
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Microscopically segregated ligand distribution in co-assembled peptide-amphiphile nanofibers†
Turash
Haque Pial
 ab,
Yang
Li
bc and
Monica
Olvera de la Cruz
*abcde
ab,
Yang
Li
bc and
Monica
Olvera de la Cruz
*abcde
aDepartment of Materials Science and Engineering, Northwestern University, 2220 Campus Drive, Evanston, IL 60208, USA. E-mail: m-olvera@northwestern.edu
bCenter of Computation and Theory of Soft Materials, Northwestern University, 2145 Sheridan Road, Evanston, IL 60208, USA
cDepartment of Chemical and Biological Engineering, Northwestern University, 2145 Sheridan Road, Evanston, IL 60208, USA
dDepartment of Physics and Astronomy, Northwestern University, 2145 Sheridan Road, Evanston, IL 60208, USA
eDepartment of Chemistry, Northwestern University, 2145 Sheridan Road, Evanston, IL 60208, USA
First published on 27th May 2024
Abstract
Peptide amphiphiles (PAs) self-assemble into cylindrical nanofibers with applications in protein purification, tissue engineering, and regenerative medicine. For these applications, functionalized PAs are often co-assembled with oppositely charged filler PAs. Finding the conditions at which these fibers are homogeneously mixed or segregated is crucial for the required application. We co-assemble negative C12VVEE fillers and positive C12VVKK–OEG4–Z33 ligands, which are important for antibody purifications. Our results show that the ligands tend to cluster and locally segregate in the fiber surfaces. The Z33s are overall neutral and form large aggregates in bulk solution due to short range attractions. However, full segregation of the C12VVKK–OEG4–Z33 is not observed in the cylindrical surface due to the electrostatic penalty of forming large domains of similarly charged molecules. This is commensurate with previous theoretical predictions, showing that the competition between short-range attractive interactions and long-range electrostatic repulsions leads to pattern formation in cylindrical surfaces. This work offers valuable insight into the design of functionalized nanofibers for various biomedical and chemical applications.
Introduction
Peptide Amphiphiles are a class of molecules consisting of peptide heads connected to hydrophobic alkyl tails that can self-organize in aqueous solutions. This self-assembly behavior is often driven by the tendency of hydrophobic domains to bury away from water while hydrophilic peptide regions to expose to the solvent. The ability of peptide amphiphiles (PAs) to self-assemble into fibers, membranes, and micelles has been widely explored for achieving functions such as mineralization, cell proliferation, molecular imaging, drug delivery, and pH sensing.1–14 One of the critical advantages of PAs is their ability to be modified with specific ligands to achieve the desired functionality. For instance, PA nanofibers modified with ligands have shown potential in purifying antibodies.15,16 Therefore, understanding the distribution of ligands on the nanofiber surface is extremely important to optimize functionalization, thereby enabling their widespread application in biomedical research and therapeutics.Peptides can carry charge, and it has been observed that oppositely charged peptides can facilitate the co-assembly process of PAs with different peptides and epitopes. Co-assembly by mixing oppositely charged PAs is a powerful way to achieve increased functionalities. Niece et al.17 first reported the importance of opposite charges on the co-assembly of PAs functionalized with different bioactive sequences, of which each bearing a different biological signal. Recently, Li et al.15,16 showed the assembly of negatively charged C12VVEE (filler) and positively charged C12VVKK–OEG4–Z33 (ligand) to capture and purify IgG antibodies. Z33 is a 33 amino acid derivative (FNMQQQRRFYEALHDPNLNEEQRNAKIKSIRDD) of protein A18 which strongly binds to the Fc domain of the antibody.19 Their experiment suggests that aggregation of Z33 can reduce the purification efficiency as the steric effect may hinder Z33 from accessing and binding effectively with antibodies.15,16 As the demand for therapeutic antibodies continues to increase,20,21 it is crucial to understand the factors affecting the mechanism to recover antibodies. Here we exploit the co-assembly and distribution of positive ligands in a nanofiber made with negative filler Pas.
Phase segregation is a common phenomenon in amphiphilic supramolecular structures involving competitions among different driving forces.22–24 Careful truing the competing forces is an effective method to design nanomaterial architecture and achieve functions.25–27 To understand the physical origin of the supramolecular structure created by competing forces, previous theoretical works have analyzed the distribution of oppositely charged molecules absorbed on flat and cylindrical surfaces.28,29 The model suggested that oppositely charged molecules with different cohesive energy or compatibility can induce the formation of charged domains or aggregation of molecules on the surface.28 This is due to the competition between the line tension, and electrostatic energy. Line tension favors the growth of domains of segregated components of similar compatibility, while electrostatic repulsion increases rapidly as the surface charge density and the size of the domain increase. However, those models although can include the curvature of cylindrical fibers,28–30 lacked the complexity of real PAs and ligands. The possibility of small clusters or aggregation of molecules with similar compatibility and lack of homogeneity in ligand distribution is also suggested by stochastic optical reconstruction microscopy in Stupp group.31 Besides these works, there is a dearth of knowledge on understanding the co-assembly of oppositely changed PAs and ligands distribution in a co-assembled surface.
