
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceMechanochromic polymer blends made with an excimer-forming telechelic sensor molecule†
Marta
Oggioni
 a,
Jess M.
Clough
ab and
Christoph
Weder
*ab
a,
Jess M.
Clough
ab and
Christoph
Weder
*ab
aAdolphe Merkle Institute, University of Fribourg, Chemin des Verdiers 4, Fribourg CH-1700, Switzerland. E-mail: christoph.weder@unifr.ch
bNational Center of Competence in Research Bio-inspired Materials, Chemin des Verdiers 4, Fribourg CH-1700, Switzerland
First published on 6th February 2024
Abstract
The ability to monitor mechanical stresses and strains in polymers via an optical signal enables the investigation of deformation processes in such materials and is technologically useful for sensing damage and failure in critical components. We show here that this can be achieved by simply blending polymers of interest with a small amount of a mechanochromic luminescent additive (Py-PEB) that can be accessed in one step by end-functionalizing a telechelic poly(ethylene-co-butylene) (PEB) with excimer-forming pyrenes. Py-PEB is poorly miscible with polar polymers, such as poly(ε-caprolactone) and poly(urethane), so that blends undergo microphase separation even at low additive concentrations (0.1–1 wt%), and the emission is excimer-dominated. Upon deformation, the ratio of excimer-to-monomer emission intensity decreases in response to the applied stress or strain. The approach appears to be generalizable, although experiments with poly(isoprene) show that it is not universal and that the (in)solubility of the additive in the polymer must be carefully tuned.
Introduction
The ability to monitor mechanical stresses and strains in polymeric materials via an easily detectable optical signal enables the investigation of deformation processes and is technologically useful, for example, in pressure-sensing films, tamper-evident packaging solutions, and damage sensing.1–3 The most widely employed approach to impart polymers with mechanochromic characteristics is the covalent incorporation of so-called mechanophores,4i.e., molecular motifs that undergo mechanically triggered chemical reactions,5–9 conformational changes,10,11 rearrangements of interacting optically active moieties,12–15 or other effects that cause reversible or irreversible changes to the motif's optical absorption and/or photoluminescence.14,16–19 This general approach has the fundamental disadvantage that chemical modifications are required to render a polymer of interest mechanochromic. Such modifications are often not possible by post-polymerization processes, and therefore the preparation of mechanochromic polymers generally requires de novo synthesis. A much simpler approach involves the blending of polymers with optically active compounds, whose optical properties change when the material is subjected to mechanical forces.20 This concept was first introduced two decades ago by Löwe and Weder, who incorporated excimer-forming, photoluminescent oligo(p-phenylene vinylene) dyes (cyano-OPVs) into polyethylene.21,22 The deformation of these blends causes the dissociation of aggregated dye molecules, which in turn leads to a pronounced change of the photoluminescence colour. Such emission changes represent an attractive readout, as they are often not only visually detectable, but can easily be measured and the recorded spectrum (unlike a change in the fluorescence intensity) is independent of sample dimensions and the acquisition geometry.21–25 This framework was subsequently applied to different excimer-forming26 and aggregachromic dyes27 and exploited in several matrix polymers.28 However, a significant limitation of using nanoscale aggregates of small-molecule dyes is that it requires that the mechanochromic additive forms nanoscale aggregates in the polymer matrix and that such aggregates can be mechanically dispersed.2,20 In many polymers, these conditions are difficult or impossible to satisfy. While the approach generally works well in semi-crystalline polymers, where nucleation effects can promote the formation of small dye aggregates, it is difficult to realize in amorphous materials, where the aggregate formation is hard to control and, if the glass transition temperature is low, the mechanical forces that the aggregates experience may be too small to cause their breakup.29,30 To address these problems, some of us recently devised a mechanochromic additive that was accessed by end-functionalizing a telechelic poly(ethylene-co-butylene) (PEB) with excimer-forming cyano-OPVs.31 This design was originally based on the hypothesis that the additive's PEB blocks might entangle with the chains of the matrix polymer. Such entanglements were thought to limit the size of the OPV