
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceA microrheological examination of insulin-secreting β-cells in healthy and diabetic-like conditions†
Lukas
Woolley
a,
Adam
Burbidge
b,
Jan
Vermant
 a and
Fotis
Christakopoulos‡
*a
a and
Fotis
Christakopoulos‡
*a
aDepartment of Materials, ETH Zurich, Vladimir-Prelog-Weg 5, 8093 Zurich, Switzerland. E-mail: christakopoulos@stanford.edu
bNestlé Research, Route de Jorat 57, vers-chez-les Blanc, 1000 Lausanne, Switzerland
First published on 22nd March 2024
Abstract
Pancreatic β-cells regulate glucose homeostasis through glucose-stimulated insulin secretion, which is hindered in type-2 diabetes. Transport of the insulin vesicles is expected to be affected by changes in the viscoelastic and transport properties of the cytoplasm. These are evaluated in situ through particle-tracking measurements using a rat insulinoma β-cell line. The use of inert probes assists in decoupling the material properties of the cytoplasm from the active transport through cellular processes. The effect of glucose-stimulated insulin secretion is examined, and the subsequent remodeling of the cytoskeleton, at constant effects of cell activity, is shown to result in reduced mobility of the tracer particles. Induction of diabetic-like conditions is identified to alter the mean-squared displacement of the passive particles in the cytoplasm and diminish its reaction to glucose stimulation.
1 Introduction
Diabetes is a major non-communicable disease affecting about 10% of the world population, and it was responsible for 1.5 million deaths in 2019. There are two types of diabetes: type-1 diabetes (T1D), which is caused by genetic predisposition, and type-2 diabetes (T2D), which is influenced by both genetic and environmental factors.1–6 T2D is the most prevalent form of diabetes, accounting for about 95% of cases. Humans are heterotrophic, meaning that they have developed a mechanism to control nutrient uptake based on environmental conditions, which allows them to switch between anabolic and catabolic states. And while these conditions have shifted very fast, from a high caloric demand with a low caloric supply situation to a low caloric demand with a surplus of the caloric supply situation, organisms have been unable to adapt to it resulting in the rise of conditions such as insulin resistance and T2D. In short, it is a pathological condition that arises from metabolic dysfunction induced by insulin resistance and dysfunction of pancreatic beta-cells (β-cells).1,5 T2D is regarded as a chronic disease where a progressive decline in β-cell functions leads first to β-cell exhaustion, and, eventually, β-cell apoptosis.Glucose-stimulated insulin secretion (GSIS) is a Ca2+ dependent process, where cytoplasmic glucose concentrations in the order of 20 mM result in increased electrical activity and Ca2+ influx.4,7–12 In response to this electrical activity, insulin-containing granules are secreted in a biphasic manner, consisting of a rapid and brief burst and a sustained second wave of secretion.13–16 The first wave lasts between five and ten minutes after stimulation with glucose and involves the readily releasable pool of granules that are in close proximity to the cell membrane, while the second wave comprises of granules from the reserve pool that need to be transported to the cell membrane.16–18 In T2D, the biphasic mechanism of insulin secretion is hindered, with mainly the first wave being impaired.12,19–24 T2D was regarded as a combination of insulin resistance and β-cell dysfunction; however, in recent years, it is more widely accepted that the reduced β-cell function is the main cause.12 While the exact aetiology of T2D is not fully understood, it is recognized as a result of several pathways acting in combination with each other.
An active area of research is in the role of the cytoskeletal proteins, like actin and tubulin, in insulin secretion, with their role not being completely understood.25–37 Earlier reports suggested that filamentous actin (F-actin) acts as a physical barrier preventing granule mobility27 and the microtubule network as tracks for granule transport towards the cell membrane.25,26 However, more recent studies have identified F-actin re-organization necessary for regulating insulin secretion,29,32,38,39 and microtubules restricting granule availability for secretion.40 These results suggest that fine tuning of the cytoskeleton is necessary for physiological insulin secretion. A substantial amount of the work attempting to correlate the effect of the cytoskeletal proteins on insulin secretion and T2D has been based on biological assays, involving pharmacological perturbation of the cytoskeleton and its visualization on fixed samples at discrete time points coupled with measurements of glucose stimulation. A complementary approach, which has been pursued by several groups, is through tracking of fluorescent tagged insulin granules and analyzing their trajectories in response to glucose stimulation.35,41,42 Tabei et al. report anomalous diffusion when tracking GFP-tagged insulin granules in MIN6 cells upon stimulation with glucose.42 Further analysis of the granule dynamics suggested that the insulin granules exhibit both the passive transport behaviour due to the viscoelastic nature of the cytoplasm and the active, directed motion, through specific coupling to cytoskeletal proteins. Similarly, Heaslip et al. tracked GFP-tagged insulin granules in INS-1 cells before and after stimulation with glucose.35 A small amount of granules behave in a sub-diffusive (stationary) manner, independent of the glucose levels, and the majority shows diffusive motion with a small amount indicating super-diffusive motion (directed). The ratio of super-diffusive to diffusive motion increased upon glucose stimulation. In addition, the importance of microtubules for the long-distance transport of the granules and the role of actin in insulin secretion was highlighted. The cytoskeleton was disrupted by various drugs and imaged, at discrete time points, on fixed samples. In the aforementioned examples, fluorescent-tagged insulin granules were used as tracer particles. Consequently, the anomalous diffusion stems not only from the viscoelasticity of the cytoplasm but also from the affinity of the insulin granules to certain cytoskeletal proteins.
