
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceBiopolymer-supramolecular polymer hybrids for photocatalytic hydrogen production†
Jacob E.
Kupferberg
a,
Zois
Syrgiannis
b,
Luka
Đorđević
b,
Eric P.
Bruckner
 a,
Tyler J.
Jaynes
b,
Hakim H.
Ha
a,
Evan
Qi
a,
Kristen S.
Wek
a,
Adam J.
Dannenhoffer
a,
Nicholas A.
Sather
a,
H. Christopher
Fry
c,
Liam C.
Palmer
bd and
Samuel I.
Stupp
*abdef
a,
Tyler J.
Jaynes
b,
Hakim H.
Ha
a,
Evan
Qi
a,
Kristen S.
Wek
a,
Adam J.
Dannenhoffer
a,
Nicholas A.
Sather
a,
H. Christopher
Fry
c,
Liam C.
Palmer
bd and
Samuel I.
Stupp
*abdef
aDepartment of Materials Science and Engineering, 2220 Campus Drive, Evanston, IL 60208, USA. E-mail: s-stupp@northwestern.edu
bDepartment of Chemistry, Northwestern University, 2145 Sheridan Road, Evanston, IL 60208, USA
cCenter for Nanoscale Materials, Argonne National Laboratory, 9700 S. Cass Avenue, Lemont, IL 60439, USA
dSimpson Querrey Institute for BioNanotechnology, Chicago, Illinois 60611, USA
eDepartment of Medicine, Northwestern University, Chicago, Illinois 60611, USA
fDepartment of Biomedical Engineering, Northwestern University, Evanston, Illinois 60208, USA
First published on 29th July 2024
Abstract
Solar generation of H2 is a promising strategy for dense energy storage. Supramolecular polymers composed of chromophore amphiphile monomers containing perylene monoimide (PMI) have been reported as crystalline light-harvesting assemblies for aqueous H2-evolving catalysts. Gelation of these supramolecular polymers with multivalent ions creates hydrogels with high diffusivity but insufficient mechanical stability and catalyst retention for reusability. We report here on using sodium alginate (SA) biopolymer to both induce supramolecular polymerization of PMI and co-immobilize them with catalysts in a robust hydrogel with high diffusivity that can also be 3D-printed. Faster mass transfer was achieved by controlling the material macrostructure by reducing gel diameter and microstructure by reducing biopolymer loading. Optimized gels produce H2 at rates rivaling solution-based PMI and generate H2 for up to 6 days. The PMI assemblies in the SA matrix create a percolation network capable of bulk-electron transfer under illumination. These PMI–SA materials were then 3D-printed on conductive substrates to create 3D hydrogel photoelectrodes with optimized porosity. The design of these versatile hybrid materials was bioinspired by the soft matter environment of natural photosynthetic systems and opens the opportunity to carry out light-to-fuel conversion within soft matter with arbitrary shapes and particular local environments.
1. Introduction
Solar fuel generation through photocatalysis is a method of converting abundant solar energy into stable chemicals for energy storage.1 Biological photosynthesis utilizes supramolecular dye assemblies to harvest light for highly endothermic reactions under ambient conditions.2 This has inspired the design of chromophores with finely tuned energy levels to achieve a similar function in synthetic systems. These chromophores can also be designed such that they self-assemble into supramolecular arrays that enable charge separation for long-range charge transport, similar to what physically takes place in the thylakoid membranes of chloroplasts.3–5 Our vision has been to create light-absorbing soft materials using these large supramolecular assemblies that can be co-localized with catalysts for solar fuel production.5–13 In our previous work, we developed the idea of using hydrogels composed of supramolecular chromophore assemblies as scaffolds for solar fuel production.10 These types of photocatalytic hydrogel materials can “heterogenize” otherwise homogeneous catalysts by entrapping them in a polymeric matrix and are composed of interconnected fluid compartments that allow for rapid diffusion.14–18Our group has previously reported on a number of negatively charged chromophore amphiphiles that self-assemble in water through π–π stacking into micron-long supramolecular assemblies with molecular thickness that are capable of intermolecular charge transport for photocatalysis.5–13,19–21 Of interest to this study is a perylene monoimide substituted by an ionizable carboxylated tail with five methylene units (PMI-L5) which has been used to photosensitize catalysts for the hydrogen evolution reaction (HER). Ionized PMI-L5 forms disordered stacks in aqueous solution through π–π interactions, but the introduction of ionic species will electrostatically screen the charged tails of the monomers, enabling denser packing, enhanced charge transport from excitons, and higher HER rates.6,19 These light-harvesting assemblies can be crosslinked by divalent cations or cationic polymers to form fragile hydrogels with the same assembled state after gelation and after photocatalysis.5,11 A more robust PMI hydrogel was created by assembling crystalline PMI-L5 stacks on a cationic polyacrylamide hydrogel scaffold using a solvent exchange procedure.10 Despite their high diffusivity for small molecules, diffusion speed has been shown to be a limiting factor in the HER rate of both types of PMI hydrogel.10,11 Recently, the highest rate of HER in a PMI-L5 system was achieved by co-assembling an anionic linear polymer with covalently attached PMI chromophores with monomeric PMI to create a suspension of water-soluble PMI assemblies.12 The enhanced rate of photocatalysis by these “hybrid bonding polymers” was attributed to the rapid diffusion rate of a solution-based system relative to a hydrogel system.
The effect of mass-transport rate on photocatalysis has mainly been explored in solid oxide systems that have low diffusion coefficients and high extinction coefficients that render only nanoscale materials viable for photocatalysis.22,23 In contrast, hydrogel-based photocatalysis systems enable the exploration of materials at a larger length scale and enable the facile alteration of both diffusivity constant and internal mass-transfer distance. Sodium alginate (SA) was chosen as a robust hydrogel matrix to immobilize PMI assemblies and catalysts because its rapid ion-gelation mechanism enabled the creation of gels with variable mass-transfer distance. Previously, sodium alginate (SA) has been used to assemble perylene chromophores into supramolecular crystals through polymer confinement over the course of hours or days, but the resulting gels were not investigated for photocatalysis applications.24 Hydrogel materials can also be 3D-printed to create bulk materials with high diffusivity and large active surface areas, both of which can benefit the terminal goal of applying these photocatalytic hydrogels to electrode systems.25–28 While some studies have investigated the use of sub-micron thick hydrogels for photocathodes, we have not found any instances of incorporating 3D printing to alter the gel architecture.29,30
We report here on a printable hydrogel ink capable of immobilizing both PMI-L5 assemblies and catalysts for aqueous hydrogen production. Utilizing the biopolymer sodium alginate (SA) PMI-L5 monomers could be charge-screened in order to induce stacking into crystalline light-harvesting assemblies. These materials could then be gelled using a CaCl2 solution to create gels with tunable diameter and polymer loading in order to explore how mass-transfer distance affected photocatalysis rates. Four different molybdenum-based catalysts were incorporated into the material to explore the effects of catalyst retention. Finally, these materials were converted into a printable ink and deposited on conductive substrates to create photocathodes with macroscopic porosity.
2. Experimental section
2.1 Materials
Synthesis of PMI-L5 followed previously published procedures.12 The SA used for this study [Protanal HF 120RBS], derived from the leaf of brown algae Laminaria hyperborea was provided by DuPont. This is expected to contain 45–55% guluronic units and have a mass average molar mass of 340–400 kg mol−1.31 HER catalysts were synthesized using methods in the literature.32–36 Synthesis methods, catalysts X-ray diffraction patterns (Fig. S20, ESI†), and catalyst absorption spectra (Fig. S21 and S22, ESI†) can be found in the ESI.† All other chemicals were purchased from Sigma-Aldrich and used without further purification unless otherwise noted.2.2 PMI–SA ink preparation
All inks were prepared using a stock solution of 11.5 mM PMI and 30 wt% (w/w) sodium alginate (SA). PMI was solubilized in deionized water using 1 molar equivalent of NaOH from a stock solution of 4 M NaOH, resulting in a solution with a pH of 8.5.11 SA powder in a sodium salt form was dissolved in deionized water using a high shear dual syringe mixing method to ensure all solids were dissolved. Given the high viscosity of the 30% SA stock solution, it was measured by mass rather than volume. PMI stock solution, catalyst stock solution, gelation solution, and water were all added at volumes relative to the initial SA solids. Catalyst solution was added to the SA solution first to ensure maximum catalyst dispersion. The PMI solution was added after the catalyst stock solution and mixed for at least 5 minutes. Gelation solution or deionized water was added last. As an example, in a 1 mL solution of PMI–SA ink [9.6 mM PMI, 2.5 wt% SA], 108 mg of 30 wt% SA stock solution (24.9 mg SA in 83 μL deionized water) was mixed with 835 μL of 11.5 mM PMI solution. 82 μL of deionized water was then added to the ink to reach a total liquid volume of 1 mL.To ensure adequate mixing, we utilize a dual syringe mixing method used by Freeman and Kelly in which the solution is extruded between two syringes via a small female–female Luer lock connector.37 After 5 minutes of mixing the sample was inspected to ensure no aggregates of SA remained. The ink was then centrifuged between 3500 rpm to 4000 rpm using a Thermo ScientificTM SorvallTM ST8 centrifuge to remove bubbles.
2.3 Sample naming convention
PMI–SA samples contain 9.6 mM PMI and if not otherwise stated contain 2.5% (w/w) SA. Tests varying the amount of SA in the gel are labeled as PMI–SA[x] where [x] represents the wt% (w/w) loading of SA in the ink. Samples mixed with a specific catalyst are labeled as PMI–SA[x]-C where C represents the catalyst used (either 1, 2, 2.5, 3, or 4). As an example, a PMI–SA sample with 2.5 wt% loading, mixed with catalyst 2, and extruded from a 27G needle would be labeled as PMI–SA[2.5]-2.2.4 PMI–SA gel extrusion
PMI–SA Ink was extruded by hand into a 0.5 M CaCl2 solution. The needle opening was placed below the surface of the gelling solution to ensure immediate gelation in the shape of the extruder. The pressure used to extrude the gel was maintained as consistent as possible. Insufficient pressure could produce defects in the gel or result in clogging, so enough pressure was applied to ensure continuous flow of gel out of the nozzle such that it matched the diameter of the needle. Gels were allowed to sit in CaCl2 solution for 15 minutes before being moved to a deionized water wash bath. Needles used in this study were acquired from SAI infusion systems and ranged from 0.191 mm to 1.372 mm in diameter. Commercial blunt needles with diameters down to 0.089 mm are available, but attempts to extrude PMI–SA[2.5] from 0.140 mm diameter needles resulted in nozzle clogging and inconsistent gel sizes.2.5 Confocal laser scanning microscopy and FRAP experiments
High-resolution confocal laser scanning microscopy (CLSM) of the samples was performed either on a Nikon A1R confocal microscope using Plan Apo VC 60× oil immersion objective or Plan Apo VC 20× air objective. For Nikon A1R, the excitation wavelength was 561 nm, and a filter cube was used to select 570–620 nm emission for detection by a photomultiplier tube. Gels soaked in 0.5 M CaCl2 were placed in silicon wells on top of a thin glass slide, backfilled with 0.5 M CaCl2, and then gently sealed with another thin glass slide to avoid evaporation. Care was taken not to excessively compress the samples.Fluorescence recovery after photobleaching (FRAP) experiments were performed on a Nikon A1R confocal microscope using Nikon Plan Apo VC 20× air objective, using the same optical configuration for excitation and emission as described above. Gels were first soaked in deionized water overnight, then soaked in 0.1 mg mL−1 fluorescein dye overnight. Fluorescein was chosen as a model dye due to its singular anionic charge and relatively similar size to sodium ascorbate. Gels were then placed in silicon wells on top of a thin glass slide, backfilled with 0.1 mg mL−1 fluorescein solution, and then gently sealed with another glass slide to prevent evaporation. FRAP was performed in the interior bulk of the gel rather than the surface.