aggregates and facilitate efficient stress transfer between the matrix and the mechanochromic additive. Blends of different polymers, including poly(ε-caprolactone), poly(isoprene), poly(styrene-b-butadiene-b-styrene), and different thermoplastic polyurethanes, and 0.1–2% w/w of the telechelic additive indeed all display mechanochromic luminescence.31,32 However, the mechanism that causes this effect was shown to be different than originally hypothesized. On the basis of confocal laser scanning microscopy experiments, it was demonstrated that mixtures of the various matrices and the telechelic additive undergo microphase separation to form discrete spherical inclusions.33 These droplets have radii in the range of 400–900 nm and consist of mixtures of the host polymer and the additive. In the unstretched state, the photoluminescence of these droplets is dominated by excimer emission. However, upon deformation of the blends, the inclusions deform,34 and this reduces the extent of excimer emission. The deformation of the inclusions follows Eshelby's inclusion theory,34 which allows the mechanical properties of the droplets to be determined by relating the change in their shape to the macroscopically applied strain. The inclusions were softer than the surrounding matrices, elongate upon deformation, and this shape change causes a decrease in excimer emission. With the goals of demonstrating that the approach is generalizable and simplifying the synthesis of the sensor molecule, we set out to prepare a pyrene-terminated PEB telechelic and investigated the mechanochromic response of polymers containing this additive.Results and discussion
From the plethora of luminophores that might be useful to create the targeted telechelic mechanochromic sensor molecules, we selected pyrene, one of the most widely investigated excimer-forming emitters.35 Pyrene has previously been used to render different polymers mechanochromic, for example by covalently attaching the dye to poly(dimethylsiloxane),36,37 to styrene-b-(ethylene-co-butylene)-b-styrene triblock copolymer,38 or to water-soluble PEG-based cross-linkers for hydrogels.39 While elegant and effective, the aforementioned approaches comprise additional synthetic steps to endow the selected polymers with mechanochromism. Our choice was in part motivated by the fact that several functionalized pyrene derivatives are commercially available, and this allowed us to access a pyrene-terminated PEB (Py-PEB, Fig. 1 and ESI,† Fig. S1) in one synthetic step. Thus, we esterified dihydroxy-terminated PEB with a number-average molecular weight (Mn) of 3100 kg mol−1 with 1-pyrenebutyric acid. This was achieved by adapting a literature protocol and using dicyclohexylcarbodiimide and 4-dimethylaminopyridine as coupling agents (see ESI† for details).40 Nuclear magnetic resonance (NMR) spectra reveal that for the isolated fraction (32% yield, no attempts to improve the yield were made), the esterification is quantitative, while size-exclusion chromatography traces acquired with different detectors confirm the covalent attachment of the pyrene moieties to the PEB backbone (Fig. S2 and S3, ESI†). The successful esterification is also confirmed by the appearance of a sharp carbonyl band at 1738 cm−1 in FTIR spectra (Fig. S4, ESI†).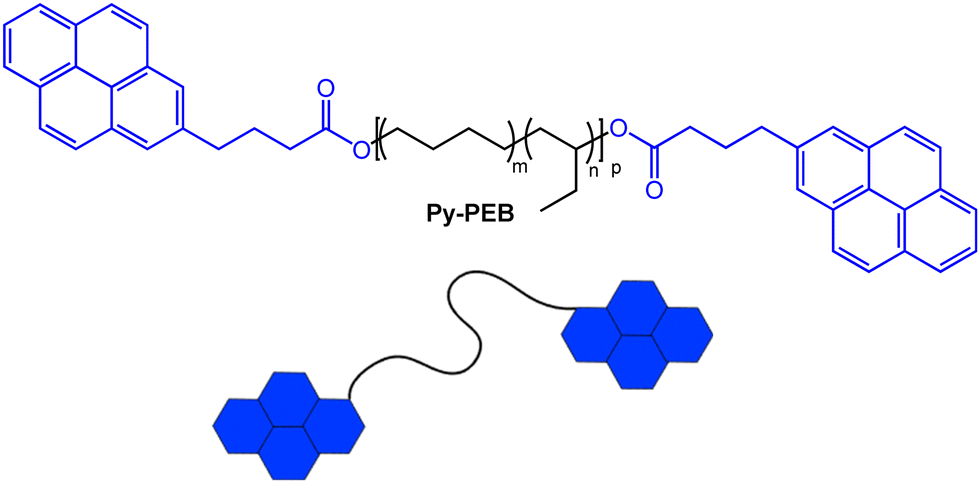 | ||
| Fig. 1 Chemical structure of the pyrene-terminated poly(ethylene-co-butylene) telechelic Py-PEB (m ≈ 0.36, n ≈ 0.64, p ≈ 55). | ||
The photoluminescence spectrum of a solution of Py-PEB (c = 1 μmol L−1) shows the characteristic monomer emission of pyrene, with well-resolved phonon bands at 376, 397, and 420 nm. The spectrum is almost identical to that of a 2 μmol L−1 solution of 1-pyrenebutanol, which we used as a reference compound that does not show any excimer emission at this concentration. By contrast, the emission spectrum of the bulk Py-PEB material, which at ambient temperature is a viscous liquid, displays primarily a broad emission band around 475 nm that is diagnostic of pyrene excimers (Fig. S5b, ESI†).35 Since intramolecular encounters of the end groups of such telechelics dominate over intermolecular interactions only in highly dilute solutions,41 the excimer emission in the Py-PEB/polymer blends must be caused by intermolecular assembly, and is possibly facilitated by microphase separation of the pyrenyl groups. Moreover, the differential scanning calorimetry (DSC) trace of Py-PEB shows only a glass transition at −44 °C, slightly higher than the parent OH-PEB (−50 °C), and is void of any other thermal transitions (Fig. S6b, ESI†). Thermogravimetric analysis (TGA) traces show that Py-PEB displays high thermal stability, with a degradation onset temperature of 326 °C (Fig. S7, ESI†).