In this study, we use molecular dynamics simulations to co-assemble oppositely charged filler and ligand PAs. We use the capability of the Martini3 coarse-grained model to simulate large time and length scales. We have also employed all-atom simulation to verify ligand aggregation. We find that ligands come close to each other in the assembled fiber and form clusters on the surface. We discussed the importance of attractive interactions between ligands and electrostatics of the fiber surface in creating local segregation of ligands.
Results and discussion
C12VVEE is an ideal candidate for forming one dimensional self-assembled structure. C12 is the hydrocarbon hydrophobic core, V (valine) provides hydrogen bonds, hydrophobicity and E (glutamic acid) is negatively charged and hydrophilic. Our previous study shows that only the outer E of the PA is deprotonated or carries −1e charge32 in a self-assembled fiber. Keeping this in mind, we also model the inner glutamic acid as protonated. K (lysin) is used as positively charged group in our simulation in C12VVKK–OEG4–Z33 ligand. Z33 acts as an epitope in the ligand with OEG4 (oligoethylene glycol) helping as the spacer group. We used Martini 3 force field33 which averaged 2–4 heavy atoms into one bead and was extensively used in many amino acid simulations.The randomly added fillers and ligands initially form micelles and then assembled into a fiber. To differentiate the effect of Z33, we start our analysis with observing co-assembly of C12VVEE and C12VVKK. Interesting differences in the distribution of C12VVKK and C12VVKK–OEG4–Z33 can be seen from the snapshot of the assembled fibers (Fig. 1) where C12VVEE act as filler chains. C12VVKK are randomly distributed in the fiber (upper panel), while C12VVKK–OEG4–Z33 ligands come together and form ligands-rich regions. No distinct clusters of C12VVKK is observed in the upper panel, but we see C12VVKK–OEG4–Z33 ligands form clusters in the fiber surface.
 | ||
| Fig. 1 Simulation snapshots of the initial and co-assembled stages. Upper panel shows the distributions of lysin (red) and glutamic acid (green) amino acids in the assembled fiber. Lower panel shows the distribution of ligands in the fiber surface. A double alpha-helix structure of Z33 is shown in the inset. Water, ions, valines are omitted from the snapshots for better visualization. In both cases the ratio between ligand and filler are 1/10 and no extra salt is added except counterions. | ||
To better understand the distribution, we have calculated the cluster size distributions of Z33. The cut-off distance of Z33 beads was set to 0.7 nm when calculating the cluster; two Z33s are considered to form a cluster when they have at least one pair of beads closer than 0.7 nm. We chose 0.7 nm as the cut-off as it is the first valley of the radial distribution function of the Martini3 beads and is also suggested in previous studies as the optimal cut-off distance for amino acid cluster analysis.34,35 Additionally, we checked the cluster size distribution of C12VVKK for comparison (the simulation snapshot is shown in Fig. 1, upper panel). We used 1.1 nm as the cut-off distance between K of different PAs for this case, as it represents the second valley of the radial distribution function of the Martini3 beads and would consider two C12VVKK to form a cluster even if they are separated by one layer of C12VVEEs.
In Fig. 2 we plot the cluster size probability distribution for various ligand-filler ratios and two salt concentrations. Comparison between C12VVKK and C12VVKK–OEG4–Z33 (salt 0.0 mM) can be observed from Fig. 2(c). A quick drop in the probability value with increasing N shows that C12VVKK does not form large clusters, they stay separated from each other. Electrostatistically this is reasonable as positively charged groups prefer to mix with oppositely charged groups to minimize the electrostatic energy. On the other hand, larger clusters are observed when we add the Z33 ligands.
 | ||
| Fig. 2 Cluster size distribution. (a) A cartoon representation of the cluster size (N) definition. (b)–(d) shows the cluster size distribution probability for ligand-filler ratio of 1/25, 1/10 and 1/5 respectively. | ||
From Fig. 2(b)–(d), we can see that the probability of finding small clusters reduces with increasing the concentration of the ligand. More interestingly, we see a non-monotonic reduction of the cluster probability with increasing the number of ligands in a cluster, N arise. This non-monotonic cluster size distribution is associated with micellar aggregation.36–38 Srebnik and Douglous also observed this kind of cluster size distribution, where positive and negative coarse-grained beads were aggregating in a neutral nanotube surface.39 A small but finite probability for a very large cluster size indicates the formation of large clusters by transient bridges between more persistent smaller structures. The effect of salt can be easily understood from these plots. The addition of 100 mM salt results in smaller clusters of ligands for all ligand concentrations.
To clearly understand how ligands are distributed in a fiber surface, we have plotted the distribution of Z33 ligands by mapping them onto a plane. To do this, we calculated the angle each Z33 forms with the fiber center and the x-axis by taking each Z33's center of mass. We then plotted these angles against the axial position of each Z33 along the fiber, which gave us a rectangular visualization of their distribution on the cylindrical surface of the fiber. The mapping is depicted in a cartoon in Fig. 3 (top panel). Red arrows indicate new positions of Z33s in the mapped plane. We multiplied the visualization twice in the radial and axial directions to account for periodicity (Fig. 3(a)–(c)).