2.6 Photocatalytic hydrogen evolution
HER catalysis conditions were based on a protocol from our group's previous work.11 Ascorbic acid (AA) was added to deionized water at a loading of 0.299 g mL−1. The solution was then pH adjusted with 4 M NaOH to pH 4. The resulting AA concentration is about 1.5 M. CaCl2 salt is added to the AA solution to reach a final concentration of 160 mM CaCl2. This ratio of Ca/Na was determined to keep SA gels sufficiently stable. AA solution was prepared fresh before each photocatalysis experiment. The Ca2+ ions did not affect the photocatalysis of Ru(bpy)32+ controls.Photocatalysis tests were performed in 17 mm diameter clear borosilicate vials with a total volume of 8.92 mL. Each vial contained 860 μL of AA solution and up to 100 μL of photosensitizer gel/solution. An additional volume of up to 40 μL was added for control experiments. PMI–SA gels were briefly dried on a Kimwipe to remove excess liquid and then massed in the photocatalysis vial. The amount of catalyst added to each vial was calculated using the measured mass of the gel and the known swelling behavior of the gel (calculations for the swelling behavior can be found in the ESI,† and Fig. S8). This method for estimating moles of catalyst was used in turnover number (TON) calculations. Care was taken to avoid the gels drying out. The vial was then sealed with a screw cap and rubber/PTFE septum. Before photocatalysis, the vials were purged with Ar for up to 45 minutes and brought to atmospheric pressure. The photocatalysis setup was custom built, consisting of a fan-cooled metal block with vials placed directly above 5000 K white LED (Cree XLamp XT-E White LEDs 5000 K) with collimators without making contact. LED lights were used to minimize heat production. The setup was placed in a fume hood and a desk fan was placed above the vials to improve air flow. The intensity of the white LEDs was evaluated at 447 nm LED at 250 W cm−2, measured using an optical power meter PM100D with optical sensor S120VC from (Thorlabs).
Vials were illuminated continuously for a specific time period (usually 18 hours) after which an aliquot of less than 1 mL was extracted from the vial headspace via autosampler and injected into the gas chromatograph (Shimadzu GC-2014) equipped with a 5 Å molecular sieve column, argon carrier gas, and a thermal conductivity detector. Vials contained some overpressure due to the gas evolution, but we did not consider the pressure buildup in the turnover number (TON) calculation. This discrepancy may have led to underestimation of the TON. Eight-point calibration curves for H2 and N2 were created using a standard gas (7% H2 balanced with N2) and integrated peak areas were used to determine the H2 concentration in the sample headspace at STP.
2.7 3D-Printing of photocathodes
3D printable PMI–SA ink consisted of a solution with 3 wt% SA, 9.6 mM PMI, established catalyst loading, and 15 mM CaCl2 concentration. The ink was extruded from a 3D Bioplotter with air pressure driven extrusion from a 0.2 mm conical blunt nozzle. Inks were extruded on the electrode substrate as a single connected layer. Substrates used included fluorine-doped tin oxide (FTO) coated glass or FTO-coated glass with a thin film of NiO. After extrusion, 0.5 M CaCl2 was dropped on the surface of the gel and allowed to sit for 10 minutes. The sample was then completely immersed in a 0.5 M CaCl2 solution and stored until use.Photocathode measurements were performed at pH 6 in 100 mM MES buffer containing an additional 160 mM CaCl2 and 1.24 M NaCl. All measurements were performed at room temperature (22 °C) under continuous purging with argon gas unless otherwise stated. A platinum counter electrode and Ag/AgCl (3 M NaCl) reference electrode were used in a three-electrode cell. Voltages were then converted to the reversible hydrogen electrode (RHE) using the equation RHE = 0.197 + 0.059 × pH + VAg/AgCl. The photocathodes were illuminated with a 5000 K white LED light with 300 mA current (about 3× sun). Representative cyclic voltammetry (CV) displayed are taken after 60 cycles run at 10 mV s−1 to ensure stabilization. CV taken under dark conditions are taken prior to CV taken under illuminated conditions.
2.8 Materials characterization
UV-vis absorption spectroscopy on solutions was performed in a 0.05 mm path length, closed demountable quartz spectrophotometer cell (Starna Cells) using an Ocean Optics QEPro Spectrophotometer equipped with a DH-2000-Bal lamp.Rheological characterization was performed using an Anton Paar Physical Modular Compact Rheometer 300 operating in a 25 mm cone-and-plate configuration with a 2° cone angle.
Scanning electron microscopy (SEM) was performed using a Hitachi SU8030 instrument with an accelerating voltage of 2 kV. Hydrogel samples were prepared by critical point drying (CPD) in a Tousimis Samdri-795. Dried samples were cleaved to expose cross-sections and coated with 8 nm of osmium (Filgen, OPC-60A).
Inductively coupled plasma mass spectrometry (ICP-MS) was performed on the used AA solutions to determine the presence of Mo from the catalyst that had leaked out of the gel.
Electrochemical experiments were performed using a Metrohm Autolab potentiostat.
Additional information on all techniques can be found in the ESI.†
3. Results and discussion
3.1 PMI-L5 sodium alginate hydrogel
Stock solutions of 11.5 mM PMI-L5 and 30 wt% (w/w) SA (Protanal HF 120RBS) were mixed with deionized water using high shear to create inks with a final concentration of 9.6 mM PMI and 0.5 wt% to 4 wt% SA (Fig. 1A). We previously showed that the negatively charged PMI-L5 supramolecular assemblies are most effective at photosensitizing HER catalysts when they form stacks with dense crystalline packing, which can be achieved through electrostatic screening.6 It was proposed that these nanoscale light-harvesting ribbons enable the formation of delocalized charge-transfer excitons that enhance the rate of photocatalysis reactions.19 This crystallization is accompanied by a color change from dark red to bright red and a broadening of the absorption spectra with blue- and red-shifted peaks relative to the absorbance peaks of the same PMIs in their non-crystalline state (Fig. S1, ESI†). Aqueous SA behaves as an anionic polyelectrolyte at neutral pH with charges provided by the carboxylate groups on polymer chains. Mixing solutions of SA and PMI produced a bright red viscous solution that displayed an absorption spectrum indicative of the formation of crystalline PMI assemblies (Fig. S1D, ESI†). The bulky SA polymers hinder the assembly of the PMI, particularly at lower ionic strengths like at 0.5 wt% SA loading where assembly took place over a day after mixing (Fig. S1E, ESI†). A minimum SA concentration of 0.75 wt% was required to induce the assembly of crystalline PMI within a few minutes of mixing (Fig. S2D, ESI†). | ||
| Fig. 1 (A) Schematic of the mixing and assembly of PMI–SA and photographs of the PMI–SA ink extruded through a 0.19 mm and 0.584 mm needle into 0.5 M CaCl2. Scale bars are 1 cm in both photographs. (B) and (C) Confocal laser scanning micrographs of PMI–SA[2.5] gels. The PMI assemblies fluoresce under confocal microscopy and are red in these images. (D) and (E) SEM imaging of the cross-section of PMI–SA[2.5] hydrogel at different magnifications. Panel D shows a higher magnification of the gel where PMI ribbons (false-colored red) can be found within the SA matrix (false-colored yellow). | ||
PMI–SA solution was ionically crosslinked by extrusion into a 0.5 M aqueous CaCl2 solution to form a robust hydrogel as shown in Fig. 1A. This process could produce PMI–SA hydrogel strings of a constant diameter controlled by the diameter of the extrusion needle. Rheology measurements show that the presence of PMI ribbons in the SA matrix increases the stiffness of partially crosslinked PMI–SA, but once fully gelled the SA dominates the viscoelastic behavior due to its storage and loss moduli being an order of magnitude greater than fully gelled PMI-L5 (Fig. S2C, ESI†). PMI–SA gels were qualitatively stable and did not dissolve in deionized water, synthetic seawater (synthetic seawater, Ricca Chemical), and highly acidic water (pH 0). PMI did not leach from the gels under aqueous conditions, likely due to its stability as an assembly and the physical immobilization of the large ribbons in the SA polymer matrix. Because the Ca2+ crosslinks of the PMI–SA gels are dynamic with other solution ions, the gels will exchange Ca2+ for other non-gelling cations like Na+ leading to dissolution.38 Any changes in the mechanical properties of the PMI–SA hydrogel are likely to correspond to changes in the gel microstructure (e.g., porosity, gel density). In order to avoid changes in the mechanical properties of the PMI–SA gels, 160 mM CaCl2 was added to the ascorbic acid (AA) catalysis solution (Fig. S3, ESI†).