Blends of three commercially available polymers and Py-PEB were prepared by solvent-casting THF solutions and solvent evaporation. Semicrystalline trans-poly(isoprene) (PI), which was blended with 0.5–10 wt% Py-PEB, poly(ε-caprolactone) (PCL) that was formulated with 0.5 or 1 wt% Py-PEB, and a poly(urethane) (PU) elastomer that was mixed with 0.1–1 wt% Py-PEB were chosen to (i) probe the effectiveness of Py-PEB in nonpolar (PI) and polar (PCL, PU) matrices, (ii) explore the capability of monitoring irreversible deformation of a semicrystalline plastic (PI, PCL) and reversible deformation of an elastomer (PU), and (iii) enable the comparison with previous studies that involved the same matrix polymers, but a cyano-OPV terminated telechelic.31,32 PI/Py-PEBx and PCL/Py-PEBx blends were processed into ca. 100 μm thin films by compression-moulding at 100 °C (subscript x denotes the Py-PEB content in wt%) whereas PU/Py-PEBx films were prepared via solvent casting.
Fig. 2a displays normalized photoluminescence spectra of films made with PI/Py-PEB blends with a Py-PEB content of 0.5–10 wt%. The data shows that the excimer emission increases with the content of the telechelic and becomes dominant at a Py-PEB content of 10 wt%. Stretching PI/Py-PEB5 films up to 150% strain does not cause a reduction of the excimer emission (Fig. 2b and Fig. S8a, b, ESI†), which contrasts with the results of a previous investigation of PI blends containing an otherwise similar cyano-OPV terminated PEB telechelic. Even at a low additive concentration of 0.5 wt% the previously studied blends display mostly excimer emission,31 as the additive phase separated from the matrix, forming spherical inclusions with sub-micrometre radii. As a result of the deformation of these inclusions, the ratio of excimer-to-monomer intensity (IE/IM) was considerably reduced upon deformation.33 The emission spectra of PI/Py-PEB blend films shown in Fig. 2a suggest that the solubility of Py-PEB in the nonpolar PI matrix is much higher than that of the corresponding cyano-OPV terminated PEB telechelic. Thus, the nature of the luminescent end groups can significantly impact the miscibility of the two components and therefore the blends’ morphology, at least in case of nonpolar matrices. Accordingly, we were unable to observe the formation of any inclusions, or any other phase-separated morphology, in PI/Py-PEB blend by widefield fluorescence microscopy, even at a Py-PEB content of 10 wt% (Fig. S11a and b, ESI†).
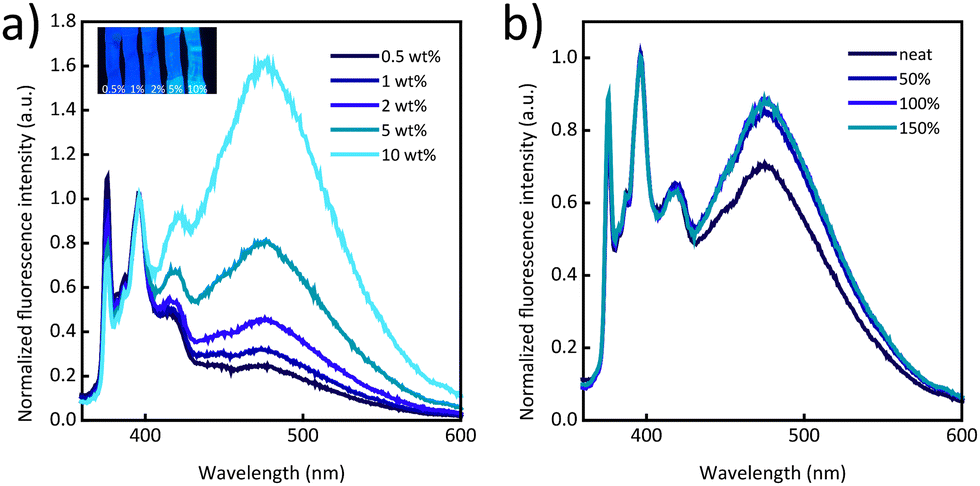 | ||
| Fig. 2 (a) Photoluminescence spectra of PI/Py-PEB blend films with a Py-PEB content of 0.5–10 wt%. The inset shows a picture of stretched films taken under illumination with 365 nm UV light. (b) Photoluminescence spectra of a PI/Py-PEB5 film that was uniaxially deformed to the indicated strain. All spectra were acquired upon excitation at 310 nm and were normalized to the intensity of the monomer peak at 397 nm. | ||
Gratifyingly, blends of Py-PEB and the more polar PCL mirror the behaviour of previously reported blends of PCL and the cyano-OPV terminated PEB telechelic more closely. In this case, the photoluminescence spectra are dominated by excimer emission at a Py-PEB content of as low as 0.5 wt% (Fig. 3a) and fluorescence microscopy images suggest the formation of spherical particles (Fig. S11e, ESI†). Photoluminescence spectra recorded after irreversibly deforming PCL/Py-PEB1 films by stretching them past the yield point reveal a clear change from excimer-dominated to monomer-dominated emission (Fig. 3b and Fig. S9, ESI†). To probe this behaviour further, we subjected PCL/Py-PEB1 films to tensile tests and simultaneously monitored IE/IM at 475 and 397 nm by in situ photoluminescence spectroscopy (Fig. 3c). The stress–strain curves reveal that the films deform elastically before they neck at a strain of ca. 15% and then plastically deform with little strain hardening. The IE/IMvs. strain trace appears to capture this behaviour well; IE/IM drops from 1.7 to 1.6 in the elastic regime, and from 1.6 to 0.53 upon plastic deformation. The fact that IE/IM continues to decrease beyond the yield point up to ca. 90% strain is likely related to experimental limitations, i.e., the photoluminescence measurement, which probes a small area of the sample (ca. 10 mm2), may initially average over necked and non-necked regions of the sample, which show different emission characteristics (Fig. S12 and S13, ESI†). A photograph of a stretched PCL/Py-PEB1 film shows the corresponding change in photoluminescence colour (Fig. 3c, inset).