A way to gain insight into cytoskeletal changes is by evaluating the effect on the mechanics inside the cell, using passive microrheological techniques. The network of the filamentous proteins of the cytoskeleton is the main contributor to the viscoelastic properties of the cytoplasm.43,44 The mechanics of the cytoplasm regulates the physiological microenvironment and intervenes in processes such as signal transduction, cell division, nutrient diffusion, and cancer metastasis, amongst others.45–47 A powerful and non-destructive tool for probing the mechanical properties of materials at such small scales is optical microscopy-based microrheology. The experimental observable is the motion of non-interacting probe particles embedded within the cells.48–51
Directly accessing the diffusion in the cytoplasm without disrupting it can provide valuable insights into the interplay between the viscoelastic properties, activity and intracellular transport. The technique has been successfully employed in measuring the viscoelastic properties of biological systems and living cells.52–58 While the aforementioned works35,41,42 look into the diffusive motion of insulin granules, analysis of their trajectories is unable to capture the mechanics of the cells due to their affinity to cytoplasmic proteins. Here, we focus on insulin-secreting β-cells to investigate how control over cytoskeletal proteins and diffusivity in the cytoplasm may modulate the regulation of the secretory insulin granules.
We probe the effect of biological events, associated with glucose-stimulated insulin secretion by studying the motion of tracer beads in the cytoplasm in situ. In the present work, a passive microrheological evaluation is carried out by following tracer particles that are subjected to thermal and mechanical noise. As the latter is a priori unknown, in contrast to systems at equilibrium, for the present systems in which active noise is also present, only apparent rheological properties are calculated, which can, however, be used for a comparative analysis. A pancreatic cell line is evaluated using passive microrheology at a single-cell level. The mechanics of the cytoplasm are evaluated by analyzing the time- and ensemble-averaged mean-squared displacement (MSD) of inert tracer particles. In addition, structural changes of the cytoskeletal filamentous proteins upon stimulation by glucose are examined. Finally, the induction of diabetic-like conditions allows assessment of the cell mechanics in different T2D-relevant conditions, in light of the relevance of differences in mass transport for diseased conditions.
2 Materials and methods
2.1 Cell culture
The rodent insulinoma INS1E cell line was kindly provided by Prof. Markus Stoffel (ETHZ) and cultured as described in a previous work.59 In brief, complete culture media contained the RPMI-1640 medium (Roswell Park Memorial Institute medium, Thermo-Fisher, cat. nr. 11530586) supplemented with FBS (10%), glucose (11.1 mM), HEPES (10 mM), sodium puryvate (1 mM), β-mercaptoethanol (50 μM), penicillin (50 U mL−1), streptomycin (50 μg mL−1), and neomycin (1000 μg mL−1). β-cell dysfunction was accomplished through palmitate (PA) induction by exposing the cells to 0.25 mM PA for 72 h.60,61 All the reagents were obtained from Thermo-Fisher.2.2 Bovine serum albumin conjugated palmitate
Bovine serum albumin (BSA)-conjugated palmitate (PA) was prepared as follows. First, 6.8 g of BSA (>97% Sigma-Aldrich) was dissolved in 100 mL of a 150 mM NaCl solution at 37 °C. After complete dissolution of the BSA, the solution was sterile filtered using a 0.2 μm filter. 44 mL of 150 mM NaCl solution was heated to 70 °C and 91.8 mg of sodium palmitate was added and stirred until the solution was clear. The solution was then quickly transferred step-wise into the 50 mL preheated BSA solution while during this process the temperature of the BSA never exceeded 40 °C. Subsequently, the solution was stirred for 1 h, followed by a pH adjustment using a 0.1 M NaOH solution until the pH reached a final value of 7.4, followed by an additional filtration step into 5 mL glass vials.2.3 Insulin secretion assay
Cells were incubated for 1 h in a low glucose (2.8 mM) concentration Krebs Ringer HEPES (KRBH) buffer solution at 37 °C, and studied first in their “fasting” state. Subsequent stimulation was carried out by adding glucose to a final concentration of 20 mM in static incubation for 1 h. The concentration (ng mL−1) of insulin released in the medium was measured through an ELISA kit (ALPCO) and normalized against the total cellular protein amount (μg). The total protein amount equaled 182 μg and 228 μg for the fasting conditions and 225 μg and 251 μg for the glucose-stimulated conditions for the control and the PA-treated cells, respectively.2.4 Poly(ethylene glycol)-functionalized polystyrene beads
Carboxylated, fluorescent, polystyrene beads with a diameter of 200 nm (Fluoresbrite YG Microspheres, Polysciences) were functionalized with poly(ethylene glycol) (PEG) by coupling to 2 kDa PEG diamine. Beads were washed three times by centrifuging at 10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 × g for 5 minutes with 50 mM (2-(N-morpholino)ethanesulfonic acid) buffer (pH 6.0) at a concentration of 2.6% v/v. 1-Ethyl-3-(3-dimethyloaminopropyl)carbodiimide hydrochloride and N-hydroxysulfosuccinimide were added to final concentrations of 2 and 5 mM, respectively, and mixed for 30 minutes at room temperature. An equal volume of 2 mM poly(ethylene-glycol) diamine, dissolved in 100 mM sodium biocarbonate (pH 8.0), was added and mixed for 30 minutes at room temperature. An equal volume of glycine was added and mixed for 30 minutes at room temperature. Finally, the PEG-coated beads were washed five times by centrifuging at 10000× for 5 minutes and resuspended in ultrapure deionized water (Millipore). All reagents were obtained from Sigma-Aldrich. The functionalization was verified by an increase of 8 nm in particle diameter, as measured through an increase in hydrodynamic radius using dynamic light scattering (ALV CGS-3).