Fig. 1B shows a confocal micrograph of PMI assemblies (in red) distributed in a PMI–SA solution. Solutions of PMI assemblies tend to look homogeneous in confocal with much smaller assemblies than those visualized in PMI–SA solutions.11 It is likely that the higher ionic strength of the SA solution and steric effects linked to the polymer cause significant bundling of the PMI assemblies, which results in the large structures observed by confocal microscopy. Despite the potential for PMI to interact with Ca2+, the gelation of PMI–SA does not appear to alter the distribution or size of the PMI assemblies (Fig. 1C). The PMI bundles also appear to align during extrusion gelation, which likely arises from the shear forces applied during nozzle extrusion.39 SEM shows the pores of the SA matrix which contain bundles of PMI ribbons (Fig. 1D). Pores containing PMI assemblies appear to be larger in diameter. We hypothesize that the SA and PMI repel one another due to their like charges leaving the PMI ribbons “floating” in a matrix of SA with maximum exposed surface area for electron transfer, but we cannot rule out the possibility of this microstructure being an artifact of CPD preparation. In particular, the solvent-exchange protocol leads to some dissolution of PMI into the ethanol solvent and dissolution of PMI may occur in the liquid CO2. However, the CPD PMI–SA gels still maintain a red color, indicating the majority of PMI is still entrapped in the SA. Despite the appearance of large PMI aggregates in confocal microscopy, an SEM cross-section of a PMI–SA gel shows a relatively homogeneous structure without large defects (Fig. 1E).
3.2 Catalyst immobilization
Immobilizing catalysts in a hydrogel matrix can greatly improve their long-term stability and reusability.16,17,40 Furthermore, co-immobilizing them in proximity to photosensitizers can enhance catalysis efficiency.41–43 To test the effect of catalyst localization, (NH4)2(Mo3S13) (1) was mixed with a PMI–SA solution containing an SA loading of 2.5 wt% (PMI–SA[2.5]). This was compared to a PMI–SA[2.5] gel with catalyst 1 added to the surrounding solution (the unmixed condition). PMI–SA solutions were extruded through a 0.191 mm diameter nozzle into a 0.5 M CaCl2 gelation bath and allowed to gel for at least 10 minutes before being transferred to vials containing catalysis solution. Photocatalysis experiments were all performed under 1 atm of Ar in pH 4 aqueous solution containing AA as a sacrificial electron donor and were irradiated for 18 hours with 5000 K white LED light. The PMI–SA[2.5] with catalyst mixed into the gel reached TON of 8000 ± 1630 at a rate of 0.30 ± 0.08 mmol H2 per ggel, which outperformed the PMI–SA[2.5] samples with catalyst only added to the surrounding solution which only produced a TON of 1600 ± 380 (Fig. S4B, ESI†). The mixed PMI–SA[2.5] system also outperformed the thermally annealed PMI-PDDA with catalyst mixed into the PMI solution prior to gelation (TON of 5040 ± 609), despite the higher density of the PMI–SA gel.To determine if these improvements were the result of greater catalyst localization near the PMI, the used AA solution was analyzed Mo with ICP-MS to determine the location of catalyst 1. When catalyst 1 was directly mixed into the PMI–SA gel, only 28% of the total Mo leaked into the solution over 18 hours, indicating the PMI–SA gel matrix slows down the diffusion of the otherwise mobile (Mo3S13)2− cluster (Fig. S4C, ESI†). In contrast, the PMI-PDDA control retained almost none of the added catalyst 1, indicating that the cationic PDDA is unable to bind the (Mo3S13)2− cluster and that the high rate of HER in the PMI-PDDA system is primarily the result of high diffusivity rather than high catalyst retention (Fig. S4A, ESI†). The low H2 TON generated by the unmixed PMI–SA[2.5] samples suggest poor penetration of catalyst 1 into the gels similar to issues noted in the PMI-APTAC system.10 However, ICP-MS analysis revealed that the PMI–SA[2.5] gels absorbed almost 50% of catalyst 1 from the solution, indicating some interaction between the catalyst and gel despite their like charges (Fig. S4, ESI†). Cross-sections of pure SA gel cubes that were allowed to soak in a solution of catalyst 1 revealed that (Mo3S13)2− clusters concentrated near the surface of the gel (Fig. S4D, ESI†), which may result in aggregation deactivation and limited catalysis in the gel interior. Conversely, the results from the unmixed samples suggest that not all of the photocatalysis is occurring at the surface of the gels. These results demonstrate that catalyst 1 can be absorbed by the gel but that the interaction is reversible, enabling diffusion of the molecule within the material. The tortuosity of the hydrogel matrix slows the diffusion of the catalyst into the solution, improving the co-localization of the catalyst and photosensitizer and thus enhancing photocatalysis. The ability to pre-mix catalyst into the gel maximizes PMI-catalyst interaction and is only possible due to the benign gelation conditions of SA. Given these results, we pre-mixed the catalyst with the ink for all subsequent samples.
The thermal annealing of PMI-L5 for use in a PMI-PDDA gel was recently reported to create extra-large ribbon structures with enhanced photocatalysis due partly to the higher diffusivity of AA.11 PMI–SA solutions could be annealed in a similar manner to induce the growth of large PMI ribbons (Fig. S5A and B, ESI†). The larger size of the PMI ribbons caused relatively large pores to form in the SA matrix during gelation, but this had no significant effect on HER (Fig. S5C and D, ESI†). These results indicate that the larger pores are likely non-continuous and diffusion is still controlled by the SA matrix. The faster diffusion previously enabled by annealing has minimal effect on a well-dispersed PMI–SA system.
To reduce the catalyst leaching from the PMI–SA gels and improve sustained hydrogen production, we decided to investigate three other Mo–S-based HER catalysts: Mo3S7(C2S2NH6)3I (2), [Mo3S4(H2O)9]Cl4 (3), and amorphous MoS3 nanoparticles (4) (Scheme 1).33,35,44 Catalyst 2 is a derivative of 1 with disulfide ligands substituted for dithiocarbamate ligands, giving the molecule air and water stability, cationic charge, and increased hydrophobicity. Catalyst 2 was solubilized in alkaline DMSO and slowly precipitated when mixed into aqueous solvent (Fig. S6, ESI†). Catalyst 3 is an incomplete cubane cluster that can host up to four cationic charges, was solubilized in methanol, and precipitated when mixed into aqueous solvent at pH 4.45 Catalyst 4 is an amorphous nanoparticle approximating a “polymer” of 1 clusters and was dispersed in an aqueous solution as an unstable suspension. Although catalysts 3 and 4 have been reported to be less efficient HER catalysts compared to 1 and 2, catalyst 3 was chosen for its potential to interact electrostatically with the carboxylate groups of the SA matrix and catalyst 4 for its larger size that should enable physical immobilization within the SA network.46 To account for their lower activities, the gels were prepared with higher catalyst loadings (see ESI,† Methods for details).
 | ||
| Scheme 1 Proposed mechanism of HER photocatalysis using PMI as a photosensitizer (PS) and ascorbic acid (AA) as a sacrificial electron donor. Electron transfer from AA to PMI produces dehydroascorbic acid (DHA) which can parasitize electron transfer from AA (catalyst structures are also shown). | ||
All catalysts were mixed into the PMI–SA prior to gelation, extruded through a 0.191 mm diameter nozzle, and irradiated under the conditions described previously. After 18 hours of light exposure, PMI–SA[2.5] containing catalyst 2 (PMI–SA[2.5]-2) produced TON of 15![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 400 ± 2160 h−1 at a rate of 0.55 mmol H2 per ggel, nearly twice the rate observed with catalyst 1. Using Ru(bpy)3Cl2 or PMI-PDDA as control photosensitizers, catalyst 2 also displayed higher activity than catalyst 1, indicating that the dramatically enhanced HER resulted from a more efficient catalyst (Table S1, ESI†). ICP-MS analysis showed only a 4.82% loss of Mo from the PMI–SA[2.5]-2 gel, indicating significant immobilization within the matrix (Fig. 2B). Dispersing insoluble but active catalyst 2 in the hydrogel heterogenizes the otherwise molecular catalyst while still maintaining excellent exposure to the aqueous environment. In addition, the cationic charge can interact with the carboxylate groups on both the PMI and the SA. PMI–SA containing catalyst 3 (PMI–SA[2.5]-3) reached a TON of 142 ± 28 at a rate of 0.054 mmol H2 per ggel, and ICP-MS showed a 10.28% loss of Mo from the gel indicating good cluster immobilization. We expect that catalyst 3 can be heterogenized due to its low solubility in the AA solution. PMI–SA[2.5] containing catalyst 4 (PMI–SA[2.5]-4) produced TON of 2.6 ± 0.6 at a rate of 0.039 mmol H2 per ggel. As expected, the nanoparticle structure of catalyst 4 ensured excellent physical immobilization in the PMI–SA network with negligible catalyst loss. By using the higher catalyst loadings in PMI–SA[2.5]-3 and PMI–SA[2.5]-4, these gels produced a volume of H2 that is comparable to PMI–SA[2.5]-1, despite generating low TON (Table S1, ESI†). Overall, PMI–SA-2 outperformed the samples containing other catalysts both in terms of total volume of H2 produced and TOF. These results show that the PMI–SA system can be used to physically immobilize a number of different catalysts. We propose that the hydrogel effectively “heterogenizes” the otherwise homogeneous catalysts and co-localizes them with our supramolecular photosensitizer to enable better transfer of electrons among the components. This characteristic of the PMI–SA gels enabled the gels to rival HER rates of solution-based systems12 despite having slower diffusion rates.
400 ± 2160 h−1 at a rate of 0.55 mmol H2 per ggel, nearly twice the rate observed with catalyst 1. Using Ru(bpy)3Cl2 or PMI-PDDA as control photosensitizers, catalyst 2 also displayed higher activity than catalyst 1, indicating that the dramatically enhanced HER resulted from a more efficient catalyst (Table S1, ESI†). ICP-MS analysis showed only a 4.82% loss of Mo from the PMI–SA[2.5]-2 gel, indicating significant immobilization within the matrix (Fig. 2B). Dispersing insoluble but active catalyst 2 in the hydrogel heterogenizes the otherwise molecular catalyst while still maintaining excellent exposure to the aqueous environment. In addition, the cationic charge can interact with the carboxylate groups on both the PMI and the SA. PMI–SA containing catalyst 3 (PMI–SA[2.5]-3) reached a TON of 142 ± 28 at a rate of 0.054 mmol H2 per ggel, and ICP-MS showed a 10.28% loss of Mo from the gel indicating good cluster immobilization. We expect that catalyst 3 can be heterogenized due to its low solubility in the AA solution. PMI–SA[2.5] containing catalyst 4 (PMI–SA[2.5]-4) produced TON of 2.6 ± 0.6 at a rate of 0.039 mmol H2 per ggel. As expected, the nanoparticle structure of catalyst 4 ensured excellent physical immobilization in the PMI–SA network with negligible catalyst loss. By using the higher catalyst loadings in PMI–SA[2.5]-3 and PMI–SA[2.5]-4, these gels produced a volume of H2 that is comparable to PMI–SA[2.5]-1, despite generating low TON (Table S1, ESI†). Overall, PMI–SA-2 outperformed the samples containing other catalysts both in terms of total volume of H2 produced and TOF. These results show that the PMI–SA system can be used to physically immobilize a number of different catalysts. We propose that the hydrogel effectively “heterogenizes” the otherwise homogeneous catalysts and co-localizes them with our supramolecular photosensitizer to enable better transfer of electrons among the components. This characteristic of the PMI–SA gels enabled the gels to rival HER rates of solution-based systems12 despite having slower diffusion rates.