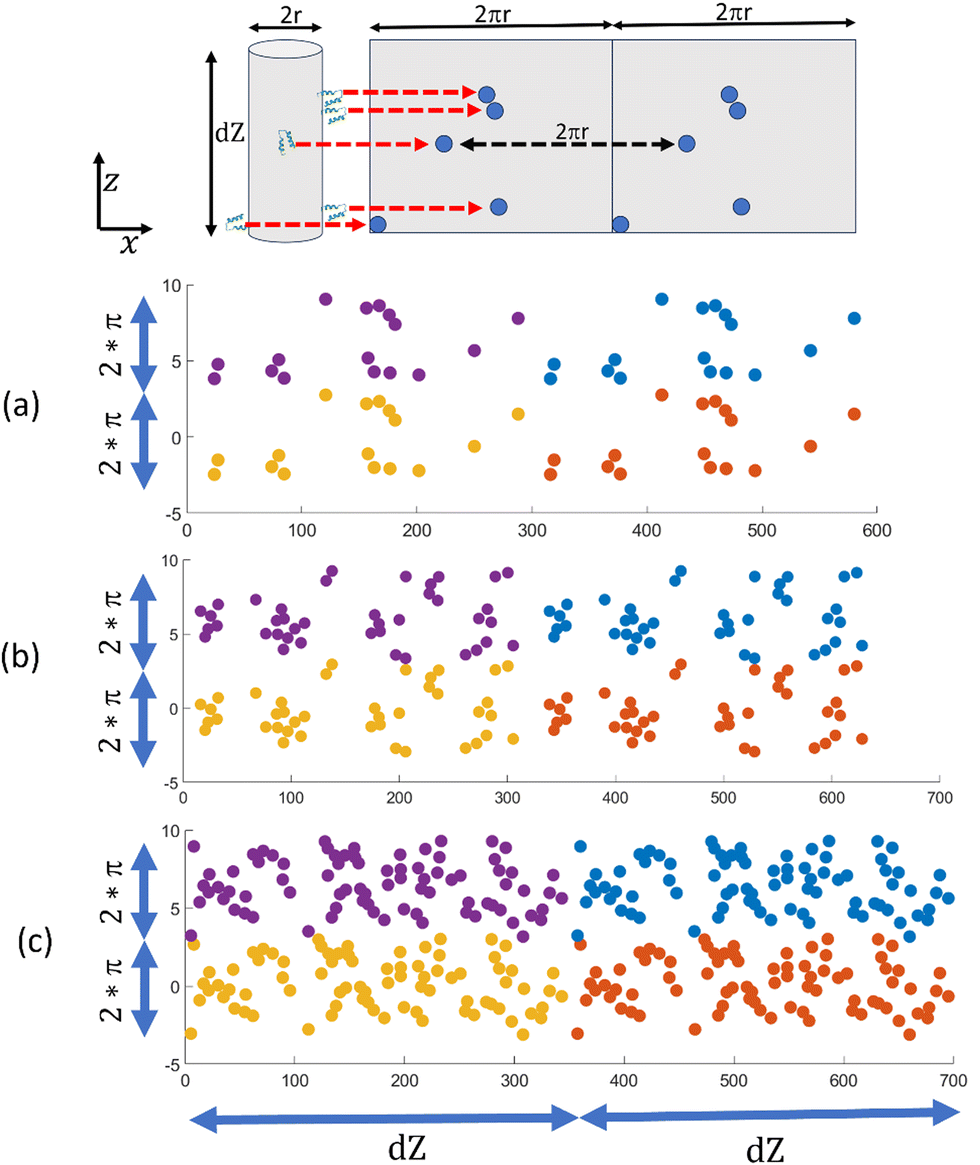 | ||
| Fig. 3 Top panel shows the cylinder (radius r) to plane (width 2πr) mapping. Red arrows indicate new positions of Z33s in the mapped plane. Mapped distribution is repeated twice in radial direction for periodicity. (a)–(c) show the mapped distribution for ligand-filler ratio of 1/25, 1/10 and 1/5 respectively. Four color indicates 4 periodic distributions. | ||
In Fig. 3, we have plotted the distribution of Z33 for ligand-filler ratios of 1/25, 1/10, and 1/5 (without any additional salt) in (a), (b), and (c), respectively. The plots clearly show that Z33s are forming clusters. What is more, all three distributions exhibit regions with high Z33 concentrations as well as areas without any Z33s. This is an example of local or microscopic segregation, which has been proposed in previous theoretical analyses.28–30 Recent studies also suggested the possibilities of similar segregation in other structures, like in charged polymer vesicles, polyelectrolyte brushes.40,41 Microscopic segregation arises from two competing forces: a short-range attractive force that encourages molecules to come close and form large-scale aggregates or macroscopic segregation, and a long-range repulsive force that opposes it. In the following sections, we will delve into the opposing factors in our case that could result in the local separation of ligands on the fiber surface.
