobenzoate end group to the PEG homopolymer – transforming it into a macroscopic reversible addition–fragmentation chain transfer agent – causes the polymer to form a small number of nanoscopic aggregates in solution. Adding PA to the PEG solution encourages further aggregation and macroscopic phase separation. During the PISA of PEG-PHPMA block copolymers, inclusion of PA in the reaction mixture promotes faster nucleation of spherical micelles. Although increasing the targeted PA loading from 0 to 20 mg mL−1 does not affect the micelle size or shape, it alters the drug spatial distribution within the PISA microenvironment. PA partitions into either PEG-PHPMA micelles, deuterium oxide, or other polymeric species – including PEG aggregates and unimer chains. Increasing the targeted PA loading changes the fraction of drug within each encapsulation site. This work indicates that the corona block plays a critical role in dictating drug encapsulation during PISA.
Introduction
Polymerization induced self-assembly (PISA) offers a facile approach for encapsulating biomedical cargo within block copolymer nanoparticles. For aqueous PISA, a water soluble polymer A is chain-extended by a second block B that is initially water soluble but grows insoluble as the degree of polymerization increases. Eventually, the growing chains nucleate into spherical micelles in which the A and B blocks constitute the hydrophilic corona and hydrophobic core, respectively. PISA yields highly concentrated block copolymer nanoparticle solutions. Beyond spherical micelles, altering the relative ratio of the corona and core block lengths generates cylindrical micelles and vesicles.1–18 Because of the ease and versatility of PISA, it serves as a potentially powerful platform for sequestering therapeutics – ranging from hydrophobic small molecule drugs to large proteins.19–26 In this work, we study the impact of the corona block on drug encapsulation during PISA.The presence of a hydrophobic drug within a PISA reaction mixture impacts the final nanostructure and encapsulation efficiency. As a seminal example, Cao et al. used PISA to physically entrap curcumin within methacrylic block copolymer nanoparticles. As the curcumin loading was increased from 0 to 0.5 mg mL−1, the nanoparticle morphology shifted from spherical micelles → cylindrical micelles → vesicles.27 Li et al. used poly(glycerol monomethacrylate)-block-poly(2-hydroxypropyl methacrylate) (PGMA-PHPMA) to sequester phenylacetic acid (PA) with drug loadings ranging from 0 to 40 mg mL−1. Consistent with Cao et al., the PGMA-PHPMA morphology transitioned from spherical micelles → cylindrical micelles → vesicles as the drug loading increased. The authors also used 1H nuclear magnetic resonance diffusion ordered spectroscopy (1H NMR DOSY) to evaluate the encapsulation efficiency of the drug. The drug loading had a significant effect, with the encapsulation efficiency reaching a maximum at a targeted loading of 32 mg mL−1. The core block molar mass, however, had virtually no impact. Based on this surprising result, the authors hypothesized that some of the PA partitioned into the corona of the PGMA-PHPMA nanoparticles, rather than the hydrophobic core.28
Although the concept of hydrophobic drugs binding to nominally hydrophilic polymers may seem counterintuitive, historical precedent supports this phenomenon. Johnson et al. found that poly(N-isopropylacrylamide) (PNIPAM) synthesized by reversible addition–fragmentation chain-transfer (RAFT) polymerization enhanced and stabilized the solubility of the hydrophobic drug phenytoin. This improvement stemmed from the RAFT agent end groups of the polymer chains, which induced the formation of PNIPAM aggregates. The drug readily partitioned into the hydrophilic PNIPAM region of these aggregates.29 Extending that idea, Li et al. sequestered hydrophobic drugs in the corona of PNIPAM-block-polystyrene spherical micelles.30 Haider et al. found that curcumin and paclitaxel interacted with the corona of block copolymer spherical micelles featuring poly(2-oxazoline) as the hydrophilic corona block. Minor alterations of the alkyl moiety on the hydrophilic block altered the affinity of the drug to the hydrophilic corona. High drug loadings dehydrated the corona region, destabilizing the micelles and causing colloidal aggregation.31
Building upon our previous work,28 we investigate the role of the hydrophilic corona block on drug encapsulation during PISA. Poly(ethylene glycol) (PEG) – ubiquitous in pharmaceutical formulations – serves as the model corona block, and PHPMA acts as the model core block. PA, the model drug, is sequestered using a PISA physical entrapment approach (see Scheme 1). PA drug loadings are varied between 0 to 20 mg mL−1 so that spherical micelles are maintained, thereby removing the impact of nanoparticle shape on encapsulation. Solutions of homopolymer PEG (prepared by direct dissolution) and block copolymer PEG-PHPMA (prepared by PISA) are investigated using a combination of dynamic light scattering, transmission electron microscopy, size-exclusion chromatography, and 1H NMR DOSY. A macroscopic RAFT (macro-RAFT) agent of PEG is prepared by attaching a 4-cyano-4-(phenylcarbonothioylthio)pentanoic acid (CPPA) end group to the polymer. This hydrophobic moiety induces PEG to form a small number of aggregates in deuterium oxide. The addition of PA drives PEG to further aggregate and, eventually, macrophase separate. For PEG-PHPMA PISA, the incorporation of drug promotes micelle nucleation. While the targeted PA loading does not affect the size and shape of the PEG-PHPMA micelles, it significantly alters the partitioning of drug throughout the PISA solution microenvironment. These findings establish the crucial role of the hydrophilic corona block in controlling encapsulation during PISA.
 | ||
| Scheme 1 (A) Synthesis of PEG macro-RAFT agent. (B) Simultaneous PEG-PHPMA synthesis and PA encapsulation via PISA. | ||
Materials and methods
Materials
2-Hydroxypropyl methacrylate (HPMA) was purchased from Thermo Fisher Scientific and purified by column chromatography (basic aluminum oxide, Brockmann I, 2 × 10 cm column dimensions) to remove inhibitor. Monomethoxy poly(ethylene glycol) (PEG, Mn = 6.14 kg mol−1, Đ = 1.04) was purchased from Fisher Scientific and used without any further purification. 4-Cyano-4-(phenylcarbonothioylthio)pentanoic acid (CPPA) was purchased from Boron Molecular. 2,2′-Azobis[2-(2-imidazolin-2-yl)propane]dihydrochloride (AIPD) was purchased from TCI America. 1-Ethyl-3-(3-dimethylaminopropyl)carbodiimide (EDC), 4-dimethylaminopyridine (DMAP), 3-(trimethylsilyl)propionic-2,2,3,3-d4 acid sodium salt, phenylacetic acid (PA), dichloromethane, dimethylformamide (DMF), and methanol were purchased from Sigma-Aldrich. Dialysis bags (molar mass cut off = 3.5 kg mol−1) were purchased from Spectrum Labs. Diethyl ether was purchased from Oakwood Chemicals. Deuterium oxide (D2O) was purchased from Cambridge Isotope Lasboratories.Synthesis of poly(ethylene glycol) (PEG) macro-RAFT agent
PEG macro-RAFT agents were synthesized using a standard esterification protocol.32 To prepare the macro-RAFT agent, PEG (10 g, 2 mmol) and CPPA (0.838 g, 3 mmol) were dissolved in 56 mL of dichloromethane to form a solution with a total solids concentration of 200 mg mL−1. The solution was transferred to a 250 mL round-bottom flask and degassed through three freeze–pump–thaw cycles, followed by backfilling with argon gas. After immersing the round-bottom flask in an ice bath, EDC (0.383 g, 2 mmol) and DMAP (0.029 g, 0.24 mmol) pre-dissolved in dichloromethane were added drop-wise to the reaction mixture. The round-bottom flask then was removed from the ice bath, and the esterification reaction proceeded at room temperature for 48 hours. The resulting reaction mixture was extracted with 2 × 750 mL of deionized water, concentrated under reduced pressure, and precipitated in 500 mL of cold diethyl ether. The polymer solid was re-dissolved in methanol, transferred to a dialysis membrane (molar mass cutoff = 3.5 kg mol−1), and dialyzed against three 700 mL volumes of methanol over a 24 h period. After dialysis, the solution was concentrated under reduced pressure, precipitated in 500 mL of cold diethyl ether, and dried for 18 h at room temperature under reduced pressure. The molar mass and composition of the PEG macro-RAFT agent was characterized using UV size-exclusion chromatography and 1H nuclear magnetic resonance spectroscopy (1H NMR), respectively. See Section S2 of the ESI† for PEG macro-RAFT agent 1H NMR spectra and peak assignments.Synthesis of poly(ethylene glycol)-block-poly(2-hydroxypropyl methacrylate) (PEG-PHPMA) spherical micelles via polymerization induced self-assembly (PISA)
PEG-PHPMA spherical micelles were prepared using PISA. To achieve spherical micelle morphologies, a PHPMA degree of polymerization (NPHPMA) of 45 was targeted. PEG macro-RAFT agent (0.2000 g, 0.0326 mmol), HPMA (0.2106 g, 1.4635 mmol) and AIPD (0.00035 g, 0.0109 mmol, [RAFT]/[AIPD] = 3![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1) were dissolved in D2O (3.727 g, 3.5 mL, 90 w/v%), and transferred to a 10 mL Schlenk flask. The solution was sparged with argon for 30 minutes and immersed into a silicon oil bath at 50 °C for 3 hours to reach ∼100% HPMA monomer conversion. The reaction was quenched by exposure to air. PEG-PHPMA samples were stored at ambient temperature and pressure. All PEG-PHPMA samples were characterized within 1 week of synthesis. See Section S2 of the ESI† for PEG-PHPMA 1H NMR spectra and peak assignments.