 | ||
| Fig. 2 (A) Rates of hydrogen evolution for PMI–SA[2.5] mixed with different catalysts measured after 18 hours of light irradiation. (B) Percentage of each catalyst in the solution for the samples in Panel A after irradiation measured by ICP-MS. | ||
3.3 Optimizing hydrogel macrostructure and microstructure
Having established that the catalysts can be immobilized within the PMI–SA hydrogel matrix, we investigated the effect of the gel macrostructure on photocatalysis. PMI–SA gels have a range of submicron pores (Fig. 1D) that slow the diffusion of small molecules like AA and its oxidized byproduct DHA, which can parasitize electron transfer from AA.47–49 A concentration gradient will form in the hydrogel as a function of the distance from hydrogel surface when reactant is consumed during photocatalysis at steady state, forming an area of minimum reaction rate in the gel interior.50 The use of SA enables control of diffusion in the gel through two parameters: mass-transfer distance and mass-transfer speed. The former can be controlled by changing the extrusion nozzle of gelation. Thinner gels will have shorter diffusion lengths and result in smaller reaction rate gradients. To probe the effects of mass-transfer distance, we generated PMI–SA[2.5] gels extruded using different diameters ranging from 0.191 mm to 1.372 mm (Fig. S7, ESI†). Care was taken to ensure that a similar mass of gel was used for all conditions and that a sufficiently small mass of gel was used to avoid gels on the bottom of the vial blocking light from penetrating the rest of the vial. All gels experienced a loss of mass upon gelation and the removal of excess surface water with greater loss of mass at smaller extrusion diameters. Because TON values were calculated using gel mass, these swelling differences were analyzed and accounted for (see Methods section of ESI,† and Fig. S8).As expected, increasing the gel diameter led to a decrease in HER efficiency regardless of the catalyst used. Fig. 3A and B shows that the trend between gel diameter and TON was nearly linear for PMI–SA[2.5]-1 and PMI–SA[2.5]-2, with both showing increases in TON over 300% as diameter decreased from 1.372 mm to 0.191 mm. This result indicates clearly how internal mass transfer can limit hydrogel photocatalysis at a macroscopic length scale. PMI–SA[2.5]-3 also showed diminished photocatalysis with increasing gel diameter, but the trend was more variable, making it much harder to establish a clear relationship between extrusion nozzle diameter and H2 production (Fig. 3C). The PMI–SA[2.5]-4 samples also showed a linear trend where TON increased by almost 400% as the nozzle diameter was decreased from 1.372 mm to 0.191 mm (Fig. 3D). This steeper increase in TON relative to the other gels may be the result of added light scattering and absorbing from the black MoS3 nanoparticle. Lower HER due to decreased light penetration would be exacerbated at thicker gel diameters. These massive increases in HER were achieved by simply decreasing the gel diameter without any change to the composition of the material.
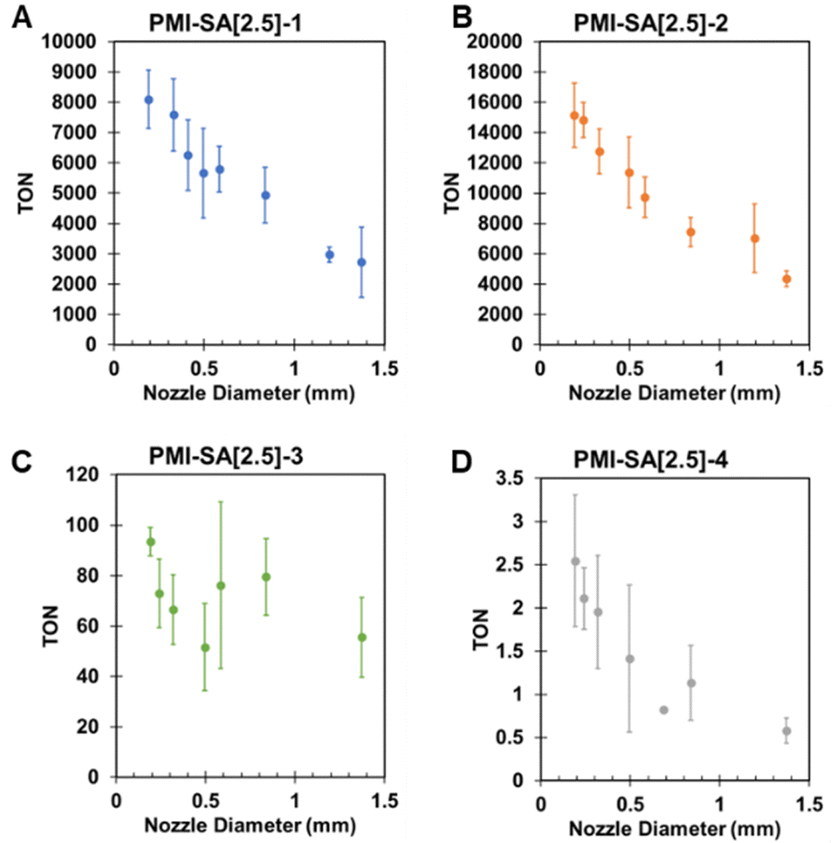 | ||
| Fig. 3 TON values for PMI–SA[2.5] extruded from different nozzle diameters with gels containing catalysts 1 (A) 2 (B) 3 (C) and 4 (D) exposed to light for 18 hours (nozzle diameters used ranged from 0.2 mm to 1.37 mm). | ||
Visually, PMI–SA appears more opaque than pure SA due to light scattering by the long PMI ribbons. The addition of light-scattering nanoparticles like catalyst 4 further limits light penetration. Maximum photocatalytic efficiency is dependent on light reaching all catalytic sites within the gel, which is a function of the gel diameter. To characterize light penetration in PMI–SA material, we collected UV-vis absorption of SA solution with varying concentrations of PMI-L5. The largest absorption peak in the visible region PMI–SA solution is 442 nm and a PMI–SA material with 9.6 mM PMI-L5 absorbs 99% of light at this wavelength at a thickness of 1.83 mm. A second absorption peak at 505 nm corresponds to the highest quantum efficiency of PMI-L5 and PMI–SA with 9.6 mM PMI-L5 absorbs 99% of light at this wavelength at a thickness around 4 mm (Fig. S9, ESI†).11 Some loss of photocatalytic efficiency is expected with increasing gel thickness, but it is difficult to decouple the effects of mass-transfer distance. However, we should be providing an excess of photons relative to the number of PMI-L5 molecules despite the low internal quantum efficiency of PMI-L5 for HER.
Seeking to probe the effects of mass-transfer speed and potentially decouple the effects of light penetration from gel thickness, we tested PMI–SA with variable SA loading. SEM showed that a gel formed from 1 wt% SA looked slightly more porous than gels formed with higher loadings of polymer, but these differences were difficult to visualize (Fig. S10, ESI†). To quantify these differences, we used fluorescence recovery after photobleaching with fluorescein as a model anion for ascorbate. The results showed that the diffusion time (t1/2) of 10.0 ± 1.8 s in solution could be slowed to 12.2 ± 0.4 s in a 1.0 wt% SA gel. Additional loading of SA increases t1/2 until a plateau of around 13.4 ± 0.4 s diffusion speed at 3 wt% (Fig. S10, ESI†). This difference represents up to a 35% decrease in diffusion speed relative to the solution state. Even small changes in diffusion rates can lead to “dead zones” of inefficient photocatalysis due to rapid local consumption of reactants and efficient delivery of fresh reactants.51 We therefore expected that increasing the SA loading of the PMI–SA gel would slow the diffusion of AA reactant by increasing the density of the resulting gel and thus diminish photocatalysis.52 Having established some effect of SA loading on diffusion speed, we prepared PMI–SA-1 and PMI–SA-2 with SA loading ranging from 1 wt% to 4 wt% which were extruded through nozzles with diameters of 0.191 mm or 0.584 mm.
The photocatalysis revealed the surprising result that at constant extrusion diameter, increased SA loading could produce enhanced photocatalysis rates. For the PMI–SA[X]-1 extruded from a 0.2 mm nozzle, TON increased from 3570 ± 609 at 1 wt% SA up to 5660 ± 374 at 3 wt% SA, with a higher loading of 4 wt% producing lower TON at 4780 ± 628 (Fig. 4A). We hypothesize that the peak in HER at 3 wt% SA represents the point at which the benefit of catalyst entrapment begins to diminish due to slower reactant diffusion. The PMI–SA[X]-2 samples extruded from a 0.191 mm nozzle also displayed enhanced HER at higher SA loading with a consistent increase in TON from 8360 ± 1880 at 1 wt% SA to 17600 ± 3280 with 4 wt% SA (Fig. 4B). Given the nearly complete entrapment of catalyst 2 in PMI–SA gels, it is unlikely that the increasing HER is the result of better catalyst immobilization. It is instead hypothesized that there is a beneficial interaction between the SA and catalyst 2 that enhances photocatalysis. Enhanced photocatalysis due to polymer–catalyst interactions has been determined to result from the prevention of aggregation deactivation, enhanced solubilization of otherwise insoluble catalysts, stabilization of catalyst intermediates by polymer functional groups (e.g. hydroxyls), and greater localization near photosensitizers.18,53,54 To determine if these beneficial interactions were the result of SA itself or the SA in the gel state, we performed photocatalysis on solution state PMI–SA-2 at various SA loadings. To avoid gelation, we simply avoided adding CaCl2 to the AA catalysis solution and agitated the PMI–SA ink until dispersed. The ungelled samples also displayed an enhancement in HER, increasing from TON of 14900 ± 2950 at 1 wt% SA to TON of 24100 ± 360 at 4 wt% SA (Fig. S11, ESI†). Despite a lack of a gelator, the PMI–SA still formed small precipitates in solution after agitation. While further testing is required to elucidate the exact nature of this interaction, it is apparent that catalyst 2 benefits from interaction with SA polymer beyond immobilization in a fully gelled hydrogel matrix. We hypothesize that catalyst 2 and the PMI assemblies remain non-covalently adsorbed to the SA polymer even after dispersion in water, providing beneficial ligand interactions and preventing aggregation deactivation.