To understand why we are seeing local segregation of Z33s or ligands, we analyzed how Z33 aggregates in the bulk. Randomly added Z33s aggregate in both Martini3 (Fig. 4(a)) and all-atom (Fig. 4(b)) force fields (we are showing two simulation boxes for better visualization). These simulations show that, in bulk solutions, Z33s form large aggregates or macroscopic segregation, suggesting strong attractive interactions between Z33s. Simulations with 100 mM added salt show a weaker aggregation behavior for both Martini3 and all-atoms (Fig. 4(c) and (d)); one or two Z33s stay separated from the aggregates. To quantify the aggregations for different cases, we calculated the number of nearest neighbors (NN) for each Z33. This number indicates how many Z33s are in close contact with another Z33 (0.7 nm is used as the cut-off). Fig. 4(e) shows the neighbor number distribution for Martini3, and all-atom have similar profiles, although Martini3 shows a slightly higher probability for large NN. A weaker aggregation with the addition of salt is apparent from the reduction of probability at higher NN. This weaker aggregation can describe the salt concentration-dependent cluster size distribution discussed in Fig. 2; the addition of salt reduces the probability of having a large cluster. This demonstrates the importance of controlling attractive interactions among ligands to regulate the cluster size in fiber surfaces.
 | ||
| Fig. 4 Aggregation of Z33 in bulk solution. (a)–(d) shows snapshots of Z33 aggregation for Martini3 (a) and (c) and all-atom simulation (b) and (d) with no added salt (a) and (b) and 100 mM salt (c) and (d). Two periodic images are shown for better visualization. (e) Nearest neighbor analysis for the simulations shown in (a)–(d). A cartoon representation is added to help understand the nearest neighbor definition. (f) Radial distribution function plot of positive and negative amino acids beads of Z33 in Martini3 simulations. Inset showing the positive (red) and negative (teal) amino acids. | ||
We know that protein aggregation in water is complicated and involves several factors, such as the interaction between hydrophobic amino acids, electrostatic interactions, hydrogen bonds, and the compactness of the protein. There are van der Waals and electrostatic energy gain associated with Z33 aggregation as some of the 33 amino acids are hydrophobic and nonpolar (see Fig. S2, ESI†), which prefer to stay away from polar water. Although the total charge of a Z33 is zero, some of the 33 amino acids are charged; positively charged amino acids are shown in red, and negatively charged amino acids are shown in teal color in the inset of Fig. 4(f). When the salt concentration is low, these positively charged and negatively charged amino acids form strong electrostatic interactions, leading to stronger attractive interactions. However, adding salt neutralizes some of the charged amino acids and reduces the electrostatic interactions, as seen in the radial distribution plot of positive and negative beads of the charged amino acids in Fig. 4(f). The addition of salt reduces the value of g(r) which indicates a less attractive electrostatic interaction between positive and negative amino acids at high salt concentrations.
We conducted three additional simulations to verify that the electrostatic penalty linked with large clusters of positively charged ligands in a negatively charged fiber might cause the local segregation of ligands in the fiber. We modified our original ligand from C12VVKK–OEG4–Z33 to C12VVEE–OEG4–Z33 by replacing the positive lysin (K) with negative glutamic acids (E) in an already co-assembled fiber. We have depicted the cylinder-to-plane mapping of the ligands in Fig. 5 using similar methodologies in as Fig. 3. We can see more connected ligand clusters are observed for all concentrations. No electrostatic penalty is associated with large clusters for these modified ligands, as all the molecules in the fiber now have similar charges. As a result, cluster formation is wholly driven by Z33–Z33 attractions, which can generate extensive, separated regions of ligand-rich and filler-rich surfaces.