:1) were dissolved in D2O (3.727 g, 3.5 mL, 90 w/v%), and transferred to a 10 mL Schlenk flask. The solution was sparged with argon for 30 minutes and immersed into a silicon oil bath at 50 °C for 3 hours to reach ∼100% HPMA monomer conversion. The reaction was quenched by exposure to air. PEG-PHPMA samples were stored at ambient temperature and pressure. All PEG-PHPMA samples were characterized within 1 week of synthesis. See Section S2 of the ESI† for PEG-PHPMA 1H NMR spectra and peak assignments.
Encapsulation of phenylacetic acid (PA) in PEG-PHPMA spherical micelles during PISA
Physical entrapment of PA in PEG-PHPMA spherical micelles was achieved via PISA. PA loadings of 10, 16, and 20 mg mL−1 and NPHPMA = 45 were targeted to maintain a spherical micelle morphology. To sequester 16 mg mL−1 of PA in PEG-PHPMA, PA (0.0552 g, 0.4053 mmol) and HPMA monomer (0.2106 g, 1.4635 mmol) were mixed in a 10 mL Schlenk flask. A solution of PEG macro-RAFT agent (0.2000 g, 0.0326 mmol) and AIPD (0.00035 g, 0.0108 mmol, [RAFT]/[AIPD] = 3:1) dissolved in D2O (3.727 g, 3.5 mL, 90 w/v%) were added to the HPMA and PA mixture. The solution was sparged with argon for 30 minutes and immersed into a silicon oil bath at 50 °C for 3 hours to reach ∼100% HPMA conversion. The reaction was quenched by exposure to air. PEG-PHPMA and PA PISA samples were stored at ambient temperature and pressure. All PEG-PHPMA and PA PISA samples were characterized within 1 week of synthesis. See Section S2 of the ESI† for PEG-PHPMA and PA PISA sample 1H NMR spectra and peak assignments.
PEG-PHPMA and PA PISA kinetics and molar mass evolution
HPMA monomer conversion and PEG-PHPMA molar mass evolution during PISA were evaluated as a function of the targeted PA loading. To study the PISA kinetics of PEG-PHPMA spherical micelles with a targeted PA loading of 16 mg mL−1, PA (0.1104 g, 0.8106 mmol) and HPMA monomer (0.4213 g, 2.927 mmol) were mixed in a 50 mL Schlenk flask. A solution of PEG macro-RAFT agent (0.4000 g, 0.0650 mmol) and AIPD (0.0070 g, 0.0217 mmol, [RAFT]/[AIPD] = 3:1) dissolved in D2O (7.4547 g, 6.9 mL, 90 w/v%) were added to the HPMA and PA mixture. After the solution was sparged with argon for 30 min, the side arm of the Schlenk flask was opened to introduce a positive pressure of argon, the top glass stopper was removed, and 0.2 mL aliquot of the reaction mixture was collected. The flask was immediately resealed and submerged in a silicone oil bath at 50 °C. Over a period of 2 hours, aliquots of the PISA mixture were collected every 20 minutes. HPMA monomer conversion was quantified by 1H NMR using 6 mg mL−1 of 3-(trimethylsilyl)propionic-2,2,3,3-d4 acid sodium salt as an internal standard, while the molar mass was determined using UV size-exclusion chromatography.
Size-exclusion chromatography (SEC) of PEG-PHPMA
SEC of PEG-PHPMA was performed using a Malvern Viscotek VE 2001 GPCmax equipped with two successive Viscotek T6000M columns (40 °C dimethylformamide and 0.05 M LiBr mobile phase). The instrument also featured a pump, degasser, autosampler, and thermostatted column chamber. Molar masses were estimated using a Viscotek UV Detector 2600. UV SEC traces were collected using a detector wavelength of 268 nm. Molar masses are based on poly(ethylene oxide) standards. For SEC traces that exhibited a bimodal distribution, the peaks were deconvoluted using the procedure described in Section S8 of the ESI.†Transmission electron microscopy
PEG-PHPMA and PA PISA solutions were diluted 100-fold by deionized water. 13 μL of solution were deposited onto a square 200 mesh copper grid with a 3–4 nm thick layer of amorphous carbon film; the grid was pre-treated with 25 s of plasma glow discharge to enhance surface energy. After 1 min, the grid was blotted with filter paper to remove excess sample solution. To improve image contrast through negative staining, 9 μL of 0.75 w/v% uranyl formate solution was added to the sample-loaded grid. After 20 s, the grid was blotted with filter paper to remove excess staining solution. TEM grids were dried at ambient temperature and pressure overnight. Bright-field TEM imaging was conducted using a Hitachi HT7800 TEM at an accelerating voltage of 120 kV. Spherical micelle radius R was determined using the ImageJ open-source software.Dynamic light scattering (DLS)
PEG and PA solutions – prepared by direct dissolution – were stirred for at least 16 h, and then passed through 0.22 μm PTFE hydrophilic filters into clean glass culture tubes. PEG-PHPMA and PA solutions – prepared by PISA – were diluted 50-fold by deionized water before passing through 0.22 μm PTFE hydrophilic filters into clean glass culture tubes. Dynamic light scattering was performed using an ALV/LSE-5004 light scattering instrument. g2 autocorrelation functions were collected at angles between 30° to 135° in increments of 15°. The scattering vector (q) is defined as | (1) |
| g2(q,τ) − 1 = β|g1(q,τ)|2 + BG | (2) |
| |g1(q,τ)|2 = (fDLSexp(−Γ1τ) + (1 − fDLS)exp(−Γ2τ))2 | (3) |
 | (4) |
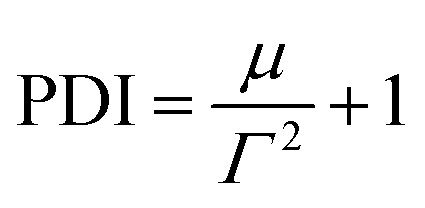 | (5) |
 | (6) |
1H nuclear magnetic resonance diffusion ordered spectroscopy (1H NMR DOSY)
Sample solutions in sealed 5 mm diameter NMR tubes were characterized using a Bruker AVIII B600 nuclear magnetic resonance spectrometer with a 600 MHz magnet and a 5 mm BBO probe with a Z-gradient. Measurements were performed at a temperature of 25.00 ± 0.01 °C. DOSY was conducted using the ledbpgp2s pulse sequence (longitudinal eddy current delay experiment acquired in 2 dimensions).Diffusion coefficients for PEG, PEG-PHPMA, and PA were obtained by collecting a series of one-dimensional spectra, each an average of 8 scans. The pulse gradient strength (g) was increased from 2 to 95% of the maximum strength using a 90° pulse angle, 3 s relaxation delay, and 10 ppm chemical shift. For all collected spectra, values of the gradient pulse Δ = 0.2 s, pulse gradient length δ = 0.005 s, and gyromagnetic ratio γ = 4258 G cm−1 were used.