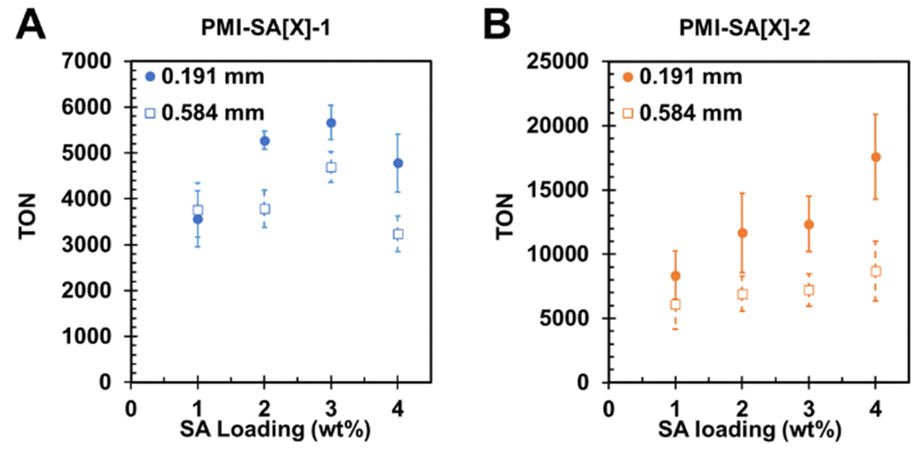 | ||
| Fig. 4 H2 TON for (A) PMI–SA-1 and (B) PMI–SA-2 prepared with different SA loading and extruded from different nozzle diameters after 18 hours of illumination. | ||
PMI–SA-1 and PMI–SA-2 at varying SA loading were also extruded through a larger 0.6 mm diameter nozzle and compared to the previous samples extruded through the smaller nozzle (Fig. 4A and B). We found that at SA loading of 1 wt%, the HER of PMI–SA[X]-1 samples was independent of the nozzle diameter, indicating that the faster diffusion in these gels dominates the photocatalysis behavior rather than the mass-transfer distance. At SA loadings above 1 wt%, the thinner PMI–SA[X]-1 gels displayed higher HER rates relative to the thicker gels (Fig. 4). While the rate of catalyst loss from the gel should be unaffected by the gel size, the thinner gels do elute more catalyst within a defined time due to the shorter distance to the solution (Fig. S4C, ESI†).
At all SA loadings, the thinner 0.2 mm PMI–SA[X]-2 gels showed enhanced HER relative to the thicker 0.6 mm gels. However, the increase of TON was within error at SA loading of 1 wt%. Similar to the previous test, the 0.6 mm gels demonstrated increased HER as SA loading was increased, though the effect was not as dramatic as in the thinner gels. The 0.2 mm gels showed an increase in TON of 2300 per 1 additional wt% SA, while the 0.6 mm gels showed a smaller increase of 650 per additional 1 wt% SA. Also noteworthy was that the difference between the HER rate of the thinner gels and the thicker gels became larger as SA loading increased, demonstrating around two-fold improvement at 4 wt% SA (Fig. S7, ESI†). These results confirm that the reduction of gel diameter improves photocatalytic rate through decreased mass transfer distance and that while diffusion can limit hydrogel photocatalysis systems, structural optimization enables gels to approach the efficiency of solution-state systems.
3.4 Long-term reusability
In previous studies using PMI and catalyst 1, PMI-PDDA hydrogels were capable of reaching TON 13500 over 108 hours of illumination while solution-based PMI assemblies were capable of reaching TON over 20000 under the same conditions.11,12 However, in these systems, the rate of HER began to slow after 36 hours of illumination. We observed that PMI–SA[2.5]-2 gels could also produce H2 for up to 108 hours, with a similar slowing in HER after 36 hours of light exposure (Fig. S12, ESI†). When the headspace of vials was purged after each 18-hour illumination cycle to avoid a buildup of H2, a similar trend was observed (Fig. S12, ESI†). The buildup of DHA limits HER at longer time points due to parasitic back reactions with AA. To test the effects of DHA buildup, PMI–SA[2.5] gels extruded through 0.191 mm diameter nozzles were utilized for consecutive 18-hour illumination cycles. At the end of each measurement, the used AA solution was replaced with fresh AA and the vial was purged again with Ar gas to return the system to the initial conditions at the beginning of each cycle. This method was only made possible through the mechanical stability of the PMI–SA gel. TON values are provided in the SI but HER is given in terms of mmol H2 per ggel to enable better comparison between catalysts.
After six exchanges over 108 hours, PMI–SA[2.5]-1 had produced 30900 TON or 1.1 mmol H2 per ggel, but the rate of HER dropped after each cycle, leading to an 87% loss in HER over 108 hours (Fig. 5A). We hypothesized that diminishing catalysis was due to both catalyst deactivation as well as catalyst leakage. ICP-MS of the used AA solution revealed a 54% total loss of Mo from the gels over six cycles, with most of the catalyst loss occurring during the first cycle. While taking catalyst loss into account stabilizes the HER rate of the gels, the overall loss of HER hinders the long-term reusability of this material.
 | ||
| Fig. 5 (A) HER rate for PMI–SA[2.5] gels extruded from 0.191 mm diameter nozzles after 18 hours of illumination for six consecutive photocatalysis cycles where the solution was replaced with fresh AA and the vial was purged with Ar and underwent standard photocatalysis conditions. (B) Concentration of each Mo catalyst in the solution measured by ICP-MS after each photocatalysis cycle. | ||
PMI–SA[2.5]-2 reached TON of 66700 or 2.3 mmol H2 per ggel after six exchanges (Fig. 5A), which is 33% greater than the TON reached by PMI–SA[2.5]-2 without any AA exchange. Despite a relatively small catalyst loss of 20%, the rate of HER dropped by almost 75% of its original value after six exchanges (Fig. 5B), indicating catalyst decomposition. This could also explain the small but consistent loss of Mo to the solution. We did not observe any loss of HER activity in PMI–SA[2.5]-2 gels stored in deionized water under air for one week (Table S2, ESI†). This observation suggests that while catalyst 2 is stable in PMI–SA gels when exposed to O2 and H2O, it is likely that either the acidic conditions or the process of photocatalysis leads to catalyst deactivation. Dithiocarbamate salts can be protonated under acidic conditions, leading to decomposition into amines and carbon disulfide.55 However, further studies are required to confirm the true nature of catalyst 2 decomposition in these samples. In an attempt to better stabilize the PMI–SA-2 gels, we performed photocatalysis at a higher pH, but the rate of HER was severely diminished above pH 4 (Fig. S13, ESI†). PMI–SA[2.5]-3 produced TON of around 130 or 0.048 mmol H2 per ggel over the first two cycles but experienced diminished HER over time such that by cycle 6 the TON was below 54 or 0.02 mmol H2 per ggel. ICP-MS data shows a significant 26% loss of catalyst during the first cycle. However, the consistency of HER over the first two cycles indicates that this initial loss of Mo was from catalysts that were loosely bound and did not participate in photocatalysis (Fig. 5). Subsequent exchanges saw minimal loss of Mo, suggesting that diminishing HER was likely the result of catalyst deactivation rather than catalyst loss.37 When PMI–SA[2.5]-3 was stored in deionized water with air headspace HER dropped substantially, indicating catalyst deactivation in aqueous conditions that are not highly acidic (Table S2, ESI†).
PMI–SA[2.5]-4 demonstrated small but consistent HER, generating an average of 1.34 TON or 0.02 mmol H2 per ggel over six cycles with negligible loss of catalyst. This nanoparticle catalyst demonstrates the potential for long-term reusability of a hydrogel photocatalysis system when coupled with a stable catalyst. PMI–SA-4 could also be stored in deionized water with air headspace for a week and be used with negligible loss of photocatalytic efficiency (Table S2, ESI†). In these reusability studies, the catalysts used have been the limiting factor in long-term stability.
3.5 PMI–SA photocathodes
The elimination of sacrificial electron donors that act as a replacement for the oxidation half-reaction of the water-splitting reaction will enable a greater range of conditions to be explored. There have been previous studies applying perylene monoimide type photosensitizers to cathodes in order to perform the HER.45,56,57 We hypothesized that the supramolecular assemblies of PMI may be utilized as both a light-absorbing and charge-transferring layer in a photocathode device. This possibility is based on the fact that supramolecular assemblies formed by π–π stacking of aromatic chromophores are known to result in soft solids that exhibit electrical conductivity.20,58 The incorporation of the SA polymer is critical to creating PMI photocathodes, as the pure PMI gels would not be mechanically strong enough to form a stable gel layer. By modifying a method for 3D-printing SA, we prepared a PMI–SA ink for extrusion-based 3D printing to prepare hydrogel photoelectrodes with control over thickness and porosity. PMI–SA ink with 3 wt% SA was partially gelled by mixing with 20 mM CaCl2, converting the PMI–SA[3] ink from a viscous solution to a self-standing material (Fig. S15, ESI†).37 Once deposited on the substrate, the PMI–SA[3] gel could be securely adhered through complete gelation with CaCl2.To measure the bulk conductivity of the gel material, chronoamperometry was performed on PMI–SA[3] gel discs roughly 0.13 mm in thickness sandwiched between two conductive FTO substrates separated by a 0.13 mm silicone spacer. While illuminated by a 5000 K white LED, the gel displayed electrical conductivity of around 33 MΩ cm (Fig. S14, ESI†). While this conductivity is on the low end of conductive polymers, it is important to remember that this material managed to conduct electrons through a bulk hydrogel at only 1 wt% PMI loading.59 Further evidence of photoconductivity in PMI–SA gels and intrinsic electrical conductivity in PMI assemblies was demonstrated using electrochemical impedance spectroscopy (Fig. S14, ESI†). A visual indication of electron transfer from FTO to PMI–SA was made using a PMI–SA[3] gel printed on a bare FTO substrate. As shown in Fig. S16 (ESI†), a single layer of PMI–SA[3] experienced a distinct color change from red to green at applied voltages beyond −0.78 V vs. RHE. This color change is associated with the formation of the anion of the PMI core and occurs at applied voltages more negative than the LUMO of PMI.60,61 The PMI–SA gel reverted to red once the applied voltage was made more positive, indicating the extraction of an electron from the PMI. It is important to note that the color change appears to take place throughout the bulk of the hydrogel, which has a thickness of at least 100 μm, indicating electron transport beyond the gel–electrode interface. Reversible electrical reduction of supramolecular perylene bisimide has been demonstrated in the dried state and reversible photoreduction has been demonstrated in the hydrogel state.62,63
We chose to use the PMI–SA[3]-4 system for our photoelectrode studies because of their high stability and good catalyst immobilization. The ink was printed as a 1 cm2 square using a tapered tip with a diameter of 0.191 mm. The resulting electrode had filaments roughly 400 μm in diameter, establishing an area coverage of around 0.42 cm2 for the porous electrode (PE) containing one layer (PE1), and around 0.73 cm2 for the porous electrode with two (PE2) and three layers (PE3) (Fig. 6B). We were able to print up to 5 layers without loss of resolution and the ink was stiff enough to bridge the gaps between filaments. The non-porous bulk electrodes (BE) were printed so that the filaments were all ordered in the same direction in each layer (Fig. 6B). With each additional layer, an increase in opacity is apparent, but the light intensity of our LED was high enough to ensure illumination throughout the gel. The substrate used was an FTO-coated glass slide with a thin, dense layer of NiO to act as a hole-accepting layer.64,65 FTO-NiO produces negligible photocurrent, so any photocurrent can be assumed to be the result of PMI–SA (Fig. S17, ESI†). In these experiments testing our materials as photoelectrodes, the buffer electrolyte was adjusted to pH 6 and had an ionic strength of 1.5 M containing 160 mM CaCl2. A platinum wire was used as the counter electrode, and all measurements were referenced to an Ag/AgCl (3 M NaCl) electrode. The system was kept under Ar atmosphere for HER experiments, and illumination was provided by a 5000 K white light LED.