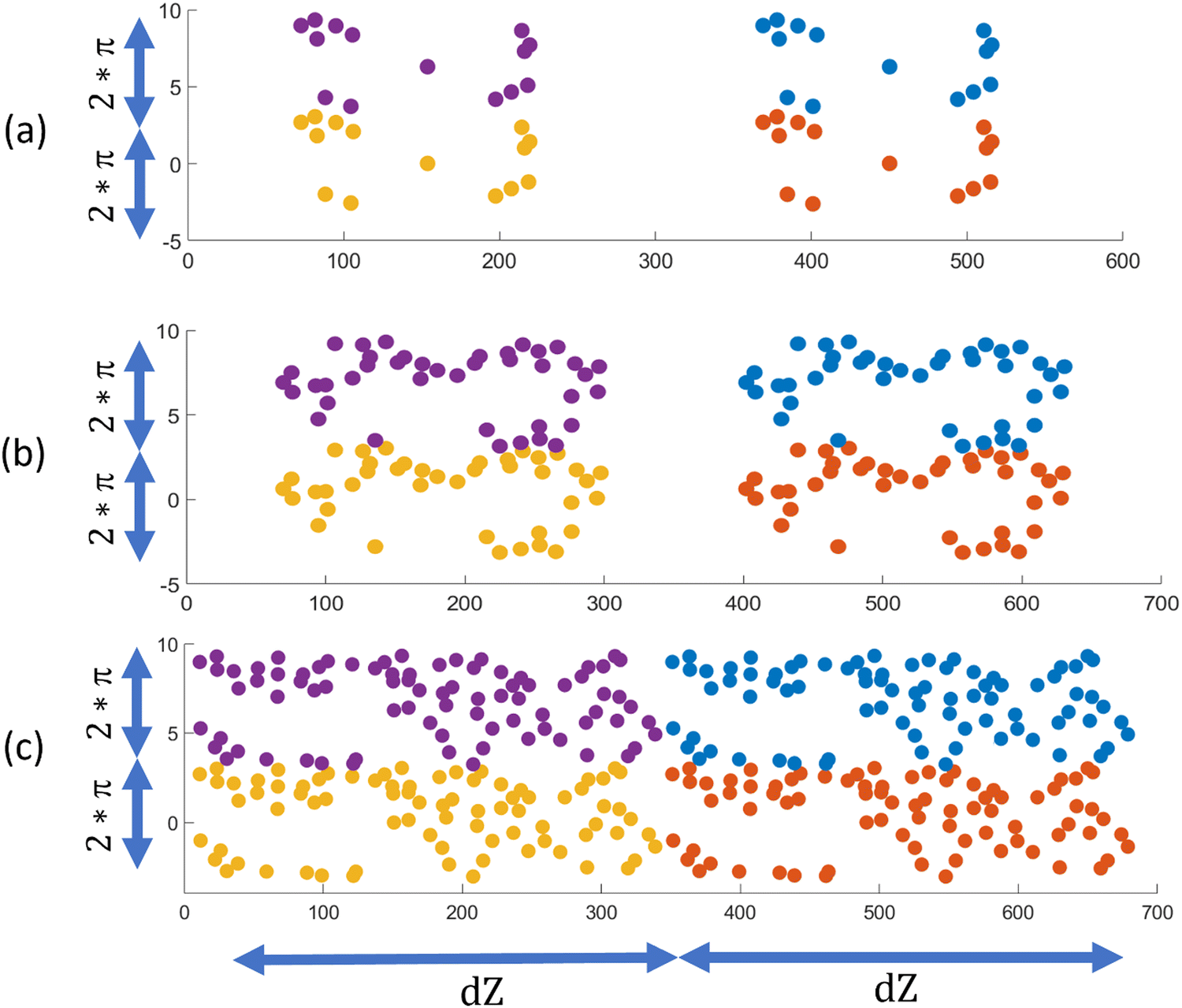 | ||
| Fig. 5 (a)–(c) shows the mapped C12VVEE–OEG4–Z33 ligands distribution for ligand-filler ratio of 1/25, 1/10 and 1/5 respectively. Four color indicates 4 periodic distributions. | ||
It is worth mentioning that we have performed our simulation with C12VVEE–OEG4–Z33 starting with a co-assembled structure to distinguish the effect of electrostatics in local segregation, we have not checked the co-assembly process itself. Because of very large differences in cohesive energy between C12VVEE and C12VVEE–OEG4–Z33, co-assembly should be less favorable without any added benefit coming from opposite charges. We have also observed some instability in the fiber when few ligands came close (Fig. S5(a), ESI†), although we did not observe any demixing of C12VVEE–OEG4–Z33 from the fiber during 10 μs of simulation time. We have performed two simulations to check the relative co-assembly of oppositely charged (C12VVEE and C12VVKK–OEG4–Z33) and similarly charged (C12VVEE and C12VVEE–OEG4–Z33) ligands and fillers. All the oppositely charged C12VVKK–OEG4–Z33 ligands entered the already assembled fiber of fillers (upper panel, Fig. S5(b), ESI†) showing good mixing or co-assembly behavior. On the other hand, only 3 of the 16 added similarly charged C12VVEE–OEG4–Z33 ligands enter the fiber after 1 μs, indicating a significantly lower propensity of co-assembly.
Method
GROMACS 2021.542 and MARTINI3 force field33,43 are combined in the coarse-grained simulations. We used similar definitions of MARTINI3 beads for C12 as defined in our previous work32 and mapped bond, angle parameters with all-atom simulation (see ESI†). Our previous work showed that Martini 3 would provide a similar fiber structure to CHARRM36 force-field. We used Martinize244 to get amino acid parameters which can define bead types, bond, angle, dihedral parameters and can constrain protein's secondary structure. Initially, we solvated 200 fillers and 8 ligands in an aqueous solution in a 14 × 14 × 14 nm3 box resulting a 1/25 ligand-filler ratio. We used regular waters beads in Martini 3 simulations. We have increased the number of ligands and box size for other ratios. We have also added required numbers of counterions and salts. Energy minimization is first performed on the system, followed by equilibration under NVT (constant number of molecules, constant volume, and constant temperature) ensemble and then NPT (constant number of molecules, constant pressure, and constant temperature) ensemble. After the co-assembly of filler and ligands, we have replicated the fiber along the axial direction of the fiber, which doubles the number of fillers, ligands and gives better statistics. We have also increased the simulation box size in the other two directions and solvated with new water and ions (we changed the ligand type for Fig. 5 in this stage). Then we performed the energy minimization and equilibration under NVT for 500 ns and semi-isotropic NPT for 10 μs. The last 3 μs was considered as the production run. 3 copies of simulations are performed to get error bars.For Z33 aggregation in bulk simulations (Fig. 4), we have added 10 Z33s in a 13 × 13 × 13 nm3 simulation box and added required water and ions. The CHARMM3645 force field was used for all-atom simulations and the recommended CHARMM TIP3P water model was applied with the SETTLE algorithm.46 Other simulation parameter details can be found in the ESI.†
We used Ovito47 for clustering (see Fig. 2) and visualizations. To calculate the nearest neighbor (see Fig. 4(e)), we got the contact map from VMD.48 This contact map contained the residue-residue minimum distance information and combined with in-house code to get nearest neighbor probability. A such contact map for all-atom, no salt system is shown in Fig. S4 (ESI†). We have also used VMD to get the g(r) shown in Fig. 4(f).