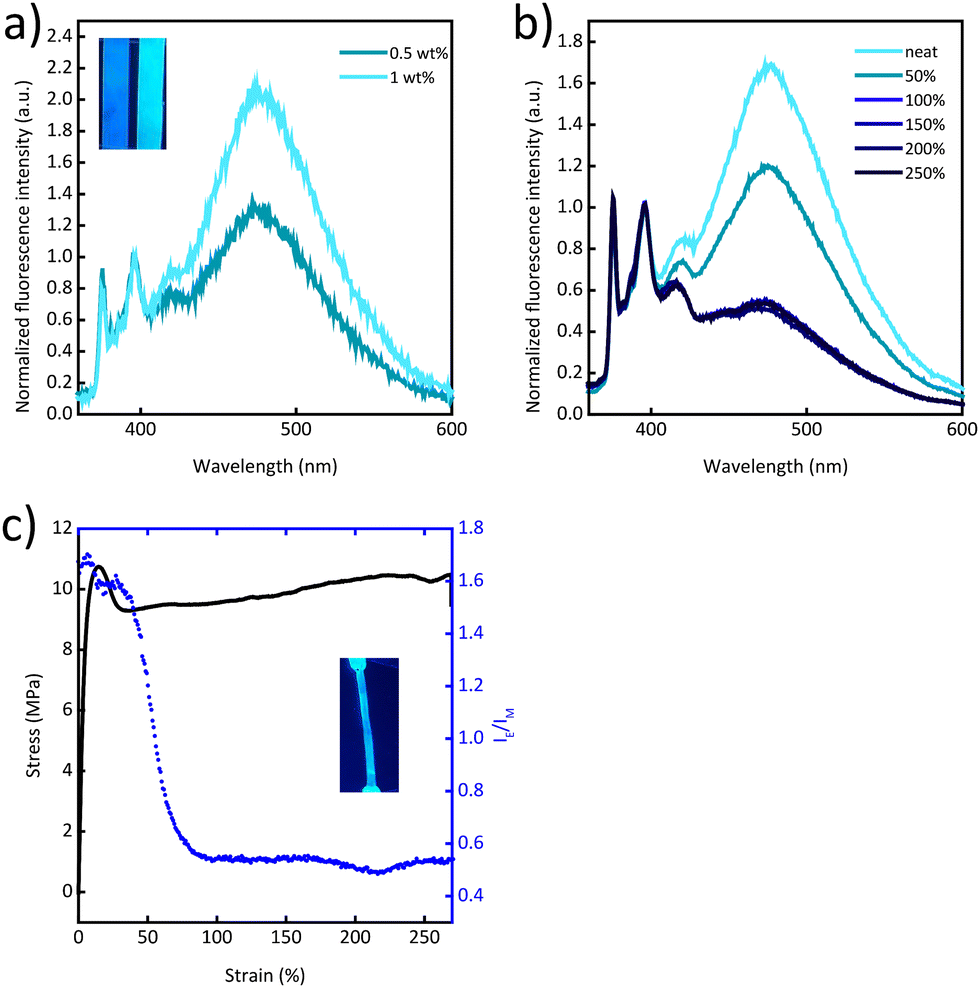 | ||
| Fig. 3 (a) Photoluminescence spectra and photograph of PCL/Py-PEB blend films with a Py-PEB content of 0.5 or 1 wt%. (b) Photoluminescence spectra of a PCL/Py-PEB1 film that was uniaxially deformed to the indicated strain. (c) Stress–strain curve and excimer-to-monomer emission intensity ratio (IE/IM) recorded at 475 and 397 nm of a PCL/Py-PEB1 film. The inset shows a picture of a deformed film under excitation at 365 nm. All spectra were acquired upon excitation at 310 nm and were normalized to the intensity of the monomer peak at 397 nm. | ||
Mechanochromic behaviour was also observed for the PU/Py-PEB blend films. In this matrix, the solubility of Py-PEB appears to be further reduced, as reflected by the fact that blends containing only 0.1 to 0.2 wt% of Py-PEB display a significant extent of excimer emission (Fig. 4a). Moreover, fluorescence microscopy images reveal unequivocally the occurrence of microphase separation and the formation of spherical particles that have diameters of the order of 1 μm (Fig. S11c and d, ESI†). The uniaxial deformation of PU/Py-PEB0.2 films is accompanied by a decrease in the excimer emission (Fig. 4b, c and Fig. S10a, b, ESI†), but in this case, the response is reversible. A cycling deformation experiment was performed, which shows that IE/IM decreases after stretching, and quickly recovers upon relaxation, over at least 4 stretch-and-release cycles (Fig. S14a, ESI†).