 | ||
| Fig. 6 (A) Schematic diagram of the photoelectocatalysis setup used in experiments. (B) photograph of the non-porous bulk electrodes (BE) and porous electrodes (PE) printed with thicknesses of 1, 2, or 3 layers. (C) CV of a single layer of PMI–SA[3]-4 printed on FTO-NiO under both dark and illuminated conditions. (D) Values of the measured photocurrent at +0.1 V vs. RHE for PMI–SA[3]-4 of printed BE and PE with 1 to 3 layers. | ||
When the photocathodes were tested using cyclic voltammetry (CV), reversible peaks appeared around +0.5 V vs. RHE and were attributed to Ni2+/Ni3+ (Fig. 6C). The electrodes also began to display photocurrent at around +0.5 V vs. RHE, the magnitude of which increases slightly with more negative voltages. The representative CV in Fig. 6C from a PE3 electrode displayed a modest photocurrent beginning at around +0.5 V vs. RHE, peaking at a magnitude of −0.6 μA cm−2 at −0.15 V vs. RHE. We attribute photocurrent to the photosensitization of catalyst 4 for HER. However, we were unable to measure H2 in the headspace due to the poor seal in our electrochemical cell and the low current density. Regardless, the PMI–SA[3]-4 photocathode was capable of producing constant negative current under an applied voltage of −0.21 V vs. RHE, indicating minimal decomposition of PMI, catalyst, or electrode (Fig. S18, ESI†). We noticed that when air leaked into an argon-purged cell, the current and the photocurrent of the PMI–SA photocathode increased. Purging the cell with argon gas caused the currents to return to their original values confirming the role of atmospheric gases. We later confirmed through colorimetric assay that the electrode was reducing O2 into H2O2, which has been shown previously with a perylene monoimide sensitized NiO cathode.66 However, the faradaic efficiency of H2O2 production was low, possibly as a result of homogeneous H2O2 decomposition, H2O2 decomposition by the platinum counterelectrode, and spontaneous oxidation of SA polymers by H2O2.67
Assuming that the entire gel is electroactive, it would be expected that increasing the thickness of the electrode would result in an increase in overall current and potentially photocurrent as more active sites were added. However, beyond a given thickness, increased resistance of the material would result in diminished current and thus inactive material closer to the gel–solution interface that would impair reactions closer to the electrode–gel interface by slowing mass transfer. Thickness limitations on photocurrent have been demonstrated in the literature using several kinds of photocathode materials; however, the optimal thickness has been limited to less than a few microns by light penetration and slow charge transport.57,68–70 We see this effect in our BE samples where photocurrent values begin to diminish beyond thicknesses of two layers, indicating a tradeoff between resistance and active surface area. However, in the PE samples, the average photocurrent increases slightly with added layers of photocatalytic hydrogel. The main limitation of this system is its poor electrical conductivity, and improvements to gel conductivity through the inclusion of dopants or conductive polymers could amend this problem in future work. However, these results have shown that printing can be used to enhance (photo)electrocatalysis of the hydrogels investigated and demonstrate that additive manufacturing can be a useful tool to improve diffusion in electrode systems.
4. Conclusions
We have developed robust photocatalytic hydrogels for solar fuel production that encapsulate visible light harvesting supramolecular assemblies of a chromophore amphiphile and catalysts within a biopolymer matrix. The hydrogel material offers the advantage of functioning as a homogeneous photocatalysis system within the fluid compartments of a highly porous biopolymer matrix, while at the same time providing stability of a solid-state heterogeneous system through its catalyst immobilization and reusability. The rapid gelation of the SA component enabled us to investigate the effect of macroscopic morphology on photocatalysis, and we determined that even in a hydrogel material with high diffusivity, HER efficiency could be increased by over 300% by reducing mass transfer length. Finally, we provided proof of principle for the printing of the hybrid photocatalytic material on electrodes for use in photoelectrocatalysis. This work showed evidence for bulk electrical conductivity and for a photosensitized hydrogen evolution reaction near neutral pH. The use of this versatile approach is bioinspired by natural photosynthetic systems such as green plants and opens the opportunity to carry out light-to-fuel conversion within soft matter of any dimension that can be shaped and localized in a specific environment.Author contributions
J. E. K., Z. S., N. A. S., and S. I. S. conceptualized this research. J. E. K, Z. S., L. Đ., E. P. B., T. J. J., H. H. H., E. Q., K. S. W., A. J. D., N. A. S., and H. C. F. performed the investigation. J. E. K, Z. S., L. Đ., E. Q., K. S. W., A. J. D., N. A. S., and H. C. F. contributed to methodology development. J. E. K., L. Đ., and K. S. W. contributed to formal analysis. J. E. K., Z. S., L. Đ., E. P. B., T. J. J., H. H. H., and A. J. D. all contributed to resources primarily in terms of material synthesis. H. C. F., L. C. P., and S. I. S all contributed to resources. J. E. K. and Z. S. wrote the original draft. S. I. S. supervised this research and acquired funding. L. C. P. supported data curation and project administration. All: writing-review & editing.Data availability
The data supporting this article have been included within the ESI.†Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was primarily supported by the Center for Bio-Inspired Energy Science (CBES), an Energy Frontier Research Center (EFRC) funded by the U.S. Department of Energy, Office of Science, Basic Energy Sciences, under Award DE-SC0000989. Additional support for 3D printing was provided by the National Science Foundation (Grant DMR-2310178). This work made use of the Northwestern MatCI Facility supported by the MRSEC program of the National Science Foundation (DMR-1720139) at the Materials Research Center of Northwestern University. This work was also supported by the Air Force Research Laboratory under agreement number FA8650-15-2-5518. The U.S. Government is authorized to reproduce and distribute reprints for Governmental purposes notwithstanding any copyright notation thereon. The views and conclusions contained herein are those of the authors and should not be interpreted as necessarily representing the official policies or endorsements, either expressed or implied, of Air Force Research Laboratory or the U.S. Government. Work on the photoelectrodes was performed, in part, at the Center for Nanoscale Materials, a U.S. Department of Energy, Office of Science, Office of Basic Energy Sciences User Facility under Contract No. DE-AC02-06CH11357. Metal analysis was performed at the Northwestern University Quantitative Bio-element Imaging Center generously supported by NASA Ames Research Center NNA06CB93G with assistance from core manager Rebecca A. Sponenburg. SEM sample preparation in this work made use of the BioCryo facility of Northwestern University's NUANCE Center, which has received support from the SHyNE Resource (NSF ECCS-2025633), the IIN, and Northwestern's MRSEC program (NSF DMR-2308691). SEM imaging in this work made use of the EPIC facility of Northwestern University's NUANCE Center, which has received support from the SHyNE Resource (NSF ECCS-2025633), the IIN, and Northwestern's MRSEC program (NSF DMR-2308691). FTIR characterization in this work made use of the Keck-II facility of Northwestern University's NUANCE Center, which has received support from the SHyNE Resource (NSF ECCS-2025633), the IIN, and Northwestern's MRSEC program (NSF DMR-2308691). The authors are grateful to Shadden Zaki from the Stupp Laboratory for assisting with XRD of the catalysts.References
- C. Acar and I. Dincer, A review and evaluation of photoelectrode coating materials and methods for photoelectrochemical hydrogen production, Int. J. Hydrogen Energy, 2016, 41(19), 7950–7959 CrossRef CAS.
- K. N. Ferreira, T. M. Iverson, K. Maghlaoui, J. Barber and S. Iwata, Architecture of the Photosynthetic Oxygen-Evolving Center, Science, 2004, 303(5665), 1831–1838 CrossRef CAS PubMed.
- M. Sykora, K. A. Maxwell, J. M. DeSimone and T. J. Meyer, Mimicking the antenna-electron transfer properties of photosynthesis, Proc. Natl. Acad. Sci. U. S. A., 2000, 97(14), 7687–7691 CrossRef CAS PubMed.
- M. Supur and S. Fukuzumi, Photodriven Electron Transport within the Columnar Perylenediimide Nanostructures Self-Assembled with Sulfonated Porphyrins in Water, J. Phys. Chem. C, 2012, 116(44), 23274–23282 CrossRef CAS.
- A. S. Weingarten, R. V. Kazantsev, L. C. Palmer, M. McClendon, A. R. Koltonow, A. P. S. Samuel, D. J. Kiebala, M. R. Wasielewski and S. I. Stupp, Self-assembling hydrogel scaffolds for photocatalytic hydrogen production, Nat. Chem., 2014, 6, 964–970 CrossRef CAS PubMed.
- A. S. Weingarten, R. V. Kazantsev, L. C. Palmer, D. J. Fairfield, A. R. Koltonow and S. I. Stupp, Supramolecular Packing Controls H2 Photocatalysis in Chromophore Amphiphile Hydrogels, J. Am. Chem. Soc., 2015, 137(48), 15241–15246 CrossRef CAS PubMed.
- A. S. Weingarten, A. J. Dannenhoffer, R. V. Kazantsev, H. Sai, D. Huang and S. I. Stupp, Chromophore Dipole Directs Morphology and Photocatalytic Hydrogen Generation, J. Am. Chem. Soc., 2018, 140(15), 4965–4968 CrossRef CAS PubMed.