000 × g for 5 minutes with 50 mM (2-(N-morpholino)ethanesulfonic acid) buffer (pH 6.0) at a concentration of 2.6% v/v. 1-Ethyl-3-(3-dimethyloaminopropyl)carbodiimide hydrochloride and N-hydroxysulfosuccinimide were added to final concentrations of 2 and 5 mM, respectively, and mixed for 30 minutes at room temperature. An equal volume of 2 mM poly(ethylene-glycol) diamine, dissolved in 100 mM sodium biocarbonate (pH 8.0), was added and mixed for 30 minutes at room temperature. An equal volume of glycine was added and mixed for 30 minutes at room temperature. Finally, the PEG-coated beads were washed five times by centrifuging at 10000× for 5 minutes and resuspended in ultrapure deionized water (Millipore). All reagents were obtained from Sigma-Aldrich. The functionalization was verified by an increase of 8 nm in particle diameter, as measured through an increase in hydrodynamic radius using dynamic light scattering (ALV CGS-3).
2.5 Live cell imaging
For the live cell imaging, cells were plated in Nunc glass-bottom microscopy dishes (Thermo-Fisher), coated with rat laminin (Millipore) in complete RPMI media as described earlier. The medium was exchanged with a fresh medium containing 208 nm diameter fluorescent beads, used as an inert fluorescent proxy for insulin granules, having similar size.62 These particles were used at a concentration of 10−5 relative to the commercial solution and then incubated overnight in the dishes. The surface of the tracer particles is coated with a PEG brush, which is a common strategy to impede interactions of the particles with the cytoskeleton.63–65Prior to imaging, cells were washed twice with PBS to remove beads that have not been incorporated into cells. Subsequently, the cells were incubated in KRBH buffer containing a low concentration of glucose (2.8 mM), to reach basal insulin secretion conditions, for 1 h at 37 °C. Live cell microscopy was then conducted using an inverted light microscope (Nikon Ti-2 A) equipped with a Hamamatsu ImageEM-X2 CCD camera, with a frame rate of 1 s−1 and an exposure time of 100 ms in an environmental chamber (Okolab) at 37 °C and 5% CO2 atmosphere. A 100× water CFI Plan objective NA 1.1 (Nikon) was used and each imaged tracer particle comprised 12 pixels. A control experiment was conducted with fluorescent beads in a Newtonian fluid (90% v/v glycerin, VWR Chemicals) under identical conditions as described above.
Bright field images were recorded before and after every fluorescent time-lapse acquisition to ensure that the analyzed beads were inside the cells. The cells were attached to the substrate in a way that makes them appear flat, which allows for analysis in a single plane. Glucose stimulation (GSIS) was carried out by the addition of glucose and palmitate at final concentrations of 20 and 0.25 mM, respectively. For consistency between each sample, imaging commenced 120 s after stimulation. A minimum of 25 individual cells were examined for each condition. The live-cell imaging of the cytoskeletal proteins was accomplished by staining the tubulin and F-actin with a SiR-tubulin and SiR-actin stain (Spirochrome), respectively.