 | ||
| Fig. 4 (a) Photoluminescence spectra of PU/Py-PEB blend films with a Py-PEB content of 0.1–1 wt%. (b) Photoluminescence spectra of a PU/Py-PEB0.2 film that was uniaxially deformed to the indicated strain. (c) Stress–strain curve and excimer-to-monomer emission intensity ratio (IE/IM) recorded at 475 and 397 nm of a PU/Py-PEB0.2 film. All spectra were acquired upon excitation at 310 nm and were normalized to the intensity of the monomer peak at 397 nm. (d) Stress relaxation experiment of a PU/Py-PEB0.2 film. The film was stretched to a strain of ca. 200% at a strain rate of 50% min−1 and the strain was then fixed and the stress and IE/IM were monitored over the course of 3 h. | ||
Finally, a stress relaxation experiment was performed, in which a PU/Py-PEB0.2 film was stretched to a strain of ca. 200% and kept under fixed strain for 3 h (Fig. 4d). Upon deforming the film at a strain rate of 50% min−1, the IE/IM value decreases, as already shown in Fig. 4c. When kept at a constant strain, stress relaxation occurs and IE/IM increases slightly. While the stress decays nonlinearly with time, the increase of IE/IM shows a linear time dependence.
Conclusions
In summary, we demonstrated that the previously reported31 excimer-forming, telechelic sensor molecule approach is, in principle, generalizable to other excimer-forming dyes. We report the facile, one-step synthesis of Py-PEB, a pyrene-capped, excimer-forming telechelic additive that is useful to render some polymeric matrices mechanochromic. However, while small quantities (0.2–1 wt%) of Py-PEB imparted the more polar PCL and PU matrices with mechanochromic behaviour, the new additive is highly miscible with the non-polar PI, and this impeded the formation of excimer-forming inclusions. The results thus suggest that the specific design of the telechelic additive is important, in particular with respect to the (in)solubility profile that the dye molecules and the telechelic core impart.Experimental section
Materials and instrumentation
All reagents and solvents were used without further purification. 1-Pyrenebutyric acid, 1-pyrenebutanol, N,N′-dicyclohexylcarbodiimide (DCC), 4-dimethylaminopyridine (DMAP), Sudan Black B, poly(caprolactone) (average Mn 80![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 kg mol−1), trans-1,4-poly(isoprene) were purchased from Sigma-Aldrich. Dry dichloromethane (DCM) (99.8%, extra dry) was purchased from Acros Organics. Tetrahydrofuran (THF) and toluene were purchased from Fisher Scientific. Polyurethane (PU) (Texin 985®, average Mn = 111000 kg mol−1) was obtained from Covestro. Bio-Beads™ SX-1 (200–400 mesh) were purchased from Bio-Rad. Dihydroxyl-terminated poly(ethylene-co-butylene) (OH-PEB) (average Mn = 3100 kg mol−1), commercial name Krasol HLBH-P 3000, was received from Cray Valley Company.
000 kg mol−1), trans-1,4-poly(isoprene) were purchased from Sigma-Aldrich. Dry dichloromethane (DCM) (99.8%, extra dry) was purchased from Acros Organics. Tetrahydrofuran (THF) and toluene were purchased from Fisher Scientific. Polyurethane (PU) (Texin 985®, average Mn = 111000 kg mol−1) was obtained from Covestro. Bio-Beads™ SX-1 (200–400 mesh) were purchased from Bio-Rad. Dihydroxyl-terminated poly(ethylene-co-butylene) (OH-PEB) (average Mn = 3100 kg mol−1), commercial name Krasol HLBH-P 3000, was received from Cray Valley Company.
Nuclear magnetic resonance (NMR) spectroscopy was carried out on a Bruker Avance DPX 400 spectrometer at a frequency of 400.19 MHz for 1H-NMR measurements and of 100.63 MHz for 13C-NMR measurements. The solvent peak of CDCl3 was used to calibrate the spectra. Data were evaluated with the MestReNova software suite (v 12.0).
Size-exclusion chromatography (SEC) measurements were performed on an Agilent 1200 series HPLC system. The column system was composed of a PLgel mixed guard column (particle size 5 μm) and two Agilent PLgel mixed-D columns (ID = 7.5 mm, L = 300 mm, particle size = 5 μm). The detectors used were a UV detector (Agilent 1200 series) and an Optilab REX interferometric refractometer. Samples for SEC were prepared in THF with a concentration of ca. 1.5 mg mL−1.