For cases in which the DOSY echo decay signal exhibited a single exponential decay, the data were analyzed using the Stejskal–Tanner model
 | (7) |
 | (8) |
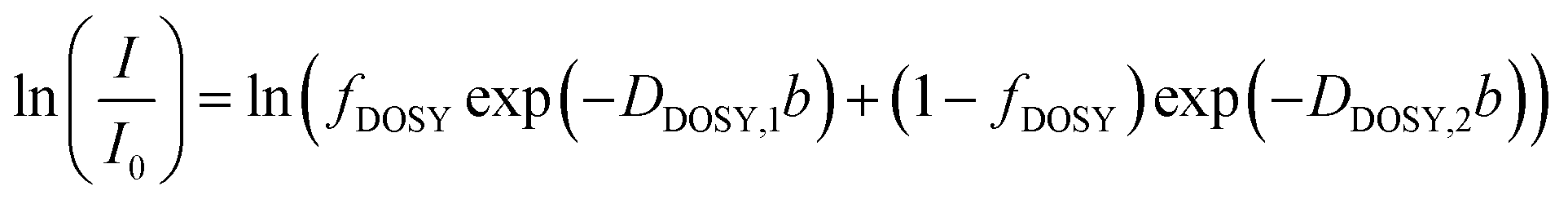 | (9) |
Results
Effect of phenylacetic acid on poly(ethylene glycol) (PEG) self-assembly
To understand how nominally hydrophilic polymers interact with hydrophobic drugs, the solution behavior of the PEG macro-RAFT agent in the presence of phenylacetic acid (PA) was evaluated. Solutions of PEG and PA in D2O were prepared via direct dissolution. In this section, samples are labeled as PEG CPA, where CPA is the targeted PA loading in units of mg mL−1.Fig. 1 depicts solutions containing PEG with varying CPA values. While the hydroxy-terminated PEG (PEG-OH) solution is clear, the PEG macro-RAFT agent solutions are pink due to the 4-cyano-4-(phenylcarbonothioylthio)pentanoic acid (CPPA) end group. The addition of phenylacetic acid (PA) alters the turbidity. PEG 10 is transparent, but samples with CPA equal to or greater than 16 mg mL−1 (the solubility of PA in D2O)28 are opaque – indicating macrophase separation.
 | ||
| Fig. 1 (A) Photograph and (B) DLS Rh distributions at a 90° scattering angle of PEG and PA solutions. | ||
Fig. 1B plots the hydrodynamic radius (Rh) distribution of filtered PEG solutions, as measured at a scattering angle of 90° by dynamic light scattering (DLS). PEG-OH has a monomodal Rh distribution. Fitting eqn (2) to the g2 autocorrelation functions produces a z-average Rh of 2 ± 1 nm (Fig. S2, ESI†). PEG 0, in contrast, has a bimodal Rh distribution (see Fig. S23, ESI† for a magnified version of the PEG 0 Rh distribution). Fitting eqn (3) to the g2 autocorrelation functions yields two distinct Rh values. The smaller Rh = 5 ± 1 nm corresponds to individual PEG unimer chains fully dissolved in D2O. The larger Rh = 70 ± 1 nm corresponds to the presence of larger PEG aggregates. The addition of PA alters the aggregate size. For PEG 10, the larger Rh is 54 ± 2 nm. For PEG 16 and PEG 20, the larger Rh grows to 88 ± 2 nm and 70 ± 1 nm, respectively. Although the larger Rh peaks dominate the distributions depicted in Fig. 1B, the number of aggregates in the PEG macro-RAFT agent solutions is likely exaggerated by the DLS measurement. In general, DLS is biased towards detecting larger structures because the scattering intensity I ∼ R6h. Consequently, larger structures will be overrepresented in the Rh distribution.34 At the same time, however, structures larger than 200 nm are removed from the analysis as DLS sample preparation requires filtration of the PEG solutions.
To obtain a more accurate representation of the relative species populations in PEG solutions, 1H nuclear magnetic resonance diffusion ordered spectroscopy (1H NMR DOSY) experiments were performed. Because PEG and PA exhibit unique peaks in 1H NMR spectra (see Section S2 of ESI†), their diffusion behavior may be deconvoluted by DOSY measurements.
Fig. 2A features echo decays of unfiltered PEG solutions. Tables S1–S3 (ESI†) contain estimated diffusion coefficients, while Tables S10 and S11 (ESI†) detail the estimated hydrodynamic radii and diffusing species. The diffusion behavior of the polymer was evaluated by tracking the echo decay of the methylene signal (Peak B in Fig. S7, ESI†). For PEG 0, the polymer echo decay may be described by a double exponential expression (eqn (9)), indicating two diffusive modes. Based on fitting eqn (9) to the data, the faster diffusion mode corresponds to DDOSY,1 = (6.5 ± 0.1) × 10−11 m2 s−1 and Rh = 3.0 ± 0.1 nm, consistent with the unimer peak identified in DLS. Conversely, the slower diffusion mode has DDOSY,2 ⪅ 1 × 10−13 m2 s−1. This small diffusion coefficient suggests the presence of aggregates in solution, though the lack of a constant slope at high b inhibits precise calculation of DDOSY,2. Nevertheless, the fractional intensity of the fast diffusion mode fDOSY = 0.99 ± 0.01, showing that aggregates constitute a negligible portion of the species population.
 | ||
| Fig. 2 1H NMR DOSY echo decays for (A) PA 16 mg mL−1, PEG 0, and PEG 10, and (B) PEG 16 solutions. (C) 1H NMR spectrum of PEG 16. | ||
The addition of PA to the PEG solution impacts the polymer phase behavior. For PEG 10, the fast diffusion mode DDOSY and Rh are comparable to PEG 0. fDOSY, however, decreases to 0.84 ± 0.01, signaling that the number of aggregates increased. For the slower diffusion mode, DDOSY,2 = (3.1 ± 0.1) × 10−11 m2 s−1 and Rh = 7.0 ± 0.2 nm.