Conclusion
In this study, molecular dynamics simulations revealed the distribution of ligands in co-assembled nanofibers of PAs with oppositely charged peptides. Our findings demonstrate that positively charged PAs (KK groups) with an attached neutral Z33 group (ligands) undergo microscopic segregation on the surface of fibers in excess of negatively charged PAs (fillers). We find that the short range attractions generated by the Z33 in PAsis responsible of the local segregation of the ligand, as evident in the segregation of Z33 in bulk solutions. The ligands came close to each other in the assembled fiber and formed clusters on the surface that grow as the overall concentration of ligand on the fiber's increases. We discuss the importance of attractive interactions between ligands and electrostatics of the fiber surface in creating local segregation of ligands. While we focus on Z33 for its utility in antibody purifications, we believe that other ligands, proteins, or epitopes may also demonstrate similar behavior in co-assembled nanofibers, as attractive interactions can be prevalent among them. This study also demonstrates the possibility of controlling distribution not only through ligand's attractive interaction modifications but also by controlling charges on the filler and ligands through chemical modifications. Overall, our results provide valuable insights into the behavior of ligands in co-assembled nanofibers, which can inform the development of more effective and efficient materials for a wide range of applications.Author contributions
M. O. d. l. C. acquired the funding and led the project. T. H. P. and Y. L. performed the simulations, T. H. P. and M. O. d. l. C. wrote the manuscript. All authors commented on the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work is supported by The National Science Foundation under Award Number 2119686.References
- A. Dasgupta and D. Das, Designer Peptide Amphiphiles: Self-Assembly to Applications, Langmuir, 2019, 35, 10704–10724 CrossRef CAS PubMed.
- R. Qi, P. Zhang, J. Liu, L. Zhou, C. Zhou, N. Zhang, Y. Han, S. Wang and W. Wang, Peptide Amphiphiles with Distinct Supramolecular Nanostructures for Controlled Antibacterial Activities, ACS Appl. Bio Mater., 2018, 1, 21–26 CrossRef CAS.
- C. Chen, Y. Chen, C. Yang, P. Zeng, H. Xu, F. Pan and J. R. Lu, High Selective Performance of Designed Antibacterial and Anticancer Peptide Amphiphiles, ACS Appl. Mater. Interfaces, 2015, 7, 17346–17355 CrossRef CAS PubMed.
- J. D. Hartgerink, E. Beniash and S. I. Stupp, Self-Assembly and Mineralization of Peptide-Amphiphile Nanofibers, Science, 2001, 294, 1684–1688 CrossRef CAS PubMed.
- L. C. Palmer, C. J. Newcomb, S. R. Kaltz, E. D. Spoerke and S. I. Stupp, Biomimetic Systems for Hydroxyapatite Mineralization Inspired by Bone and Enamel, Chem. Rev., 2008, 108, 4754–4783 CrossRef CAS PubMed.
- L.-L. Li, G.-B. Qi, F. Yu, S.-J. Liu, H. Wang, L. Li, S. Liu, G. Qi, H. Wang and F. Yu, An Adaptive Biointerface from Self-Assembled Functional Peptides for Tissue Engineering, Adv. Mater., 2015, 27, 3181–3188 CrossRef CAS PubMed.
- D. Kumar, V. L. Workman, M. O’Brien, J. McLaren, L. White, K. Ragunath, F. Rose, A. Saiani and J. E. Gough, Peptide Hydrogels—A Tissue Engineering Strategy for the Prevention of Oesophageal Strictures, Adv. Funct. Mater., 2017, 27, 1702424 CrossRef.
- A. T. Preslar, F. Tantakitti, K. Park, S. Zhang, S. I. Stupp and T. J. Meade, 19F Magnetic Resonance Imaging Signals from Peptide Amphiphile Nanostructures Are Strongly Affected by Their Shape, ACS Nano, 2016, 10, 7376–7384 CrossRef CAS PubMed.
- S. R. Bull, M. O. Guler, R. E. Bras, P. N. Venkatasubramanian, S. I. Stupp and T. J. Meade, Magnetic Resonance Imaging of Self-Assembled Biomaterial Scaffolds, Bioconjug. Chem., 2005, 16, 1343–1348 CrossRef CAS PubMed.
- H. Sai, G. C. Lau, A. J. Dannenhoffer, S. M. Chin, L. Dorddević, L. Dorddević and S. I. Stupp, Imaging Supramolecular Morphogenesis with Confocal Laser Scanning Microscopy at Elevated Temperatures, Nano Lett., 2020, 20, 4234–4241 CrossRef CAS PubMed.
- E. B. Peters, M. R. Karver, K. Sun, D. C. Gillis, S. Biswas, T. D. Clemons, W. He, N. D. Tsihlis, S. I. Stupp and M. R. Kibbe, Self-Assembled Peptide Amphiphile Nanofibers for Controlled Therapeutic Delivery to the Atherosclerotic Niche, Adv. Ther., 2021, 4, 2100103 CrossRef CAS PubMed.
- S. Martin, D. A. Harrington, S. Ohlander, S. I. Stupp, K. T. McVary and C. A. Podlasek, Peptide Amphiphile Nanofiber Hydrogel Delivery of Sonic Hedgehog Protein to the Penis and Cavernous Nerve Suppresses Intrinsic and Extrinsic Apoptotic Signaling Mechanisms, Which Are an Underlying Cause of Erectile Dysfunction. Nanomedicine, Nanotechnology, Biol. Med., 2021, 37, 102444 CrossRef CAS PubMed.