UV-Vis absorption spectra were acquired on a JASCO V670 spectrometer. Data were evaluated on the Spectra Manager software. Quartz cuvettes (1 cm diameter) were used to measure absorption from 250 to 750 nm. UV-Vis spectra of solid samples were acquired on films of ca. 100 μm thickness placed over quartz slides.
Liquid phase photoluminescence spectra were acquired on a Horiba Fluorolog 3 spectrometer. The light source was a 450 W Xenon lamp, and the detector was a FL-1030-UP photomultiplier. The excitation wavelength was 310 nm, and the fluorescence intensity was measured from 360 to 600 nm.
In situ photoluminescence spectra were acquired on an Ocean Optics USB4000-FL spectrometer, with an Ocean Optics LS-310 LED light source at an excitation wavelength of 310 nm, and an Ocean Optics QR230-7-XRS SMA 905 optical fibre. The spectra were acquired by measuring films that were either placed on a black piece of paper or during uniaxial tensile tests carried out in a LINKAM TST350 microtensile stage with a 20 N loading cell. The stage was controlled by the LINK software. A pre-load of ca. 0.1 N was applied prior to tests to ensure that the sample was well loaded on the tensile stage. Rectangular samples of ca. 30 × 5 × 0.1 mm (length × width × thickness) were used and the strain rate was 50% min−1. In order to allow the comparison of different emission spectra shown within a given figure, in particular the fractions of monomer and excimer emission, in each graph the spectra were normalized to the intensity of the monomer peak at 397 nm, which is the monomer band that is least affected by possible internal absorption effects.
Thermogravimetric analysis (TGA) curves were measured in air on a Mettler–Toledo STAR thermogravimetric analyser from room temperature to 600 °C, using a heating rate of 10 °C min−1.
Differential scanning calorimetry (DSC) measurements were recorded in air on a Mettler–Toledo STAR system, with heating/cooling rates of 10 °C min−1, from −80 to 200 °C. The glass transition temperature (Tg) was reported as the midpoint of the step change in heat capacity.
Fluorescence microscopy images were acquired at 20× magnification using an Olympus BX51 microscope equipped with an Olympus DP72 high-resolution camera. Images were recorded with a wideband UV filter (λex = 330–385 nm; λem = 420–900 nm, Olympus U-MWU2).
Elemental analysis (EA) was performed by the Molecular and Biomolecular Analysis Service MoBiAS at ETH Zurich, Switzerland, and used to determine the C and H content of Py-PEB.
Methods
Solvent-casting of films of polymer/Py-PEB blends
Films of blends of the different host polymers and Py-PEB (0.5–10 wt% in the case of PI, 0.5 and 1 wt% in the case of PCL, and 0.1–1 wt% in the case of PU) were solvent-cast from THF solutions. The respective polymer (ca. 300 mg) and a solution of the appropriate amount of Py-PEB in 15 mL of THF were added to a 20 mL glass vial equipped with a magnetic stir bar and the mixture was stirred in the capped vial on a stirring plate for 1–3 days. If the polymer had not completely dissolved at this point, the vial was heated with a heat gun for a few seconds. The solutions were then cast into poly(tetrafluoroethylene) (PTFE) dishes (6 cm diameter) and the solvent was evaporated overnight in a fume hood. The resulting films were then transferred to a vacuum oven and dried overnight at 40 °C under reduced pressure.Compression-moulding of polymer/Py-PEB films
The solvent-cast films were compression-moulded using a Carver 3851-0 hydraulic hot press (Indiana, USA). The films were cut up into small pieces, which were sandwiched between 2 PTFE sheets, using 150 μm spacers. The PI/Py-PEB and PCL/Py-PEB films were processed at 100 °C with a final pressure of 2 metric tons for 10 min. The PU films were not compression-moulded.Synthesis of Py-terminated telechelic poly(ethylene-co-butylene) (Py-PEB)