Fig. 2A also illustrates the PEG 10 echo decay of the drug, determined from Peak G. The spectral noise baseline was subtracted from the echo decay signal. For PEG 10, the drug exhibits a single exponential decay that may be described by eqn (7). The corresponding diffusion coefficient DDOSY = (4.9 ± 0.1) × 10−10 m2 s−1 is smaller than the diffusion coefficient of PA in D2O without polymer (DPA,D2O = (6.7 ± 0.1) × 10−10 m2 s−1). This discrepancy is not due to changes in the bulk viscosity, as the residual H2O diffusion coefficient is unaffected by PEG and PA (Table S7, ESI†). Rather, the observed DDOSY represents the weighted average of the diffusion of PA freely dissolved in D2O and drug bound to PEG aggregates. The appearance of a single decay mode indicates that the time scale of PA exchange between the two phases (τex) is faster than the diffusion time of the measurement, i.e., τex ≪ Δ.28,35 The fraction of PA bound to the PEG aggregates (pagg) may be estimated by
 | (10) |
Fig. 2C and Fig. S10 (ESI†) depict the 1H NMR spectra of PEG 16 and PEG 20, respectively. In addition to the PEG methylene peaks also seen in other samples (Peak B), a second set of methylene peaks appears slightly upfield (Peak B′). Because these samples are macrophase separated, Peak B and B′ represent PEG chains within D2O rich and D2O poor regions, respectively. The variance in chemical shifts between the two peaks results from the different magnetic susceptibilities of the macrophases.36 Comparison of the peak area integrations suggests that 12 and 17 mol% of the total PEG chains reside in the D2O poor regions of PEG 16 and PEG 20. Filtering the solutions removes the B′ peak completely (Fig. S9 and S11, ESI†), further supporting the idea that this signal corresponds to PEG chains within the D2O poor macrophase.
Fig. 2B and Fig. S18 (ESI†) detail the 1H NMR DOSY echo decays for PEG 16 and PEG 20, respectively. While the echo decays of polymer in the D2O rich macrophase are similar to the polymer decays observed for lower drug loadings, the echo decays of polymer in the D2O poor macrophase are markedly different. Fitting eqn (9) to the Peak B′ echo decays produces two apparent diffusion coefficients. For both PEG 16 and PEG 20, the fast diffusion mode renders DDOSY,1 ≈ 6 × 10−11 m2 s−1. The slow diffusion mode generates DDOSY,2 ≈ 1.4 × 10−12 m2 s−1.
For PEG 16 and PEG 20, macrophase separation creates two sets of PA peaks corresponding to drug in D2O rich and D2O poor regions (Peak G and G′). Based on the peak areas, 22 and 31 mol% of the total PA inhabit the D2O poor regions of PEG 16 and PEG 20. The single exponential Peak G echo decays, comparable to that of PEG 10, produce pagg ≈ 0.30. The Peak G′ echo decays, however, decay much more slowly, and their double exponential shapes produce DDOSY,1 ≈ 2.4 × 10−10 and DDOSY,2 ≈ 1.4 × 10−11 m2 s−1.
To summarize, DLS and 1H NMR DOSY were used to evaluate the solution behavior of PEG and PA solutions. PEG macro-RAFT agent forms a small number of aggregates in D2O. Addition of PA induces more PEG aggregation and, at high concentrations, macrophase separation. For PEG 10, the PA echo decays suggests that some drug binds to PEG aggregates. For PEG 16 and PEG 20, drug within D2O rich regions also binds to PEG aggregates. Drug within D2O poor regions, however, exhibits more complex diffusion behavior.
Impact of PA on PEG-PHPMA PISA kinetics, molar mass distribution, and spherical micelle nanostructure
PEG-PHPMA spherical micelles loaded with PA were prepared using PISA. All measurements were conducted at room temperature, below the glass transition temperature of water-swollen PHPMA (47 °C). For this reason, all observed PEG-PHPMA morphologies are assumed to be kinetically trapped.37 Samples are labeled as PEG-PHPMA CPA, where CPA is the targeted PA loading in units of mg mL−1.Fig. 3 depicts the PEG-PHPMA PISA solutions before and after polymerization. Prior to PISA, the solutions are similar in appearance to their PEG-only counterparts; i.e., PEG-PHPMA 0 and 10 are transparent, while PEG-PHPMA 16 and 20 are turbid. After PISA, however, all solutions have similar transparent appearances.
 | ||
| Fig. 3 PEG-PHPMA solutions (A) before and (B) after PISA. | ||
Fig. 4A and Table S8 (ESI†) detail the molar mass characterization of PEG-PHPMA 0, which does not contain any drug. In the UV SEC trace, the peak centered around a retention time of 19.8 min represents the PEG-PHPMA chains produced during the PISA reaction. The smaller peak centered around 20.5 min is due to unreacted PEG macro-RAFT agent. The appearance of this peak in the UV SEC trace indicates the chains possess the CPPA end group, rather than the hydrogen end group expected for chains that have undergone termination by disproportionation. Additional experiments also suggest that the small peak does not represent terminated chains (see Section S12 of ESI†). Deconvolution of the PEG-PHPMA 0 trace estimates that approximately 10 mol% of the PEG macro-RAFT chains are unreacted (Fig. S20 and Table S8, ESI†).
 | ||
| Fig. 4 (A) UV SEC traces of PEG and PEG-PHPMA samples with varying targeted PA loading. The UV detector was set to a wavelength of 268 nm. (B) HPMA monomer conversion vs. time and (C) first order kinetics plot of PEG-PHPMA PISA reaction. (D) Spherical micelle nucleation time (tnuc) vs. targeted PA loading. | ||
Fig. 4B and C characterize the PISA kinetics for PEG-PHPMA 16. For this sample, the polymerization reaches quantitative conversion after 120 minutes. An inflection point occurs near 60 min due to an acceleration in the polymerization rate. Commonly observed during PISA,38,39 this acceleration corresponds to the time at which PEG-PHPMA nucleates into spherical micelles (tnuc). Fig. S21 (ESI†) depicts PISA kinetics for the other PEG-PHPMA samples.
Fig. 4D plots PEG-PHPMA tnuc as a function of CPA. tnuc decreases as CPA increases, demonstrating that the presence of the drug prompts faster PEG-PHPMA micelle nucleation. As seen in Fig. 3A and Table S8 (ESI†), PEG-PHPMA 20 retains a higher fraction of unreacted PEG macro-RAFT agent during PISA.
Fig. 5 features the nanostructure characterization of the PEG-PHPMA micelles. Dry-state TEM reveals that all samples have similar morphologies and particle radii Rh ≈ 15 nm (see Fig. S25 (ESI†) for histograms). For diluted samples, DLS demonstrates that the PEG-PHPMA micelles have Rh ≈ 12 nm.