- R. V. Kazantsev, A. J. Dannenhoffer, T. Aytun, B. Harutyunyan, D. J. Fairfield, M. J. Bedzyk and S. I. Stupp, Molecular Control of Internal Crystallization and Photocatalytic Function in Supramolecular Nanostructures, Chem, 2018, 4(7), 1596–1608 CAS.
- A. Dannenhoffer, H. Sai, D. Huang, B. Nagasing, B. Harutyunyan, D. J. Fairfield, T. Aytun, S. M. Chin, M. J. Bedzyk, M. Olvera de la Cruz and S. I. Stupp, Impact of charge switching stimuli on supramolecular perylene monoimide assemblies, Chem. Sci., 2019, 10(22), 5779–5786 RSC.
- H. Sai, A. Erbas, A. Dannenhoffer, D. Huang, A. Weingarten, E. Siismets, K. Jang, K. Qu, L. C. Palmer, M. Olvera de la Cruz and S. I. Stupp, Chromophore amphiphile–polyelectrolyte hybrid hydrogels for photocatalytic hydrogen production, J. Mater. Chem. A, 2020, 8, 158–168 RSC.
- A. J. Dannenhoffer, H. Sai, B. Harutyunyan, A. Narayanan, N. E. Powers-Riggs, A. N. Edelbrock, J. V. Passarelli, S. J. Weigand, M. R. Wasielewski, M. J. Bedzyk, L. C. Palmer and S. I. Stupp, Growth of Extra-Large Chromophore Supramolecular Polymers for Enhanced Hydrogen Production, Nano Lett., 2021, 21(9), 3745–3752 CrossRef CAS PubMed.
- E. P. Bruckner, T. Curk, L. Đorđević, Z. Wang, Y. Yang, R. Qiu, A. J. Dannenhoffer, H. Sai, J. Kupferberg, L. C. Palmer, E. Luijten and S. I. Stupp, Hybrid Nanocrystals of Small Molecules and Chemically Disordered Polymers, ACS Nano, 2022, 16(6), 8993–9003 CrossRef CAS PubMed.
- O. Dumele, L. Đorđević, H. Sai, T. J. Cotey, M. H. Sangji, K. Sato, A. J. Dannenhoffer and S. I. Stupp, Photocatalytic Aqueous CO2 Reduction to CO and CH4 Sensitized by Ullazine Supramolecular Polymers, J. Am. Chem. Soc., 2022, 144(7), 3127–3136 CrossRef CAS PubMed.
- A. Thomas and M. Driess, Bridging the Materials Gap in Catalysis: Entrapment of Molecular Catalysts in Functional Supports and Beyond, Angew. Chem., Int. Ed., 2009, 48(11), 1890–1892 CrossRef CAS PubMed.
- J. Rodon Fores, M. Criado-Gonzalez, A. Chaumont, A. Carvalho, C. Blanck, M. Schmutz, C. A. Serra, F. Boulmedais, P. Schaaf and L. Jierry, Supported Catalytically Active Supramolecular Hydrogels for Continuous Flow Chemistry, Angew. Chem., Int. Ed., 2019, 58(52), 18817–18822 CrossRef CAS PubMed.
- J. Yoon, S. H. Lee, F. Tieves, M. Rauch, F. Hollmann and C. B. Park, Light-Harvesting Dye–Alginate Hydrogel for Solar-Driven, Sustainable Biocatalysis of Asymmetric Hydrogenation, ACS Sustainable Chem. Eng., 2019, 7(6), 5632–5637 CrossRef CAS.
- Z. Jiang, X. Zhang, G. Yang, Z. Yuan, X. Ji, F. Kong, B. Huang, D. D. Dionysiou and J. Chen, Hydrogel as a miniature hydrogen production reactor to enhance photocatalytic hydrogen evolution activities of CdS and ZnS quantum dots derived from modified gel crystal growth method, Chem. Eng. J., 2019, 373, 814–820 CrossRef CAS.
- J.-X. Jian, Q. Liu, Z.-J. Li, F. Wang, X.-B. Li, C.-B. Li, B. Liu, Q.-Y. Meng, B. Chen, K. Feng, C.-H. Tung and L.-Z. Wu, Chitosan confinement enhances hydrogen photogeneration from a mimic of the diiron subsite of [FeFe]-hydrogenase, Nat. Commun., 2013, 4(1), 2695 CrossRef PubMed.
- N. J. Hestand, R. V. Kazantsev, A. S. Weingarten, L. C. Palmer, S. I. Stupp and F. C. Spano, Extended-Charge-Transfer Excitons in Crystalline Supramolecular Photocatalytic Scaffolds, J. Am. Chem. Soc., 2016, 138(36), 11762–11774 CrossRef CAS PubMed.
- L. Đorđević, H. Sai, Y. Yang, N. A. Sather, L. C. Palmer and S. I. Stupp, Heterocyclic Chromophore Amphiphiles and their Supramolecular Polymerization, Angew. Chem., Int. Ed., 2023, 62(17), e202214997 CrossRef PubMed.
- A. Dannenhoffer, H. Sai, E. P. Bruckner, L. Đorđević, A. Narayanan, Y. Yang, X. Ma, L. C. Palmer and S. I. Stupp, Metallurgical alloy approach to two-dimensional supramolecular materials, Chem, 2023, 9(1), 170–180 CAS.
- L. Forni, Mass and heat transfer in catalytic reactions, Catal. Today, 1999, 52(2), 147–152 CrossRef CAS.
- M. D. L. M. Ballari, R. Brandi, O. Alfano and A. Cassano, Mass transfer limitations in photocatalytic reactors employing titanium dioxide suspensions: II. External and internal particle constrains for the reaction, Chem. Eng. J., 2008, 136(2), 242–255 CrossRef CAS.
- S. Dutta, X. Sui, H. Weissman, V. Kalchenko, H. D. Wagner and B. Rybtchinski, Composites of hydrophilic polymers and organic nanocrystals enable enhanced robustness, Polym. Adv. Technol., 2019, 30(10), 2549–2557 CrossRef CAS.
- C.-Y. Lee, A. C. Taylor, S. Beirne and G. G. Wallace, 3D-Printed Conical Arrays of TiO2 Electrodes for Enhanced Photoelectrochemical Water Splitting, Adv. Energy Mater., 2017, 7(21), 1701060 CrossRef.
- T. M. Benedetti, A. Nattestad, A. C. Taylor, S. Beirne and G. G. Wallace, 3D Printed Electrodes for Improved Gas Reactant Transport for Electrochemical Reactions, 3D Print. Addit. Manuf., 2018, 5(3), 215–219 CrossRef.
- J. Ahn, S. Lee, J. H. Kim, M. Wajahat, H. H. Sim, J. Bae, J. Pyo, M. Jahandar, D. C. Lim and S. K. Seol, 3D-printed Cu2O photoelectrodes for photoelectrochemical water splitting, Nanoscale Adv., 2020, 2(12), 5600–5606 RSC.
- Y. J. Kim, A. Lim, J. M. Kim, D. Lim, K. H. Chae, E. N. Cho, H. J. Han, K. U. Jeon, M. Kim, G. H. Lee, G. R. Lee, H. S. Ahn, H. S. Park, H. Kim, J. Y. Kim and Y. S. Jung, Highly efficient oxygen evolution reaction via facile bubble transport realized by three-dimensionally stack-printed catalysts, Nat. Commun., 2020, 11(1), 4921 CrossRef CAS PubMed.
- P. Liao, Y. Hu, Z. Liang, J. Zhang, H. Yang, L.-Q. He, Y.-X. Tong, J.-M. Liu, L. Chen and C.-Y. Su, Porphyrin-based imine gels for enhanced visible-light photocatalytic hydrogen production, J. Mater. Chem. A, 2018, 6(7), 3195–3201 RSC.
- W. Zha, Q. Ruan, L. Kong, X. Xi, M. A. Turgunov, W. Zhang, K. Chang and Z. Sun, A suspension-mimicking hydrogel-based n-type polymer photocathode for solar-driven water splitting, Cell Rep. Phys. Sci., 2022, 3(5), 100863 CrossRef CAS.
- W. Schmid and K. M. Picker-Freyer, Tableting and tablet properties of alginates: characterisation and potential for Soft Tableting, Eur. J. Pharm. Biopharm., 2009, 72(1), 165–172 CrossRef CAS PubMed.
- J. Kibsgaard, T. F. Jaramillo and F. Besenbacher, Building an appropriate active-site motif into a hydrogen-evolution catalyst with thiomolybdate [Mo3S13]2− clusters, Nat. Chem., 2014, 6, 248 CrossRef CAS PubMed.
- P. R. Fontenot, B. Shan, B. Wang, S. Simpson, G. Ragunathan, A. F. Greene, A. Obanda, L. A. Hunt, N. I. Hammer, C. E. Webster, J. T. Mague, R. H. Schmehl and J. P. Donahue, Photocatalytic H2-Evolution by Homogeneous Molybdenum Sulfide Clusters Supported by Dithiocarbamate Ligands, Inorg. Chem., 2019, 58(24), 16458–16474 CrossRef CAS PubMed.
- T. Shibahara, M. Yamasaki, G. Sakane, K. Minami, T. Yabuki and A. Ichimura, Syntheses and electrochemistry of incomplete cubane-type clusters with M3S4 cores (M = molybdenum, tungsten). X-ray structures of [W3S4(H2O)9](CH3C6H4SO3)4·cntdot·9H2O, Na2[W3S4(Hnta)3]·cntdot·5H2O, and (bpyH)5[W3S4(NCS)9]·cntdot·3H2O, Inorg. Chem., 1992, 31(4), 640–647 CrossRef CAS.
- T. F. Jaramillo, J. Bonde, J. Zhang, B.-L. Ooi, K. Andersson, J. Ulstrup and I. Chorkendorff, Hydrogen Evolution on Supported Incomplete Cubane-type [Mo3S4]4+ Electrocatalysts, J. Phys. Chem. C, 2008, 112(45), 17492–17498 CrossRef CAS.
- C.-H. Lee, S. Lee, Y.-K. Lee, Y. C. Jung, Y.-I. Ko, D. C. Lee and H.-I. Joh, Understanding the Origin of Formation and Active Sites for Thiomolybdate [Mo3S13]2− Clusters as Hydrogen Evolution Catalyst through the Selective Control of Sulfur Atoms, ACS Catal., 2018, 8(6), 5221–5227 CrossRef CAS.
- F. E. Freeman and D. J. Kelly, Tuning Alginate Bioink Stiffness and Composition for Controlled Growth Factor Delivery and to Spatially Direct MSC Fate within Bioprinted Tissues, Sci. Rep., 2017, 7(1), 17042 CrossRef PubMed.