The synthesis of Py-PEB was adapted from a published procedure.40 An excess of OH-terminated poly(ethylene-co-butylene) (OH-PEB) was dried in a glass vial in a vacuum oven overnight at 60 °C. After removal from the oven and cooling to ambient temperature, 1.84 g (0.61 mmol) of OH-PEB were added to a 10 mL round-bottomed flask equipped with a magnetic stir bar. Dry DCM (5 mL) was added under N2 flow, and the mixture was stirred under an inert atmosphere until OH-PEB had fully dissolved. 1-Pyrenebutyric acid (591 mg, 2.05 mmol), N,N′-dicyclohexylcarbodiimide (DCC, 488 mg, 2.37 mmol), and 4-dimethylaminopyridine (DMAP, 165 mg, 1.35 mmol) were added to a 250 mL round-bottomed flask under N2 flow, and dissolved in 120 mL of dry DCM. The OH-PEB/DCM solution was added dropwise to the same flask. The reaction mixture was stirred for 24 h at room temperature under an inert atmosphere. At this point, 1H NMR spectra showed that about 63% of the OH groups (the degree of functionalization was calculated via the resonance of the protons alpha to the OH groups) had reacted with 1-pyrenebutyric acid. Therefore, another aliquot of DCC (499 mg, 2.42 mmol), dissolved in 5 mL of dry DCM under N2, was added. After stirring the reaction mixture for another 4 h, the reaction was stopped by cooling the reaction mixture to −20 °C. The mixture was kept at −20 °C for 60 h, to induce precipitation of dicyclohexyl urea, which was then filtered off. Subsequently, DCM was removed in vacuo, and the crude product was dispersed in a few mL of toluene. A small amount of Sudan Black (Mn = 456.54 g mol−1) was added to colour the mixture and facilitate purification via size-exclusion column chromatography (Bio-Beads™ SX-1, 200–400 mesh; toluene). Toluene was evaporated in vacuo, and the product was dried in a vacuum oven overnight at 70 °C, which yielded Py-PEB (680 mg, 0.019 mmol, 32% yield) as a dark yellow, blue fluorescent, viscous liquid, which was characterized by 1H NMR spectroscopy, 13C-NMR spectroscopy, FTIR spectroscopy and elemental analysis. 1H-NMR (400 MHz, CDCl3): δ = 8.32–7.85 (m, 18H, ArH), 4.14–4.07 (m, 4H, CH2–O), 3.40 (t, 4H, CH2), 2.46 (t, 4H, CH2), 2.20 (tt, 4H, CH2), 2–0.5 (m, CH2, CH3, backbone). 13C-NMR (101 MHz, CDCl3): δ = 173.71 (C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O, ester), 135.92–123.49 (aromatic carbons), 39.05–26.05 (CH2, polymer backbone), 11.04–10.70 (CH3, polymer backbone). FTIR (cm−1): 843, 1146–1262, 1382, 1461, 1738, 2853, 1922, 2960. Elemental analysis: anal. calcd for (C20H15O2)2(C4H8)55: C = 84.97, H = 12.80. Found: C = 85.16 ± 0.01, H = 12.84 ± 0.11.
O, ester), 135.92–123.49 (aromatic carbons), 39.05–26.05 (CH2, polymer backbone), 11.04–10.70 (CH3, polymer backbone). FTIR (cm−1): 843, 1146–1262, 1382, 1461, 1738, 2853, 1922, 2960. Elemental analysis: anal. calcd for (C20H15O2)2(C4H8)55: C = 84.97, H = 12.80. Found: C = 85.16 ± 0.01, H = 12.84 ± 0.11.
Author contributions
C. W., M. O., and J. M. C. contributed to the conceptualization of the present study and designed the experiments. M. O. carried out the experiments. M. O. processed the data under the supervision of C. W. and J. M. C. C. W., M. O., and J. M. C. wrote the manuscript. C. W. and J. M. C. supervised and funded the project.Data availability
The source data of this study are available from the Zenodo repository at DOI: 10.5281/zenodo.10276391.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors gratefully acknowledge financial support from the Swiss National Science Foundation (SNSF) through grants to J. M. C. (PRIMA PR00P2_208616), C. W. (200020_207796) and the National Centre of Competence in Research (NCCR) Bio-inspired Materials (51NF40-182881), and thank the Adolphe Merkle Foundation for funding. The authors are also grateful to Dr Derek Kiebala for sharing the Python script used to translate the photoluminescence spectra recorded into IE/IM values and correlating these to the stress/strain curves.References
- Y. Chen, C. J. Yeh, Q. Guo, Y. Qi, R. Long and C. Creton, Chem. Sci., 2021, 12, 1693 RSC.
- Y. Huang, S. Huang and Q. Li, ChemPlusChem, 2023, 88, e202300213 CrossRef CAS PubMed.
- Y. Chen, M. Sommer and C. Weder, Macromol. Rapid Commun., 2021, 42, 2000685 CrossRef CAS PubMed.
- G. De Bo, Macromolecules, 2020, 53, 7615 CrossRef CAS.
- D. A. Davis, A. Hamilton, J. Yang, L. D. Cremar, D. Van Gough, S. L. Potisek, M. T. Ong, P. V. Braun, T. J. Martínez, S. R. White, J. S. Moore and N. R. Sottos, Nature, 2009, 459, 68 CrossRef CAS PubMed.
- Y. Chen, A. J. H. Spiering, S. Karthikeyan, G. W. M. Peters, E. W. Meijer and R. P. Sijbesma, Nat. Chem., 2012, 4, 559 CrossRef CAS PubMed.
- K. Imato, A. Irie, T. Kosuge, T. Ohishi, M. Nishihara, A. Takahara and H. Otsuka, Angew. Chem., 2015, 127, 6266 CrossRef.
- R. Göstl and R. P. Sijbesma, Chem. Sci., 2016, 7, 370 RSC.
- J. R. Hemmer, C. Rader, B. D. Wilts, C. Weder and J. A. Berrocal, J. Am. Chem. Soc., 2021, 143, 18859 CrossRef CAS PubMed.
- M. Raisch, W. Maftuhin, M. Walter and M. Sommer, Nat. Commun., 2021, 12, 4243 CrossRef CAS PubMed.
- R. Kotani, S. Yokoyama, S. Nobusue, S. Yamaguchi, A. Osuka, H. Yabu and S. Saito, Nat. Commun., 2022, 13, 303 CrossRef CAS PubMed.