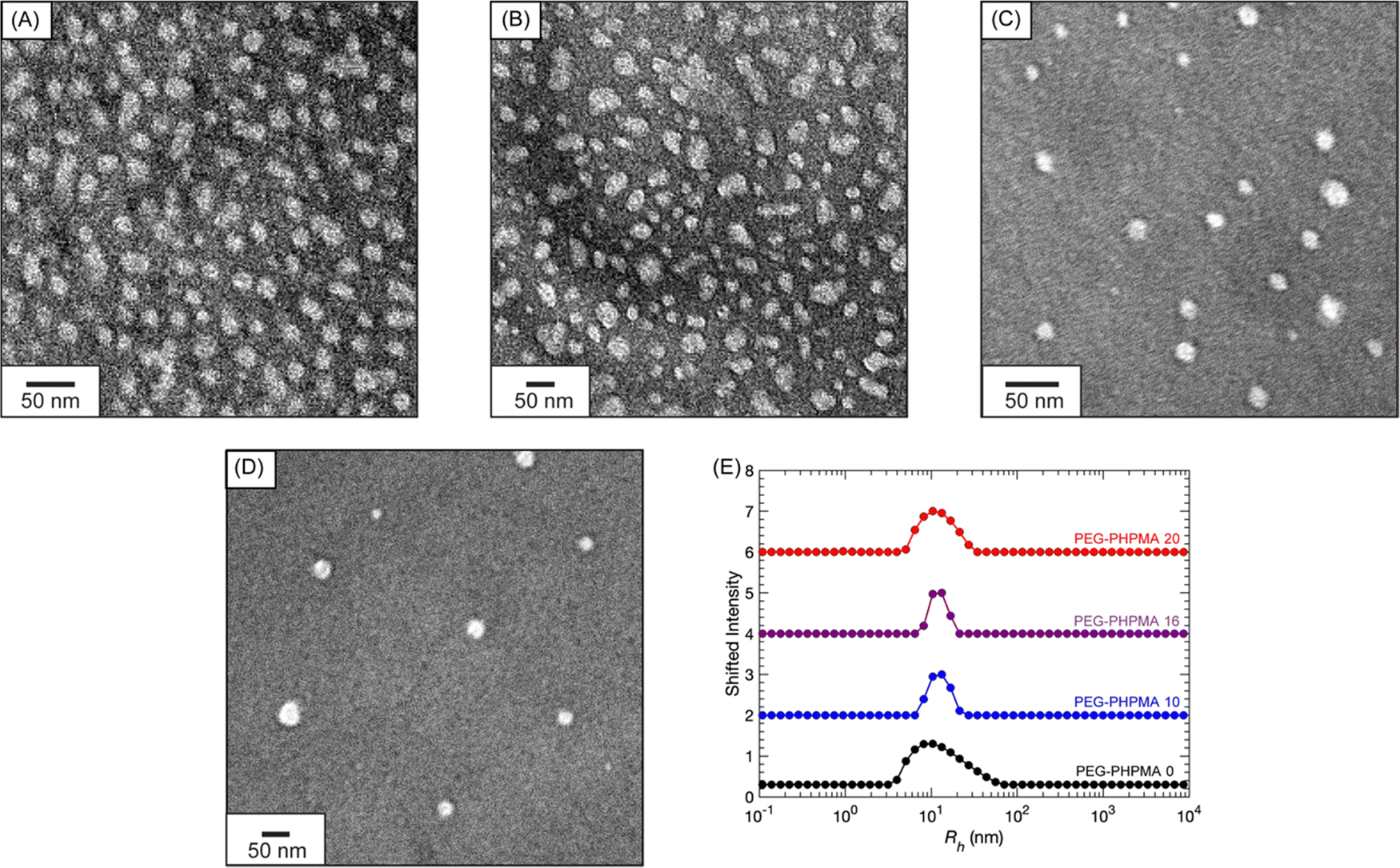 | ||
| Fig. 5 Dry-state TEM images of (A) PEG-PHPMA 0, (B) PEG-PHPMA 10, (C) PEG-PHPMA 16, and (D) PEG-PHPMA 20. (E) DLS Rh distributions at a 90° scattering angle of PEG-PHPMA and PA solutions. | ||
To summarize, prior to polymerization the addition of PA alters the turbidity of the PEG-PHPMA PISA solutions. Afterwards, the solutions contain a mixture of PEG-PHPMA block copolymers and unreacted PEG chains. The addition of PA induces earlier nucleation of PEG-PHPMA micelles. The size and shape of the micelles, however, remain essentially constant between CPA = 0 to 20 mg mL−1.
1H NMR DOSY of PEG-PHPMA micelle and PA PISA solutions
Fig. 6A depicts 1H NMR DOSY echo decays of PEG-PHPMA (Peak B). Tables S4–S6 (ESI†) contain estimated diffusion coefficients, while Table S12 (ESI†) details the estimated hydrodynamic radii and diffusing species. Due to the double exponential shape, eqn (9) was fit to each echo decay (see Table S4 (ESI†) for fit parameters). For the fast diffusion mode, DDOSY,1 ranges from 2.5 × 10−11 to 3.7 × 10−11 m2 s−1, which agrees with the spherical micelle diffusion coefficient determined by DLS. fDOSY hovers around 0.60. However, the addition of PEG-OH to the PEG-PHPMA 0 solution increases fDOSY to 0.90 (see Fig. S23 and Table S9, ESI†). This effect suggests that the fast diffusion mode observed by 1H NMR DOSY describes not only PEG-PHPMA micelles, but also unimers (PEG, PHPMA, and PEG-PHPMA) and PEG aggregates. The slow diffusion mode generates DDOSY,2 ≈ 1 × 10−13 m2 s−1. | ||
| Fig. 6 1H NMR DOSY of PEG-PHPMA PISA solutions. (A) Polymer echo decays. Inset displays eqn (9) fit to PEG-PHPMA 16 polymer data. (B) Drug echo decays. Inset displays eqn (9) fit to PEG-PHPMA 16 drug data, in which the PA DDOSY,2 = PEG-PHPMA DDOSY,1 constraint was applied. | ||
Fig. 6B details the echo decays of PA (Peak G). Compared to the polymer signal, the PA echo decays are noisy because Peak G is relatively lower in intensity compared to Peak B. For all PISA samples containing PA, the echo decays exhibit bimodal diffusion behavior well-described by eqn (9). If fDOSY, DDOSY,1, and DDOSY,2 are all treated as adjustable parameters, fitting eqn (9) to the PA echo decay produces a PA DDOSY,2 ≈ PEG-PHPMA DDOSY,1 (see Table S5, ESI†). This suggests that the slower diffusion mode of PA correlates to drug bound to PEG-PHPMA micelles. In this case, a second diffusion mode is observed because the drug is tightly bound to the micelles, such that τex > Δ. To push this idea further, eqn (9) was re-fit to the PA echo decays, treating only fDOSY and DDOSY,1 as adjustable parameters. For each sample, the PA DDOSY,2 was set equal to the corresponding PEG-PHPMA DDOSY,1 value.
The solid lines featured in the Fig. 6B inset and Fig. S19A–C (ESI†) represent the eqn (9) fits that employed the PA DDOSY,2 = PEG-PHPMA DDOSY,1 constraint. This approach produces fDOSY values that decrease as CPA increases (Table S6, ESI†), hinting that more PA binds to PEG-PHPMA micelles at higher drug concentrations. The model analysis also produces PA DDOSY,1 values that increase from 2.2 × 10−10 m2 s−1 to 3.0 × 10−10 m2 s−1 as CPA increases. Even after accounting for the relatively high bulk solution viscosity (see Section S7 of ESI†), the measured DDOSY,1 values were lower than expected for drug fully dissolved in D2O. Similar to the case for the PEG and PA solutions, this inequality was interpreted as evidence that the fast diffusion mode represents the exchange of drug between D2O and polymeric species that are not PEG-PHPMA micelles (i.e., PEG aggregates and unimers of PEG, PHPMA, and PEG-PHPMA). This interpretation is further evaluated in the Discussion section.
To summarize, both the PEG-PHPMA and PA echo decay curves exhibit bimodal diffusion behavior. The fast diffusion mode of the polymer and slow diffusion mode of the drug are correlated to one another, suggesting that PA binds to the PEG-PHPMA micelles. The fast diffusion mode of PA represents the weighted average of drug either dissolved in D2O or bound to other polymeric species that are not PEG-PHPMA micelles.