2.6 Microrheological analysis
Image analysis was performed using the adapted open-access Matlab (The Mathworks) particle tracking code written by Blair and Dufresne,66 based on the works of Crocker and Grier55,67 and Parthasarathy.68 The particle trajectories were extracted from a time-lapse fluorescent acquisition with a frame rate of 2 s−1 and tracks of 500 s. The cells were first imaged in their fasting state. Subsequently, after glucose stimulation, imaging commenced immediately for 900 s. The same procedure was carried out for both healthy (control) cells and diabetic-like (PA-treated) cells. For the image analysis, the region of interest (ROI) for each video was selected based on the position of the cells and the fluorescent beads confined inside the cells. Trajectories that were smaller than 30 frames were eliminated. Prior to analyzing the particle trajectory, a linear uniform and time-segmented drift correction was performed using a Gaussian fit of the velocity distribution. The mean value was taken as the correction for each time segment point consisting of multiple particles over 100 frames and as an initial start for the following time step. Drift correction is required since the cells can move around on the substrate and we are only interested in the particle dynamics within the cells. Drift is identified by a collective movement of the beads in one direction and manifested in a shift of the mean value of the Gaussian distribution (Fig. 1A). To correct for the drift, a linear uniform and time-segmented, as the drift might not be constant in time, drift correction is performed.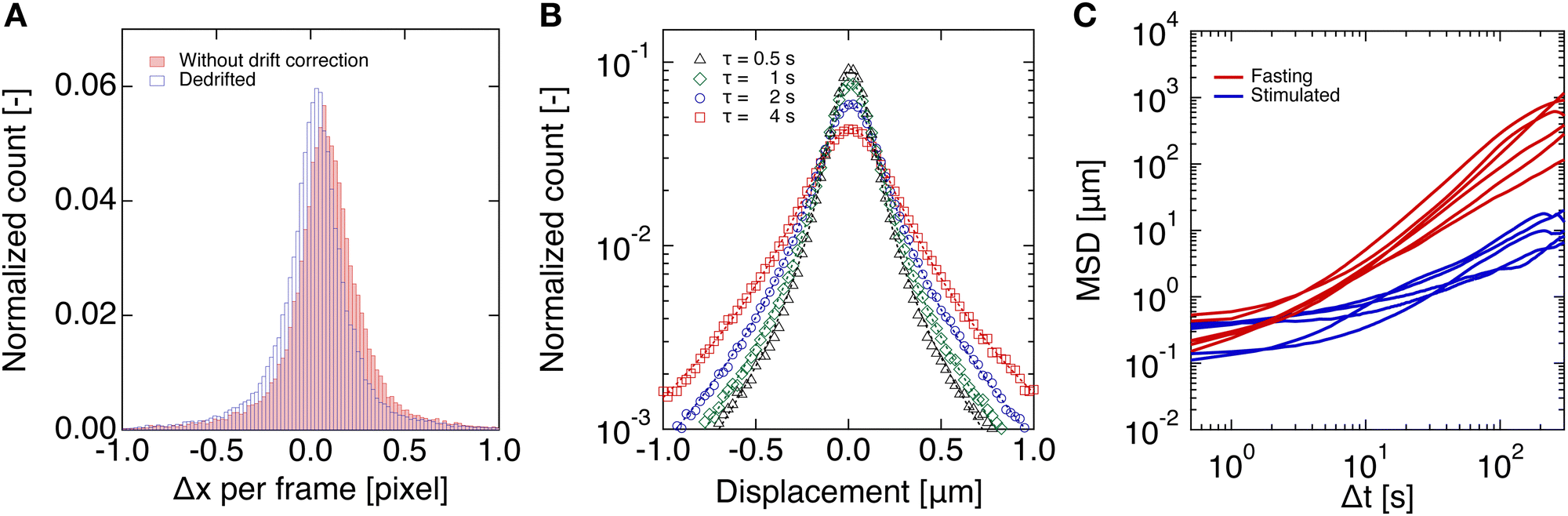 | ||
| Fig. 1 (A) Displacement per pixel of the tracer particles with (unfilled blue bars) and without (filled red bars) drift correction. (B) Distribution of the particle displacements per time step between two frames (lag time, τ), obtained from the de-drifted trajectory data, showing a Gaussian distribution, and a widening of the distribution as a function of lag time, τ. The distributions in both (A) and (B) are normalized by the total number of particle displacements. (C) Examples of mean-squared displacements before and after glucose stimulation for the control condition. | ||
From the de-drifted trajectories, the time-averaged MSD was calculated, assuming a quasi-static condition:
| 〈Δr2(τ)〉 = 〈Δr|(t0 + τ) − r(t0)|2〉n. | (1) |
In equilibrium, the relationship between the MSD of a microsphere in a viscoelastic medium and the shear creep compliance, J(t), of the said fluid is given through a simple linear relationship:
 | (2) |
 | (3) |
Another approach to analyze the trajectories is reported in the work of Heaslip et al.35 The individual tracks are analyzed and categorized according to their MSD scaling exponent m, in such a way as the MSD data is:
| m ∝ ln〈Δr2(t)〉. | (4) |
3 Results and discussion
3.1 Glucose-stimulated insulin secretion
The glucose-stimulated insulin secretion (GSIS) levels were measured for both the control and PA-exposed rodent insulinoma (INS1E) cells. In agreement with the literature, chronic PA treatment resulted in decreased GSIS levels (Fig. 2).60,61 Consequently, in the scope of the present work, the control and PA-exposed cells were used to represent the healthy and diabetic-like conditions, respectively. | ||
| Fig. 2 Glucose-stimulated insulin secretion (GSIS) in the control and exposed to 0.25 mM palmitate for 72 h INS1E cells. The statistical significance was evaluated between several groups using one-way ANOVA, where P < 0.005 was considered statistically significant. ***: P < 0.001, ****: P < 0.0001. | ||
3.2 Scaling exponents
The scaling exponent of each trajectory of the particle displacement was evaluated and sorted into sub-diffusive, diffusive and super-diffusive categories, using the criteria indicated above. Diffusive modes are of course the regular or normal modes of transport by unhindered Brownian motion. Sub-diffusive modes refer to the hindered diffusive motion of particles due to the caging or confinement of particles (or granules) in the filamentous network of the cytoskeleton (Fig. 3) on the observed timescale.75 In contrast, the super-diffusive regime is linked to the active transport of particles. In living cells, this can be achieved through coupled transport along the cytoskeletal filaments of molecules specifically bound to motor proteins. For example, in the work of Heaslip et al.,35 where the motion of fluorescent-tagged insulin vesicles is tracked overtime, a significant amount of the analyzed tracks is found to fall in the super-diffusive category, due to the affinity of the vesicles to molecular motor proteins.76 The binning of the scaling exponents for the control condition is presented in Fig. 4A and B, where it can be seen that most particles tracks fall into the subdiffusive and diffusive regimes. In addition, a Newtonian fluid was examined with the vast majority of particle trajectories showing a diffusive behaviour, as expected (Fig. 4C).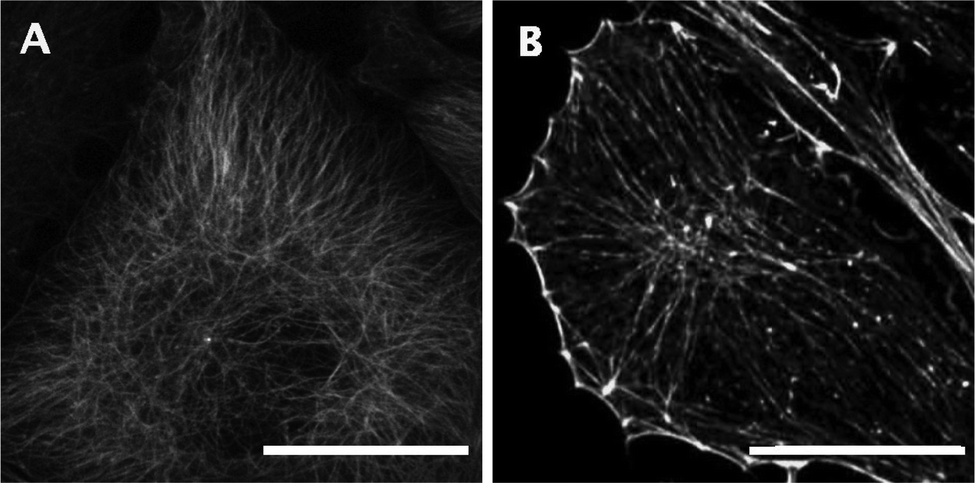 | ||
| Fig. 3 Cytoskeletal filamentous proteins, tubulin (A) and F-actin (B) in fasting INS1E cells. Scale bar corresponds to 10 μm. | ||