- Y. Sagara, M. Karman, E. Verde-Sesto, K. Matsuo, Y. Kim, N. Tamaoki and C. Weder, J. Am. Chem. Soc., 2018, 140, 1584 CrossRef CAS PubMed.
- Y. Sagara, H. Traeger, J. Li, Y. Okado, S. Schrettl, N. Tamaoki and C. Weder, J. Am. Chem. Soc., 2021, 143, 5519 CrossRef CAS PubMed.
- H. Traeger, D. J. Kiebala, C. Weder and S. Schrettl, Macromol. Rapid Commun., 2021, 42, 2000573 CrossRef CAS PubMed.
- H. Traeger, Y. Sagara, D. J. Kiebala, S. Schrettl and C. Weder, Angew. Chem. Int. Ed., 2021, 60, 16191 CrossRef CAS PubMed.
- J. Li, C. Nagamani and J. S. Moore, Acc. Chem. Res., 2015, 48, 2181 CrossRef CAS PubMed.
- C. Calvino, L. Neumann, C. Weder and S. Schrettl, J. Polym. Sci., Part A: Polym. Chem., 2017, 55, 640 CrossRef CAS.
- N. Deneke, M. L. Rencheck and C. S. Davis, Soft Matter, 2020, 16, 6230 RSC.
- Y. Chen, G. Mellot, D. Van Luijk, C. Creton and R. P. Sijbesma, Chem. Soc. Rev., 2021, 50, 4100 RSC.
- F. Ciardelli, G. Ruggeri and A. Pucci, Chem. Soc. Rev., 2013, 42, 857 RSC.
- C. Löwe and C. Weder, Adv. Mater., 2002, 14, 1625 CrossRef.
- C. Löwe and C. Weder, Color Tunable Photoluminescent Blends. US Pat., 7223988, 2007 Search PubMed.
- F. Cellini, L. Zhou, K. Sachin, S. D. Peterson and M. Porfiri, Mech. Mater., 2016, 93, 145 CrossRef.
- T. Wang, N. Zhang, Y. Ge, C. Wang, Z. Hang and Z. Zhang, Macromol. Chem. Phys., 2020, 221, 1900463 CrossRef CAS.
- A. Pucci, Sensors, 2019, 19, 4969 CrossRef CAS PubMed.
- Y. Sagara, S. Yamane, M. Mitani, C. Weder and T. Kato, Adv. Mater., 2016, 28, 1073 CrossRef CAS PubMed.
- J. Kunzelman, B. R. Crenshaw, M. Kinami and C. Weder, Macromol. Rapid Commun., 2006, 27, 1981 CrossRef CAS.
- M. Kinami, B. R. Crenshaw and C. Weder, Chem. Mater., 2006, 18, 946 CrossRef CAS.
- B. R. Crenshaw and C. Weder, Macromolecules, 2006, 39, 9581 CrossRef CAS.
- F. Cellini, S. Khapli, S. D. Peterson and M. Porfiri, Appl. Phys. Lett., 2014, 105, 061907 CrossRef.
- C. Calvino, Y. Sagara, V. Buclin, A. P. Haehnel, A. del Prado, C. Aeby, Y. C. Simon, S. Schrettl and C. Weder, Macromol. Rapid Commun., 2019, 40, 1800705 CrossRef PubMed.
- D. J. Kiebala, Z. Fan, C. Calvino, L. Fehlmann, S. Schrettl and C. Weder, Org. Mater., 2020, 02, 313 CrossRef CAS.
- D. J. Kiebala, R. Style, D. Vanhecke, C. Calvino, C. Weder and S. Schrettl, Adv. Funct. Mater., 2023, 2304938 CrossRef CAS.
- J. D. Eshelby, Proc. R. Soc. London, Ser. A, 1957, 241, 376 Search PubMed.
- J. B. Birks, D. J. Dyson and I. H. Munro, Proc. R. Soc. London, Ser. A, 1963, 275, 575 CAS.
- N. A. A. Rossi, E. J. Duplock, J. Meegan, D. R. T. Roberts, J. J. Murphy, M. Patel and S. J. Holder, J. Mater. Chem., 2009, 19, 7674 RSC.
- D. R. T. Roberts, M. Patel, J. J. Murphy and S. J. Holder, Sens. Actuators, B, 2012, 162, 43 CrossRef CAS.
- C. Micheletti, V. A. Dini, M. Carlotti, F. Fuso, D. Genovese, N. Zaccheroni, C. Gualandi and A. Pucci, ACS Appl. Polym. Mater., 2023, 5, 1545 CrossRef CAS.
- D. Rasch and R. Göstl, Org. Mater., 2022, 4, 170 CrossRef CAS.
- S. Philip and S. Kuriakose, J. Fluoresc., 2019, 29, 387 CrossRef CAS PubMed.
- Z. Ji, Y. Li, Y. Ding, G. Chen and M. Jiang, Polym. Chem., 2015, 6, 6880 RSC.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3sm01489d |
| This journal is © The Royal Society of Chemistry 2024 |