Discussion
PEG macro-RAFT agent self-assembly
PEG and its longer-chain analogue poly(ethylene oxide) are the primary polymers of choice for therapeutic delivery applications because they are biocompatible and commercially available in a wide range of molar masses.40–44 While PEG has the reputation of being hydrophilic, its aqueous phase behavior actually hinges on its end group chemistry, even at biologically relevant temperatures. Specifically, PEG with hydroxyls at the α and ω end group positions forms hydrogen bonds with water, resulting in a relatively large second virial coefficient with aqueous solvent. Replacing one or both hydroxy end groups with methoxy moieties disrupts hydrogen bond formation and reduces the second virial coefficient.45,46 Consequently, water is essentially a theta solvent for dimethyl PEG,47 while longer alkyl end groups cause PEG to aggregate.48–50 Homopolymer micelle formation may even result from the addition of a RAFT end group, especially for those containing aromatic components.29,51 Thus, the aromatic CPPA end group likely drives the PEG macro-RAFT agent aggregation observed in this study.The addition of the aromatic drug PA clearly alters the PEG solution behavior, both in size and number of aggregates. Based on the pagg values estimated from 1H NMR DOSY, PEG aggregates are only able to absorb ≈30 mol% of the total available drug in solution. CPA ≥ 16 mg mL−1 not only saturates the aggregates, but also destabilizes the entire solution such that D2O rich and poor macrophases form. Within the D2O poor region, PA exhibits bimodal diffusion behavior. The fast diffusion mode exhibits DDOSY,1 < DPA,D2O. In this case, however, we do not interpret DDOSY,1 as the weighted average of DPA,D2O and Dagg (see eqn (10)), as the D2O poor nature of this macrophase probably inhibits diffusion due to a higher viscosity. Instead, the bimodal diffusion is reminiscent of restricted diffusion within heterogeneous systems. In this type of scenario, the DDOSY,1 and DDOSY,2 for PA represent the diffusion of drug in the interior and exterior of some sort of PEG aggregate within the D2O poor region.52–54 However, precise elucidation of diffusion in macrophase separated systems lies outside the scope of this manuscript.
Polymer and drug self-assembly in PEG-PHPMA PISA solutions
Similar to the PEG solutions, the addition of PA greatly affects the turbidity of the PEG-PHPMA solutions prior to PISA. After PISA, however, the solutions have similar appearances. The PEG-PHPMA solutions produced by PISA contain both spherical micelles of block copolymer and a significant fraction of unreacted PEG macro-RAFT agent. As demonstrated in Section S11 of the ESI,† these chains are not “dead” as they still possess the CPPA end group. Unreacted macro-RAFT agent is commonly observed in other PISA systems and emulsion polymerizations involving PEG.55–57 While the origin of this impurity is not well understood, we speculate that the presence of unreacted macro-RAFT agent is related to the tendency of the CPPA end group to aggregate in solution. The steric bulk from this aggregation possibly prevents degenerative chain transfer between chains from occurring. As suggested by the increase in unreacted PEG chains as CPA increases, the fraction of unreacted macro-RAFT agent potentially correlates to the number of aggregated chains. The decrease in tnuc potentially may be related to macro-RAFT agent aggregation, as well. Additional studies, however, are needed to fully examine this hypothesis.In addition to PEG homopolymer, PHPMA homopolymer and PEG-PHPMA unimers also exist in solution.58,59 Thus, the PISA mixture offers several potential encapsulation sites for PA. Based on the 1H NMR DOSY echo decays of PA in the PISA mixture, the encapsulation sites may be organized broadly into three categories (see Fig. 7A): (I) PEG-PHPMA micelles, (II) D2O, and (III) everything else (PEG aggregates and unimers of PEG, PHPMA, and PEG-PHPMA). Quantifying the partitioning of a drug among various encapsulation sites is crucial for pharmaceutical applications, as each site releases the drug at a different rate.60
 | ||
| Fig. 7 (A) Schematic of drug partitioning throughout PEG-PHPMA PISA solution. Green triangles represent PA molecules. For the PEG-PHPMA micelle, the core is composed of PHPMA. For the PEG aggregate, the core is composed of the CPPA end group. Schematic is not drawn to scale. (B) Estimated concentration of PA in encapsulation sites I, II, and III. | ||
To estimate the mass concentration of PA within each site (i.e., CI, CII, and CIII), we apply the following analysis. CI is estimated directly from the slow diffusion mode of the PA echo decays depicted in Fig. 6B, i.e., CI = CPA(1 − fDOSY) corresponds to the amount of tightly drug bound to the micelle. Conversely, CII, and CIII are estimated from the fast diffusion mode:
 | (11) |
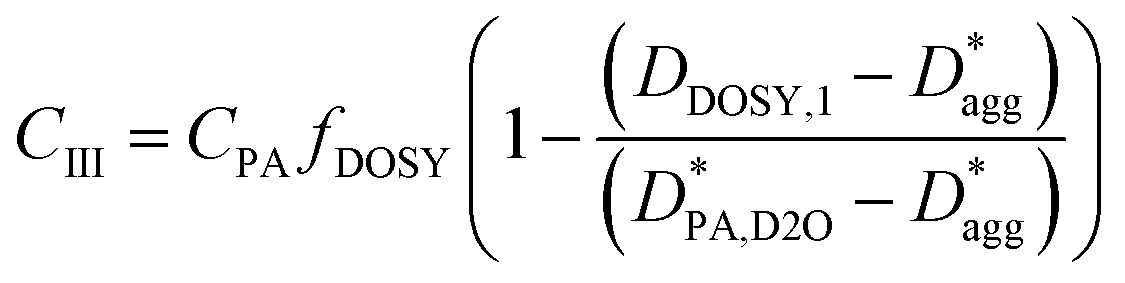 | (12) |
 and
and  are the diffusion coefficients of PA in D2O without polymer and PEG aggregates, each adjusted for the increase of bulk viscosity in the PISA solution (see eqn (S1) and (S2) in the ESI†).
are the diffusion coefficients of PA in D2O without polymer and PEG aggregates, each adjusted for the increase of bulk viscosity in the PISA solution (see eqn (S1) and (S2) in the ESI†).
Fig. 7B illustrates CI, CII, and CIIIvs. CPA. Over the evaluated CPA range, the amount of drug bound to PEG-PHPMA micelles (CI) or dissolved in D2O (CII) increases. In contrast, the concentration of drug bound to other polymeric species (CIII) plateaus at approximately 5 mg mL−1, implying that they are saturated with drug. The trends observed for CI, CII, and CIII demonstrate that the PISA microenvironment significantly impacts encapsulation, as the drug may partition among several competing sites.