 | ||
| Fig. 4 Scaling exponent distribution of individual tracks with a bin size of 0.1 between 0 and 2 for fasting (A) and stimulated (B) control cells, and a Newtonian fluid (C). The roman numerals indicate the diffusion regime (I: sub-diffusive, II: diffusive, and III: super-diffusive). | ||
While the tracer particles used in the present study have a similar size as the insulin granules, they are inert to their surrounding and so should not have a specific binding interaction with the cytoskeletal network. Even though non-specific interactions cannot be completely ruled out, the small amount of super-diffusive motion supports this notion. The advantage of this approach is that the MSDs mainly reflect the changes in the viscoelastic properties of the matrix within the cell, which is the focus of the present study. However, it should be pointed out that errors in the drift correction can lead to artifacts that can manifest as an increase of super-diffusive motion, for samples where the drift takes place during the measurement.77
3.3 Effect of diabetic-like conditions on the mechanical properties of the cytoplasm
The mechanical properties of the cytoplasm were probed through microrheological measurements in live cells using particle tracking of 208 nm fluorescent, PEG-functionalized, microbeads and analysis of their MSD.50,51,53,56,57 Different experimental conditions were evaluated, starting with the control and PA-exposed (0.25 mM) cells, both at low glucose levels (fasting) and after GSIS (stimulated) conditions, as described earlier. The calculated apparent creep compliance is presented in Fig. 5. For the control cells, the apparent creep compliance, Ja(t), of the fasting and stimulated cells follows a similar characteristic evolution over the time window studied, with Ja increasing with t (Fig. 5A). Upon GSIS, Ja(t) increases by a factor of almost two at short timescales compared to the fasting state. In addition, GSIS is accompanied by a significant delay in the retardation spectrum, resulting in the Ja(t) of stimulated cells being about half, compared to that of fasting ones, at the larger timescales. This suggests that the average movement of the tracer particles is more restricted upon stimulation with glucose and that the fasting cells are more compliant at longer times. Glucose stimulation has a similar effect on the PA-exposed cells, with Ja(t) decreasing on average by half, but without having a significant effect on the retardation spectrum (Fig. 5B), which is not readily observed in neither the fasting nor the stimulated state. Further comparing the compliance of control and PA-exposed cells, the Ja(t) values for the two different cell treatments are comparable at smaller timescales. However, they strongly deviate at larger timescales, with the PA-exposed cells being less compliant, in agreement with an observation that the microtubulin network becomes more dense in diabetic rat islets.40 | ||
| Fig. 5 Apparent creep compliance (Ja(t)) from averaged particle tracks for INS1E cells under different experimental conditions as a function of time for the (A) control and (B) PA-exposed cells. | ||
The viscoelasticity of the cytoplasm stems principally from the cytoskeleton that comprises filamentous proteins, mainly F-actin, tubulin, and intermediate filaments forming a dynamic entangled network.43,54,56,78,79 Wirtz56 presented a rheological description, based on the classical scaling theory of de Gennes.80–82 In short, for short timescales, the filamentous network is expected to relax through lateral fluctuations, depicted in an increase in Ja(t) scaling with t3/4. These short timescales were not observable in our experimental window but have been observed in previous works.56 For intermediate timescales, Ja(t) exhibits a quasi-plateau, as seen in Fig. 5A for the glucose-stimulated cells and lag time <10 s, as no net filament motions takes place with the elastic modulus having the major contribution. For timescales longer than the retardation time, at the end of the quasi-plateau, Ja(t) is observed to increase linearly with time, and viscous behaviour dominates, allowing filaments to diffuse out of their confining tubes. We assume that the dominant changes in Ja(t) are mainly caused by the underlying changes in the true mechanical properties. For large timescales, the cytoplasm behaves as a viscoelastic liquid with the proportionality indicating viscous diffusion.56 The changes in apparent compliance and the evolution of the retardation spectrum after glucose stimulation are caused by cytoskeletal reorganization that has been known to take place for the F-actin and microtubulin network upon GSIS.25,27,32,35,38,40,83 An accurate control of the cytoskeletal re-organization ensures the regulation of granule availability for secretion under physiological conditions. In contrast to other works that looked into images of fixed samples and the activity of proteins involved in cytoskeletal reorganization at discrete time points, here we qualitatively evaluate the effect of the reorganization on the mechanical properties of the