- J. C. Barrett and M. V. Tirrell, Peptide Amphiphile Micelles for Vaccine Delivery, Methods Mol. Biol., 2018, 1798, 277–292 CrossRef CAS PubMed.
- N. Singha, P. Gupta, B. Pramanik, S. Ahmed, A. Dasgupta, A. Ukil and D. Das, Hydrogelation of a Naphthalene Diimide Appended Peptide Amphiphile and Its Application in Cell Imaging and Intracellular PH Sensing, Biomacromolecules, 2017, 18, 3630–3641 CrossRef CAS PubMed.
- Y. Li, L. L. Lock, J. Mills, B. S. Ou, M. Morrow, D. Stern, H. Wang, C. F. Anderson, X. Xu, S. Ghose, Z. J. Li and H. Cui, Selective Capture and Recovery of Monoclonal Antibodies by Self-Assembling Supramolecular Polymers of High Affinity for Protein Binding, Nano Lett., 2020, 20, 6957–6965 CrossRef CAS PubMed.
- Y. Li, L. L. Lock, Y. Wang, S. H. Ou, D. Stern, A. Schön, E. Freire, X. Xu, S. Ghose, Z. J. Li and H. Cui, Bioinspired Supramolecular Engineering of Self-Assembling Immunofibers for High Affinity Binding of Immunoglobulin G, Biomaterials, 2018, 178, 448–457 CrossRef CAS PubMed.
- K. L. Niece, J. D. Hartgerink, J. J. J. M. Donners and S. I. Stupp, Self-Assembly Combining Two Bioactive Peptide-Amphiphile Molecules intoNanofibers by Electrostatic Attraction, J. Am. Chem. Soc., 2003, 125, 7146–7147 CrossRef CAS PubMed.
- A. C. Braisted and J. A. Wells, Minimizing a binding domain from protein A, Proc. Natl. Acad. Sci. U. S. A., 1996, 93, 5688–5692 CrossRef CAS PubMed.
- M. B. van Eldijk, F. C. M. Smits, J. C. Thies, J. Mecinovic and J. C. M. van Hesta, Thermodynamic investigation of Z33-antibody interaction leads to selective purification of human antibodies, J. Biotech., 2014, 179, 32–41 CrossRef CAS PubMed.
- M. A. Cook and G. D. Wright, The past, present, and future of antibiotics, Sci. Transl. Med., 2022, 14, eabo7793 CrossRef CAS PubMed.
- Monoclonal Antibodies Market Worth $494.53 Billion By 2030 https://www.grandviewresearch.com/press-release/global-monoclonal-antibodies-market.
- E. Canepa, S. Salassi, A. L. de Marco, C. Lambruschini, D. Odino, D. Bochicchio, F. Canepa, C. Canale, S. Dante, R. Brescia, F. Stellacci, G. Rossi and A. Relini, Amphiphilic gold nanoparticles perturb phase separation in multidomain lipid membranes, Nanoscale, 2020, 12, 19746–19759 RSC.
- Z. Dongsheng and M. Olvera de La Cruz, Nanopatterns in tethered membranes of weakly charged chains with hydrophobic backbones, Macromolecules, 2008, 41, 6612–6614 CrossRef.
- C. Lamar, Y. Liu, C. Yi and Z. Nie, Entropy-driven segregation and budding in hybrid vesicles of binary nanoparticle amphiphiles, Giant, 2020, 1, 100010 CrossRef.
- M. Lu, et al., Phase-Separation-Induced Porous Hydrogels from Amphiphilic Triblock Copolymer with High Permeability and Mechanical Strength, Chem. Mater., 2022, 34, 10995–11006, DOI:10.1021/acs.chemmater.2c03004.
- P. Hebbeker, A. A. Steinschulte, S. Schneider and F. A. Plamper, Balancing Segregation and Complexation in Amphiphilic Copolymers by Architecture and Confinement, Langmuir, 2017, 33, 4091–4106, DOI:10.1021/acs.langmuir.6b04602.
- S. Honda, M. Koga, M. Tokita, T. Yamamoto and Y. Tezuka, Phase separation and self-assembly of cyclic amphiphilic block copolymers with a main-chain liquid crystalline segment, Polym. Chem., 2015, 6, 4167–4176, 10.1039/C5PY00346F.
- F. J. Solis, S. I. Stupp and M. Olvera de la Cruz, Charge induced pattern formation on surfaces: Segregation in cylindrical micelles of cationic-anionic peptide amphiphiles, J. Chem. Phys., 2005, 122, 054905 CrossRef CAS PubMed.
- K. L. Kohlstedt, F. J. Solis, G. Vernizzi and M. Olvera de la Cruz, Spontaneous Chirality via Long-Range Electrostatic Forces, PRL, 2007, 99, 030602 CrossRef PubMed.
- F. J. Solis, G. Vernizzi and M. Olvera de la Cruz, Electrostatic driven pattern formation in fibers, nanotubes and pores, Soft Matter, 2011, 7, 1456–1466 RSC.
- R. M. P. da Silva, D. van der Zwaag, L. Albertazzi, S. S. Lee, E. W. Meijer and S. I. Stupp, Super-resolution microscopy reveals structural diversity in molecular exchange among peptide amphiphile nanofibres, Nat. Commun., 2016, 7, 11561 CrossRef CAS PubMed.