Conclusion
We investigated the role of the PEG corona block in governing drug encapsulation and phase behavior during aqueous PEG-PHPMA PISA. The PEG macro-RAFT agent partially aggregates in deuterium oxide. The addition of PA drug promotes further aggregation and macrophase separation. During PEG-PHPMA PISA, a minor fraction of the PEG macro-RAFT agent remains unreacted. The presence of PA in the reaction mixture prompts faster nucleation of spherical micelles. Within the PISA microenvironment, 1H NMR DOSY measurements reveals that the drug partitions among three encapsulation sites: (I) PEG-PHPMA micelles, (II) D2O, and (III) other polymeric species – including PEG aggregates and unimers of PEG, PHPMA, and PEG-PHPMA. As the targeted PA loading increases, the amount of drug in sites (I) and (II) grows while the concentration in (III) remains constant.This work illustrates that drugs in a PISA microenvironment not only bind to block copolymer nanoparticles, but also interact with other polymeric species in the solution. This finding has important implications for the use of drug-loaded PISA formulations in biological media – rich in macromolecular and colloidal species – as the competition between encapsulation sites will intensify. Subsequent release behavior from a PISA formulation will depend on the spatial distribution of drug within the solution, underscoring the need to further resolve the molecular principles that control drug partitioning. Future efforts should pinpoint the specific interactions among the drug, hydrophilic corona block, and hydrophobic core block. These same interactions also will dictate whether the drug binds to the corona, core, or corona–core interface of the nanoparticles. In the PEG-PHPMA and PA systems described in this study, potential mechanisms for drug binding to the polymer include hydrogen bonding between the carboxylic acid of PA and the polyether backbone of PEG, as well as possible π–π stacking interactions between the PA and the PEG RAFT agent end group. Additionally, exploring the encapsulation of drugs more hydrophobic than PA is especially important, as many pharmaceutical actives have solubilities well below 16 mg mL−1.61 Further studies on other block copolymer and drug pairings are needed to identify the important interactions that govern encapsulation in PISA formulations. Finally, identifying the consequences of the solution morphology prior to polymerization on the drug spatial distribution in the final PISA mixture remains a critical goal. A deeper understanding of encapsulate behavior during PISA is crucial for designing effective block copolymer therapeutics.
Author contributions
Guanrui Li: conceptualization (lead); data curation (lead); formal analysis (lead); investigation (lead); methodology (lead); writing – original draft (lead); writing – review and editing (lead). Cassie Duclos: data curation (supporting); formal analysis (supporting); writing – review and editing (supporting). Ralm Ricarte: conceptualization (lead); formal analysis (lead); project administration (lead); writing – original draft (lead); writing – review and editing (lead).Data availability
The data supporting this article have been included as part of the ESI.†Conflicts of interest
The authors declare no competing financial interest.Acknowledgements
We are very grateful for helpful discussions with Antonia Statt, Devin Grabner, Brian Collins, and Michelle Calabrese. We are very grateful to Daniel Hallinan, Michael Patrick Blatt, and the Hallinan Laboratory for use of their size-exclusion chromatography instrument. We are very grateful to Hedi Mattoussi and the Mattoussi Laboratory for use of their dynamic light scattering instrument. We are very grateful to the Florida State University Department of Chemistry and Biochemistry and Banghao Chen for use of the Nuclear Magnetic Resonance Spectroscopy Facility. We thank Daniel Barzycki, Dana Ezzeddine, and Marie Chmara for help with experiments. This work was supported by funds provided by the Oak Ridge Associated Universities Foundation, ORAU-Directed Research and Development Program. This work was partially supported by funds provided by the 3M Non-Tenured Faculty award. This work also was supported using start-up funds from Florida State University and the Florida A&M University – Florida State University College of Engineering. The work was also supported by the Florida State University Adelaide Wilson Fellowship. Instruments at the Biological Science Imaging Resource are supported by Florida State University. The Hitachi HT7800 was funded from NSF grant 2017869.References
- N. J. Warren, O. O. Mykhaylyk, D. Mahmood, A. J. Ryan and S. P. Armes, J. Am. Chem. Soc., 2014, 136, 1023–1033 CrossRef CAS.
- C. A. Figg, A. Simula, K. A. Gebre, B. S. Tucker, D. M. Haddleton and B. S. Sumerlin, Chem. Sci., 2014, 6, 1230–1236 RSC.
- J. R. Lovett, L. P. D. Ratcliffe, N. J. Warren, S. P. Armes, M. J. Smallridge, R. B. Cracknell and B. R. Saunders, Macromolecules, 2016, 49, 2928–2941 CrossRef CAS.
- S. L. Canning, G. N. Smith and S. P. Armes, Macromolecules, 2016, 49, 1985–2001 CrossRef CAS PubMed.
- C. A. Figg, R. N. Carmean, K. C. Bentz, S. Mukherjee, D. A. Savin and B. S. Sumerlin, Macromolecules, 2017, 50, 935–943 CrossRef CAS.
- L. D. Blackman, K. E. B. Doncom, M. I. Gibson and R. K. O’Reilly, Polym. Chem., 2017, 8, 2860–2871 RSC.
- J. C. Foster, S. Varlas, B. Couturaud, J. R. Jones, R. Keogh, R. T. Mathers and R. K. O’Reilly, Angew. Chem., 2018, 130, 15959–15963 CrossRef.
- S. Varlas, J. C. Foster and R. K. O’Reilly, Chem. Commun., 2019, 55, 9066–9071 RSC.
- N. J. W. Penfold, J. R. Whatley and S. P. Armes, Macromolecules, 2019, 52, 1653–1662 CrossRef.
- L. Romero-Azogil, N. J. W. Penfold and S. P. Armes, Polym. Chem., 2020, 11, 5040–5050 RSC.
- F. D’Agosto, J. Rieger and M. Lansalot, Angew. Chem., 2020, 59, 8368–8392 CrossRef PubMed.
- J. Y. Rho, G. M. Scheutz, S. Häkkinen, J. B. Garrison, Q. Song, J. Yang, R. Richardson, S. Perrier and B. S. Sumerlin, Polym. Chem., 2021, 12, 3947–3952 RSC.
- A. W. Fortenberry, P. E. Jankoski, E. K. Stacy, C. L. McCormick, A. E. Smith and T. D. Clemons, Macromol. Rapid Commun., 2022, 43, 2200414 CrossRef PubMed.
- S. Jimaja, S. Varlas, J. C. Foster, D. Taton, A. P. Dove and R. K. O’Reilly, Polym. Chem., 2022, 13, 4047–4053 RSC.
- G. Liao, M. J. Derry, A. J. Smith, S. P. Armes and O. O. Mykhaylyk, Angew. Chem., Int. Ed., 2024, 63, e202312119 CrossRef CAS PubMed.
- S. R. Mane and A. S. Carlini, Polym. Chem., 2024, 15, 1043–1051 RSC.
- G. M. Scheutz, J. I. Bowman, S. Mondal, J. Y. Rho, J. B. Garrison, J. Korpanty, N. C. Gianneschi and B. S. Sumerlin, ACS Macro Lett., 2023, 12, 454–461 CrossRef CAS.
- J. I. Bowman, C. B. Eades, J. Korpanty, J. B. Garrison, G. M. Scheutz, S. L. Goodrich, N. C. Gianneschi and B. S. Sumerlin, Macromolecules, 2023, 56, 3316–3323 CrossRef CAS.
- B. Karagoz, L. Esser, H. T. Duong, J. S. Basuki, C. Boyer and T. P. Davis, Polym. Chem., 2013, 5, 350–355 RSC.
- J. Tan, H. Sun, M. Yu, B. S. Sumerlin and L. Zhang, ACS Macro Lett., 2015, 4, 1249–1253 CrossRef CAS PubMed.
- W. Zhang, C. Hong and C. Pan, Macromol. Rapid Commun., 2019, 40, e1800279 CrossRef.
- H. Sun, W. Cao, N. Zang, T. D. Clemons, G. M. Scheutz, Z. Hu, M. P. Thompson, Y. Liang, M. Vratsanos, X. Zhou, W. Choi, B. S. Sumerlin, S. I. Stupp and N. C. Gianneschi, Angew. Chem., 2020, 132, 19298–19304 CrossRef.
- H. Phan, V. Taresco, J. Penelle and B. Couturaud, Biomater. Sci., 2020, 9, 38–50 RSC.