cytoplasm. This shows that this reorganization upon GSIS results in a generally less compliant, stiffer, and slower relaxing network. Interestingly, a time shift is observed for both conditions upon glucose stimulation, which could reflect the formation of longer actin filaments and microtubules or a decrease in the re-organization rate of the cytoskeleton. Moreover, a vertical shift is observed between the two PA-treated conditions, pointing towards an increase in crosslinking density upon glucose stimulation for this case. The exact functionality of the cytoskeletal network on insulin regulation is still an area of active research with its role being more complex than thought earlier.84 For example, the microtubule network is understood to be acting both as a barrier preventing secretion under basal glucose conditions, while at the same time serving as tracks for vesicle transport and being responsible for their accurate positioning prior to exocytosis.35,36,40,85,86 Similarly, F-actin remodeling removes steric hindrance between the insulin granules and the plasma membrane, together with assisting in short-distance granule transport and supplying mechanical force for their secretion.32,87–89 In this context, the lowering of the apparent compliance and slower relaxation kinetics of the PA-exposed cells’ cytoplasm compared to that of the control at basal glucose levels could be hypothesized to translate in a denser network with limited re-organization that possesses a decreased control fidelity, necessary for transport, accumulation, and accurate positioning of insulin granules at the plasma membrane prior to glucose stimulation. Moreover, the assessment of an effectively stiffer and slower relaxing cytoplasm could stem from lower ATP production, as similar behavior has also been reported for ATP depleted bacteria and yeast.90,91 In agreement with our results, reduced mitochondrial metabolism and ATP synthesis have been found to result from chronic hyperglycemia and were suggested as a contributing factor to progressive failure of β-cells in T2D. Although unable to unveil the exact mechanism, the differences in apparent compliance between the healthy and diabetic-like cells suggest structural changes in the cytoplasm, resulting in a loss of the precise control of its mechanical properties and the fine tuning required for the accurate regulation of insulin secretion and in turn blood sugar levels in the latter. These observations could facilitate further investigation of the effect of the mechanical properties of the cytoplasm in T2D.
4 Conclusions
Pancreatic, insulin-secreting, β-cells are probed through microrheology, with particles that mainly probe the mechanical properties of the cytoplasm. The measurements are carried out under different experimental conditions that resemble healthy and diabetic-like cells, both before and after stimulation with glucose. For the control case, the Ja(t) values have similar progression and reflect the reorganization of the F-actin and microtubulin filaments. The induction of diabetic-like conditions results in structural changes of the network giving rise to a less compliant and significantly slower relaxing cytoplasm. In addition, it shows a less pronounced re-organization upon GSIS, while remaining more compliant than the control case. The results suggest that PA-treatment hampers the precise control of the cytoskeletal reorganization and could hinder its ability to accurately position insulin granules in the proximity of the voltage-dependent Ca2+ channels, whose depolarization induces vesicle exocytosis. It has been suggested that the location of the Ca2+ channels relative to the ready-releasable pool of insulin granules rather than the magnitude of depolarization determines the capacity of the cell to induce exocytosis.61 Thus, the lack of precise positioning of insulin granules prior to GSIS could have an effect on the first wave of insulin secretion, which is relevant to the timescale of our experiments and mostly impaired in T2D.Further analysis could include the evaluation of the mechanical properties in primary cells at different stages of disease progression to examine whether there is a correlation between the two. An evaluation of the mesh size might be achieved by conducting a microrheological examination using a series of tracer beads with varying diameter.92 In addition, simultaneous tracking of inert particles and tagged-insulin granules could unveil the differences between the diffusion kinetics of the two populations, depending on the mechanical properties of the cytoskeleton and the changes in the active transport of the granules, respectively. Moreover, such experiments can be coupled with live imaging of cytoskeletal proteins, for a greater understanding of their re-organization process. We provide an insight into the compliance of β-cells and explore the structural changes in conditions relevant to T2D. Our data suggest that PA-exposed cells have an altered transport behavior and a less adaptable cytoskeletal network, which could contribute to the decreased ability of these cells to effectively control insulin secretion.