- Y. Li, M. Kim, T. H. Pial, Y. Lin, H. Cui and M. Olvera de la Cruz, Aggregation-Induced Asymmetric Charge States of Amino Acids in Supramolecular Nanofibers, J. Phys. Chem. B, 2023, 127, 8176–8184 CrossRef CAS PubMed.
- P. C. T. Souza, R. Alessandri, J. Barnoud, S. Thallmair, I. Faustino, F. Grünewald, I. Patmanidis, H. Abdizadeh, B. M. H. Bruininks, T. A. Wassenaar, P. C. Kroon, J. Melcr, V. Nieto, V. Corradi, H. M. Khan, J. Domański, M. Javanainen, H. Martinez-Seara, N. Reuter, R. B. Best, I. Vattulainen, L. Monticelli, X. Periole, D. P. Tieleman, A. H. de Vries and S. J. Marrink, Martini 3: A General Purpose Force Field for Coarse-Grained Molecular Dynamics, Nat. Methods, 2021, 18, 382–388 CrossRef CAS PubMed.
- Z. Jarin, O. Agolini and R. W. Pastor, Finite-Size Effects in Simulations of Peptide/Lipid Assembly, J. Membr. Biol., 2022, 255, 437–449, DOI:10.1007/s00232-022-00255-9.
- A. Majumder and J. E. Straub, Addressing the Excessive Aggregation of Membrane Proteins in the MARTINI Model, J. Chem. Theory Comput., 2021, 17, 2513–2521, DOI:10.1021/acs.jctc.0c01253.
- B. Smit, K. Esselink, P. A. J. Hilbers, N. M. Van Os, L. A. M. Rupert and I. Szleifer, Computer Simulations of Surfactant Self-Assembly, Langmuir, 1993, 9, 9–11, DOI:10.1021/la00025a003.
- R. Behjatmanesh-Ardakani and M. Farsad, On the Difference between Self-Assembling Process of Monomeric and Dimeric Surfactants with the Same Head to Tail Ratio: A Lattice Monte Carlo Simulation, J. Chem., 2012, 2013, e525948, DOI:10.1155/2013/525948.
- X. Zhang, J. G. Arce Nunez and J. T. Kindt, Derivation of Micelle Size-Dependent Free Energies of Aggregation for Octyl Phosphocholine from Molecular Dynamics Simulation, Fluid Phase Equilib., 2019, 485, 83–93, DOI:10.1016/j.fluid.2018.12.001.
- S. Srebnik and J. F. Douglas, Self-assembly of charged particles on nanotubes and the emergence of particle rings, chains, ribbons and chiral sheets, Soft Matter, 2011, 7, 6897 RSC.
- K. Chakraborty, P. Khatua, W. Shinoda and S. M. Loverde, Domain Formation in Charged Polymer Vesicles, Macromolecules, 2021, 54, 9258–9267 CrossRef CAS.
- K. Miliou, L. N. Gergidis and C. Vlahos, Mixed brushes consisting of oppositely charged Y-shaped polymers in salt free, monovalent, and divalent salt solutions, J. Polym. Sci., 2020, 1–14 Search PubMed.
- M. J. Abraham, T. Murtola, R. Schulz, S. Páll, J. C. Smith, B. Hess and E. Lindah, GROMACS: High Performance Molecular Simulations through Multi Level Parallelism from Laptops to Supercomputers, SoftwareX, 2015, 1–2, 19–25 CrossRef.
- R. Alessandri, J. Barnoud, A. S. Gertsen, I. Patmanidis, A. H. deVries, P. C. T. Souza and S. J. Marrink, Martini 3 Coarse-Grained Force Field: Small Molecules, Adv. Theory Simul., 2022, 5, 2100391 CrossRef CAS.
- P. C. Kroon, F. Grunewald, J. Barnoud, M. van Tilburg, P. C. T. Souza, T. A. Wassenaar and S. J. Marrink, Martinize2 and Vermouth: Unified Framework for Topology Generation, arXiv, 2022, preprint, arXiv: 2212.01191 DOI:10.48550/arXiv.2212.01191.
- J. Huang, S. Rauscher, G. Nawrocki, T. Ran, M. Feig, B. L. De Groot, H. Grubmüller and A. D. MacKerell, CHARMM36m: An Improved Force Field for Folded and Intrinsically Disordered Proteins, Nat. Methods, 2016, 14, 71–73 CrossRef PubMed.
- S. Miyamoto and P. A. Kollman, Settle: An Analytical Version of the SHAKE and RATTLE Algorithm for Rigid Water Models, J. Comput. Chem., 1992, 13, 952–962, DOI:10.1002/JCC.540130805.
- A. Stukowski, Visualization and Analysis of Atomistic Simulation Data with OVITO—The Open Visualization Tool, Modell. Simul. Mater. Sci. Eng., 2010, 18, 015012 CrossRef.
- W. Humphrey, A. Dalke and K. Schulten, VMD: Visual Molecular Dynamics, J. Mol. Graph., 1996, 14(1), 33–38, DOI:10.1016/0263-7855(96)00018-5.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4sm00315b |
| This journal is © The Royal Society of Chemistry 2024 |