- A. B. Korpusik, Y. Tan, J. B. Garrison, W. Tan and B. S. Sumerlin, Macromolecules, 2021, 54, 7354–7363 CrossRef CAS.
- H. Phan, M. Cossutta, C. Houppe, C. L. Cœur, S. Prevost, I. Cascone, J. Courty, J. Penelle and B. Couturaud, J. Colloid Interface Sci., 2022, 618, 173–184 CrossRef CAS.
- E. G. Hochreiner and B. G. P. Ravensteijn, J. Polym. Sci., 2023, 61, 3186–3210 CrossRef CAS.
- C. Cao, F. Chen, C. J. Garvey and M. H. Stenzel, ACS Appl. Mater. Interfaces, 2020, 12, 30221–30233 CrossRef CAS PubMed.
- G. Li, D. C. Barzycki and R. G. Ricarte, AIChE J., 2023, 69, e18014 CrossRef CAS.
- L. M. Johnson, Z. Li, A. J. LaBelle, F. S. Bates, T. P. Lodge and M. A. Hillmyer, Macromolecules, 2017, 50, 1102–1112 CrossRef CAS.
- Z. Li, T. I. Lenk, L. J. Yao, F. S. Bates and T. P. Lodge, Macromolecules, 2018, 51, 540–551 CrossRef CAS.
- M. S. Haider, M. M. Lübtow, S. Endres, S. Förster, V. J. Flegler, B. Böttcher, V. Aseyev, A.-C. Pöppler and R. Luxenhofer, ACS Appl. Mater. Interfaces, 2020, 12, 24531–24543 CrossRef CAS.
- R. Palao-Suay, M. R. Aguilar, F. J. Parra-Ruiz, S. Maji, R. Hoogenboom, N. A. Rohner, S. N. Thomas and J. S. Román, Polym. Chem., 2015, 7, 838–850 RSC.
- C. Zhang, Z. Jin, B. Zeng, W. Wang, G. Palui and H. Mattoussi, J. Phys. Chem. B, 2020, 124, 4631–4650 CrossRef CAS PubMed.
- T. P. Lodge and P. C. Hiemenz, Polymer Chemistry, 3rd edn, CRC Press, Boca Raton, 2020 Search PubMed.
- X. Li, V. V. S. Uppala, T. J. Cooksey, M. L. Robertson and L. A. Madsen, ACS Appl. Polym. Mater., 2020, 2, 3749–3755 CrossRef CAS.
- P. W. Kuchel, B. E. Chapman, W. A. Bubb, P. E. Hansen, C. J. Durrant and M. P. Hertzberg, Concepts Magn. Reson., Part A, 2003, 18A, 56–71 CrossRef CAS.
- A. Blanazs, A. J. Ryan and S. P. Armes, Macromolecules, 2012, 45, 5099–5107 CrossRef CAS.
- D. L. Beattie, O. O. Mykhaylyk and S. P. Armes, Chem. Sci., 2020, 11, 10821–10834 RSC.
- A. Czajka and S. P. Armes, J. Am. Chem. Soc., 2021, 143, 1474–1484 CrossRef CAS PubMed.
- C. T. Greco, T. H. Epps and M. O. Sullivan, ACS Biomater. Sci. Eng., 2016, 2, 1582–1594 CrossRef CAS PubMed.
- T. H. Epps and R. K. O’Reilly, Chem. Sci., 2016, 7, 1674–1689 RSC.
- C. T. Greco, V. G. Muir, T. H. Epps and M. O. Sullivan, Acta Biomater., 2017, 50, 407–416 CrossRef CAS.
- C. T. Greco, J. C. Andrechak, T. H. Epps and M. O. Sullivan, Biomacromolecules, 2017, 18, 1814–1824 CrossRef CAS.
- T. H. Epps, T. Vi and M. O. Sullivan, Polym. J., 2018, 50, 711–723 CrossRef.
- E. E. Dormidontova, Macromolecules, 2002, 35, 987–1001 CrossRef CAS.
- E. E. Dormidontova, Macromolecules, 2004, 37, 7747–7761 CrossRef CAS.
- J. Mondal, E. Choi and A. Yethiraj, Macromolecules, 2014, 47, 438–446 CrossRef CAS.
- E. Alami, M. Almgren, W. Brown and J. François, Macromolecules, 1996, 29, 2229–2243 CrossRef CAS.
- O. Vorobyova, A. Yekta, M. A. Winnik and W. Lau, Macromolecules, 1998, 31, 8998–9007 CrossRef CAS.
- O. Vorobyova, W. Lau and M. A. Winnik, Langmuir, 2001, 17, 1357–1366 CrossRef CAS.
- J. Du, H. Willcock, J. P. Patterson, I. Portman and R. K. O’Reilly, Small, 2011, 7, 2070–2080 CrossRef CAS PubMed.
- J. Kärger, Adv. Colloid Interface Sci., 1985, 23, 129–148 CrossRef.
- S. So and T. P. Lodge, J. Phys. Chem. C, 2014, 118, 21140–21147 CrossRef CAS.
- S. So, L. J. Yao and T. P. Lodge, J. Phys. Chem. B, 2015, 119, 15054–15062 CrossRef CAS PubMed.
- M. Siauw, B. S. Hawkett and S. Perrier, J. Polym. Sci., Part A: Polym. Chem., 2012, 50, 187–198 CrossRef CAS.
- N. J. Warren, O. O. Mykhaylyk, D. Mahmood, A. J. Ryan and S. P. Armes, J. Am. Chem. Soc., 2014, 136, 1023–1033 CrossRef CAS.
- R. A. E. Richardson, T. R. Guimarães, M. Khan, G. Moad, P. B. Zetterlund and S. Perrier, Macromolecules, 2020, 53, 7672–7683 CrossRef CAS.
- B. E. Kidd, X. Li, R. C. Piemonte, T. J. Cooksey, A. Singh, M. L. Robertson and L. A. Madsen, Macromolecules, 2017, 50, 4335–4343 CrossRef.
- T. J. Cooksey, A. Singh, K. M. Le, S. Wang, E. G. Kelley, L. He, S. V. Kesava, E. D. Gomez, B. E. Kidd, L. A. Madsen and M. L. Robertson, Macromolecules, 2017, 50, 4322–4334 CrossRef CAS.
- J. Yoo and Y.-Y. Won, ACS Biomater. Sci. Eng., 2020, 6, 6053–6062 CrossRef CAS.
- R. G. Ricarte, N. J. V. Zee, Z. Li, L. M. Johnson, T. P. Lodge and M. A. Hillmyer, Mol. Pharmaceutics, 2019, 16, 4089–4103 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Dynamic light scattering plots, 1H NMR spectra, 1H NMR DOSY spectra waterfall plots for PEG 10 and PEG-PHPMA 10, 1H NMR DOSY fit parameters for PEG solutions, 1H NMR DOSY plots and fit parameters for PEG-PHPMA PISA solutions, 1H NMR DOSY fit parameters for water, PEG-PHPMA PISA solution viscosity adjustment, PEG-PHPMA UV SEC trace deconvolution, PEG-PHPMA PISA kinetics, impact of initiator concentration on PEG-PHPMA PISA, impact of free PEG-OH chains on PEG-PHPMA PISA, PEG 0 DLS hydrodynamic radius distribution, eqn (10) derivation, estimated hydrodynamic radii and diffusing species, and transmission electron microscopy particle size distributions. See DOI: https://doi.org/10.1039/d4sm00654b |
| This journal is © The Royal Society of Chemistry 2024 |