Author contributions
Conceptualization: A. B. and F. C.; data: L. W. and F. C.; formal analysis: L. W., J. V., and F. C.; investigation: L. W. and F. C.; methodology: L. W., A. B., J. V., and F. C., supervision: A. B., J. V., and F. C., writing – original draft: L. W. and F. C.; and writing – review and editing: L. W., A. B., J. V., and F. C.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors would like to thank Dr El-Hadji M. Dioum (PepsiCo) for help during the initial stage of the project, Prof. Markus Stoffel and Svenja Godbersen (ETHZ) for providing the cell line used in the experiments, and Prof. Cornelia Halin and Katharina Blatter (ETHZ) for access and help with the cell culture facility. The authors acknowledge Dr Robert Style (ETHZ) and Dr Jordan Taylor Moore (Stanford University) for the helpful discussion.Notes and references
- R. A. Guthrie and D. W. Guthrie, Crit. Care Nurs. Q., 2004, 27, 113–125 Search PubMed.
- P. Rorsman and E. Renström, Diabetologia, 2003, 46, 1029–1045 Search PubMed.
- E. U. Alejandro, B. Gregg, M. Blandino-Rosano, C. Cras-Méneur and E. Bernal-Mizrachi, Mol. Aspects Med., 2015, 42, 19–41 Search PubMed.
- P. Rorsman and F. M. Ashcroft, Physiol. Rev., 2018, 98, 117–214 Search PubMed.
- L. I. Hudish, J. E. Reusch and L. Sussel, J. Clin. Invest., 2019, 129, 4001–4008 Search PubMed.
- A. J. Kreutzberger, V. Kiessling, C. A. Doyle, N. Schenk, C. M. Upchurch, M. Elmer-Dixon, A. E. Ward, J. Preobraschenski, S. S. Hussain, W. Tomaka, P. Seelheim, I. Kattan, M. Harris, B. Liang, A. K. Kenworthy, B. N. Desai, N. Leitinger, A. Anantharam, J. D. Castle and L. K. Tamm, eLife, 2020, 9, 1–41 Search PubMed.
- L. Orci, F. Malaisse-Lagae, M. Ravazzola, M. Amherdt and A. E. Renold, Science, 1973, 181, 561–562 Search PubMed.
- C. B. Wollheim and G. W. Sharp, Physiol. Rev., 1981, 61, 914–973 Search PubMed.
- M. Prentki and F. M. Matschinsky, Physiol. Rev., 1987, 67, 1185–1248 Search PubMed.
- F. M. Ashcroft and P. Rorsman, Prog. Biophys. Mol. Biol., 1989, 54, 87–143 Search PubMed.
- C. S. Olofsson, S. O. Göpel, S. Barg, J. Galvanovskis, X. Ma, A. Salehi, P. Rorsman and L. Eliasson, Pfluegers Arch., 2002, 444, 43–51 Search PubMed.
- F. M. Ashcroft and P. Rorsman, Cell, 2012, 148, 1160–1171 Search PubMed.
- D. L. Curry, L. L. Bennett and G. M. Grodsky, Endocrinology, 1968, 83, 572–584 Search PubMed.
- P. E. Lacy, S. L. Howell, D. A. Young and C. J. Fink, Nature, 1968, 219, 1177–1179 Search PubMed.
- J. C. Hou, L. Min and J. E. Pessin, Insulin Granule Biogenesis, Trafficking and Exocytosis, Elsevier Inc., 1st edn, 2009, vol. 80, pp. 473–506 Search PubMed.
- M. G. Pedersen and A. Sherman, Proc. Natl. Acad. Sci. U. S. A., 2009, 106, 7432–7436 Search PubMed.
- T. K. Bratanova-Tochkova, H. Cheng, S. Daniel, S. Gunawardana, Y. J. Liu, J. Mulvaney-Musa, T. Schermerhorn, S. G. Straub, H. Yajima and G. W. Sharp, Diabetes, 2002, 51, S83–S90 Search PubMed.
- J. C. Henquin, M. Nenquin, P. Stiernet and B. Ahren, Diabetes, 2006, 55, 441–451 Search PubMed.
- E. Cerasi, Diabetologia, 1975, 11, 1–13 Search PubMed.
- J. D. Brunzell, R. P. Robertson, R. L. Lerner, W. R. Hazzard, J. W. Ensinck, E. L. Bierman and D. Porte, J. Clin. Endocrinol. Metab., 1976, 42, 222–229 Search PubMed.