- A. Martinsen, G. Skjåk-Bræk and O. Smidsrød, Alginate as immobilization material: I. Correlation between chemical and physical properties of alginate gel beads, Biotechnol. Bioeng., 1989, 33(1), 79–89 CrossRef CAS PubMed.
- S. Zhang, M. A. Greenfield, A. Mata, L. C. Palmer, R. Bitton, J. R. Mantei, C. Aparicio, M. Olvera de la Cruz and S. I. Stupp, A self-assembly pathway to aligned monodomain gels, Nat. Mater., 2010, 9(7), 594–601 CrossRef CAS PubMed.
- H. Gao, E. Khera, J.-K. Lee and F. Wen, Immobilization of Multi-biocatalysts in Alginate Beads for Cofactor Regeneration and Improved Reusability, J. Visualized Exp., 2016, 110, 53944 Search PubMed.
- Y. Chen, P. Li, J. Zhou, C. T. Buru, L. Đorđević, P. Li, X. Zhang, M. M. Cetin, J. F. Stoddart, S. I. Stupp, M. R. Wasielewski and O. K. Farha, Integration of Enzymes and Photosensitizers in a Hierarchical Mesoporous Metal–Organic Framework for Light-Driven CO2 Reduction, J. Am. Chem. Soc., 2020, 142(4), 1768–1773 CrossRef CAS PubMed.
- V. Kunz, V. Stepanenko and F. Würthner, Embedding of a ruthenium(II) water oxidation catalyst into nanofibers via self-assembly, Chem. Commun., 2015, 51(2), 290–293 RSC.
- K. Gottschling, G. Savasci, H. Vignolo-González, S. Schmidt, P. Mauker, T. Banerjee, P. Rovó, C. Ochsenfeld and B. V. Lotsch, Rational Design of Covalent Cobaloxime–Covalent Organic Framework Hybrids for Enhanced Photocatalytic Hydrogen Evolution, J. Am. Chem. Soc., 2020, 142(28), 12146–12156 CrossRef CAS PubMed.
- M. Yin, F. Jia, F. Qiao, P. Zheng, W. Zhang and Y. Fan, Facile wet-chemical synthesis and efficient photocatalytic hydrogen production of amorphous MoS3 sensitized by Erythrosin B, Mater. Charact., 2017, 128, 148–155 CrossRef CAS.
- K. A. Click, D. R. Beauchamp, Z. Huang, W. Chen and Y. Wu, Membrane-Inspired Acidically Stable Dye-Sensitized Photocathode for Solar Fuel Production, J. Am. Chem. Soc., 2016, 138(4), 1174–1179 CrossRef CAS PubMed.
- A. Bibi, S.-U. Rehman and A. Yaseen, Alginate-nanoparticles composites: kinds, reactions and applications, Mater. Res. Express, 2019, 6(9), 092001 CrossRef CAS.
- J. Klein, J. Stock and K. D. Vorlop, Pore size and properties of spherical Ca-alginate biocatalysts, Eur. J. Appl. Microbiol. Biotechnol., 1983, 18(2), 86–91 CrossRef CAS.
- J. Øyaas, I. Storrø, H. Svendsen and D. W. Levine, The effective diffusion coefficient and the distribution constant for small molecules in calcium-alginate gel beads, Biotechnol. Bioeng., 1995, 47(4), 492–500 CrossRef PubMed.
- M. Golmohamadi and K. J. Wilkinson, Diffusion of ions in a calcium alginate hydrogel-structure is the primary factor controlling diffusion, Carbohydr. Polym., 2013, 94(1), 82–87 CrossRef CAS PubMed.
- H.-Y. Liu, C.-C. Lin and A. Diffusion-Reaction, Model for Predicting Enzyme-Mediated Dynamic Hydrogel Stiffening, Gels, 2019, 5(1), 17 CrossRef CAS PubMed.
- D. F. Ollis, Kinetic Disguises in Heterogeneous Photocatalysis, Top. Catal., 2005, 35(3), 217–223 CrossRef CAS.
- K. Yamagiwa, T. Kozawa and A. Ohkawa, Effects of Alginate Composition and Gelling Conditions on Diffusional and Mechanical Properties of Calcium-Alginate Gel Beads, J. Chem. Eng. Jpn., 1995, 28(4), 462–467 CrossRef CAS.
- L. Xie, J. Tian, Y. Ouyang, X. Guo, W. Zhang, U.-P. Apfel, W. Zhang and R. Cao, Water-Soluble Polymers with Appending Porphyrins as Bioinspired Catalysts for the Hydrogen Evolution Reaction, Angew. Chem., Int. Ed., 2020, 59(37), 15844–15848 CrossRef CAS PubMed.
- M. Cheng, M. Wang, S. Zhang, F. Liu, Y. Yang, B. Wan and L. Sun, Photocatalytic H2 production using a hybrid assembly of an [FeFe]-hydrogenase model and CdSe quantum dot linked through a thiolato-functionalized cyclodextrin, Faraday Discuss., 2017, 198(0), 197–209 RSC.
- E. Humeres, N. A. Debacher, M. M. D. S. Sierra, J. D. Franco and A. Schutz, Mechanisms of Acid Decomposition of Dithiocarbamates. 1. Alkyl Dithiocarbamates, J. Org. Chem., 1998, 63(5), 1598–1603 CrossRef CAS.
- R. J. Kamire, M. B. Majewski, W. L. Hoffeditz, B. T. Phelan, O. K. Farha, J. T. Hupp and M. R. Wasielewski, Photodriven hydrogen evolution by molecular catalysts using Al2O3-protected perylene-3,4-dicarboximide on NiO electrodes, Chem. Sci., 2017, 8(1), 541–549 RSC.
- S. Zhang, X. Li, K. Yun, F. Yu and J. Hua, Effects of Electrolytes on the Photocurrent of N-Annulated Perylene-Sensitized Photoelectrochemical Cells Based on NiO as Photocathode, ChemElectroChem, 2018, 5(21), 3198–3205 CrossRef CAS.
- C. L. Smith, L. L. E. Mears, B. J. Greeves, E. R. Draper, J. Doutch, D. J. Adams and A. J. Cowan, Gelation enabled charge separation following visible light excitation using self-assembled perylene bisimides, Phys. Chem. Chem. Phys., 2019, 21(48), 26466–26476 RSC.
- M. Solazzo, F. J. O'Brien, V. Nicolosi and M. G. Monaghan, The rationale and emergence of electroconductive biomaterial scaffolds in cardiac tissue engineering, APL Bioeng., 2019, 3(4), 041501 CrossRef PubMed.
- A. S. Jalilov, L. G. Nilewski, V. Berka, C. Zhang, A. A. Yakovenko, G. Wu, T. A. Kent, A.-L. Tsai and J. M. Tour, Perylene Diimide as a Precise Graphene-like Superoxide Dismutase Mimetic, ACS Nano, 2017, 11(2), 2024–2032 CrossRef CAS PubMed.
- E. Shirman, A. Ustinov, N. Ben-Shitrit, H. Weissman, M. A. Iron, R. Cohen and B. Rybtchinski, Stable Aromatic Dianion in Water, J. Phys. Chem. B, 2008, 112(30), 8855–8858 CrossRef CAS PubMed.
- E. R. Draper, J. J. Walsh, T. O. McDonald, M. A. Zwijnenburg, P. J. Cameron, A. J. Cowan and D. J. Adams, Air-stable photoconductive films formed from perylene bisimide gelators, J. Mater. Chem. C, 2014, 2(28), 5570–5575 RSC.
- E. R. Draper, R. Schweins, R. Akhtar, P. Groves, V. Chechik, M. A. Zwijnenburg and D. J. Adams, Reversible Photoreduction as a Trigger for Photoresponsive Gels, Chem. Mater., 2016, 28(17), 6336–6341 CrossRef CAS.
- A. Sápi, A. Varga, G. F. Samu, D. Dobó, K. L. Juhász, B. Takács, E. Varga, Á. Kukovecz, Z. Kónya and C. Janáky, Photoelectrochemistry by Design: Tailoring the Nanoscale Structure of Pt/NiO Composites Leads to Enhanced Photoelectrochemical Hydrogen Evolution Performance, J. Phys. Chem. C, 2017, 121(22), 12148–12158 CrossRef PubMed.
- P. Ho, S. Thogiti, L. Q. Bao, R. Cheruku, K.-S. Ahn and J. Hong Kim, Enhanced efficiency via blocking layers at photocathode interfaces in cobalt-mediated tandem dye-sensitized solar cells, Sol. Energy, 2018, 161, 9–16 CrossRef CAS.
- J. Sun, Y. Yu, A. E. Curtze, X. Liang and Y. Wu, Dye-sensitized photocathodes for oxygen reduction: efficient H2O2 production and aprotic redox reactions, Chem. Sci., 2019, 10(21), 5519–5527 RSC.
- X. Li, A. Xu, H. Xie, W. Yu, W. Xie and X. Ma, Preparation of low molecular weight alginate by hydrogen peroxide depolymerization for tissue engineering, Carbohydr. Polym., 2010, 79(3), 660–664 CrossRef CAS.
- T. Zhang, Y. Hou, V. Dzhagan, Z. Liao, G. Chai, M. Löffler, D. Olianas, A. Milani, S. Xu, M. Tommasini, D. R. T. Zahn, Z. Zheng, E. Zschech, R. Jordan and X. Feng, Copper-surface-mediated synthesis of acetylenic carbon-rich nanofibers for active metal-free photocathodes, Nat. Commun., 2018, 9(1), 1140 CrossRef PubMed.
- L. Yao, A. Rodríguez-Camargo, M. Xia, D. Mücke, R. Guntermann, Y. Liu, L. Grunenberg, A. Jiménez-Solano, S. T. Emmerling, V. Duppel, K. Sivula, T. Bein, H. Qi, U. Kaiser, M. Grätzel and B. V. Lotsch, Covalent Organic Framework Nanoplates Enable Solution-Processed Crystalline Nanofilms for Photoelectrochemical Hydrogen Evolution, J. Am. Chem. Soc., 2022, 144(23), 10291–10300 CrossRef CAS PubMed.
- B. P. G. Silva, B. Tosco, D. Z. de Florio, V. Stepanenko, F. Würthner, J. F. Q. Rey and S. Brochsztain, Efficient Electronic Coupling in Perylenediimide Multilayered Films on Indium Tin Oxide, J. Phys. Chem. C, 2020, 124(10), 5541–5551 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4sm00373j |
| This journal is © The Royal Society of Chemistry 2024 |