- M. A. Pfeifer, J. B. Halter and D. Porte, Am. J. Med., 1981, 70, 579–588 Search PubMed.
- L. C. Groop, K. Ratheiser, L. Luzi, A. Melander, D. C. Simonson, A. Petrides, R. C. Bonadonna, E. Widén and R. A. DeFronzo, Acta Diabetol., 1991, 28, 162–168 Search PubMed.
- S. Del Prato, P. Marchetti and R. C. Bonadonna, Diabetes, 2018, 51, S109–S116 Search PubMed.
- J. E. Gerich, Diabetes, 2002, 51, S117–S121 Search PubMed.
- W. J. Malaisse, F. Malaisseâ Lagae, E. Van Obberghen, G. Somers, G. Devis, M. Ravazzola and L. Orci, Ann. N. Y. Acad. Sci., 1975, 253, 630–652 Search PubMed.
- Y. Kanazawa, S. Kawazu, M. Ikeuchi and K. Kosaka, Diabetes, 1980, 29, 953–959 Search PubMed.
- D. Aunis and M. F. Bader, J. Exp. Biol., 1988, 139, 253–266 Search PubMed.
- M. Hao, X. Li, M. A. Rizzo, J. V. Rocheleau, B. M. Dawant and D. W. Piston, J. Cell Sci., 2005, 118, 5873–5884 Search PubMed.
- N. I. Mourad, M. Nenquin and J. C. Henquin, Am. J. Phys., 2010, 299, C389–C398 Search PubMed.
- D. Balamatsias, A. M. Kong, J. E. Waters, A. Sriratana, R. Gurung, C. G. Bailey, J. E. Rasko, T. Tiganis, S. L. Macaulay and C. A. Mitchell, J. Biol. Chem., 2011, 286, 43229–43240 Search PubMed.
- J. C. Henquin, N. I. Mourad and M. Nenquin, FEBS Lett., 2012, 586, 89–95 Search PubMed.
- M. A. Kalwat and D. C. Thurmond, Exp. Mol. Med., 2013, 45, e37 Search PubMed.
- E. Uenishi, T. Shibasaki, H. Takahashi, C. Seki, H. Hamaguchi, T. Yasuda, M. Tatebe, Y. Oiso, T. Takenawa and S. Seino, J. Biol. Chem., 2013, 288, 25851–25864 Search PubMed.
- N. Porat-Shliom, O. Milberg, A. Masedunskas and R. Weigert, Cell. Mol. Life Sci., 2013, 70, 2099–2121 Search PubMed.
- A. T. Heaslip, S. R. Nelson, A. T. Lombardo, S. B. Previs, J. Armstrong and D. M. Warshaw, PLoS One, 2014, 9, 27–29 Search PubMed.
- K. H. Ho, X. Yang, A. B. Osipovich, O. Cabrera, M. L. Hayashi, M. A. Magnuson, G. Gu and I. Kaverina, Diabetes, 2020, 69, 1936–1947 Search PubMed.
- A. Müller, D. Schmidt, C. S. Xu, S. Pang, J. V. D'Costa, S. Kretschmar, C. Münster, T. Kurth, F. Jug, M. Weigert, H. F. Hess and M. Solimena, J. Cell Biol., 2021, 220, e202010039 Search PubMed.
- D. C. Thurmond, C. Gonelle-Gispert, M. Furukawa, P. A. Halban and J. E. Pessin, Mol. Endocrinol., 2003, 17, 732–742 Search PubMed.
- Z. Wang and D. C. Thurmond, J. Cell Sci., 2009, 122, 893–903 Search PubMed.
- X. Zhu, R. Hu, M. Brissova, R. W. Stein, A. C. Powers, G. Gu and I. Kaverina, Dev. Cell, 2015, 34, 656–668 Search PubMed.
- L. Digiacomo, M. A. Digman, E. Gratton and G. Caracciolo, Acta Biomater., 2016, 42, 189–198 Search PubMed.
- S. M. Tabei, S. Burov, H. Y. Kim, A. Kuznetsov, T. Huynh, J. Jureller, L. H. Philipson, A. R. Dinner and N. F. Scherer, Proc. Natl. Acad. Sci. U. S. A., 2013, 110, 4911–4916 Search PubMed.
- S. Sivaramakrishnan, J. V. DeGiulio, L. Lorand, R. D. Goldman and K. M. Ridge, Proc. Natl. Acad. Sci. U. S. A., 2008, 105, 889–894 Search PubMed.
- A. F. Pegoraro, P. Janmey and D. A. Weitz, Cold Spring Harbor Perspect. Biol., 2017, 9, a022038 Search PubMed.
- K. Simons and E. Ikonen, Nature, 1997, 387, 569–572 Search PubMed.
- C. Angelucci, G. Maulucci, G. Lama, G. Proietti, A. Colabianchi, M. Papi, A. Maiorana, M. de Spirito, A. Micera, O. B. Balzamino, A. Di Leone, R. Masetti and G. Sica, PLoS One, 2012, 7, e50804 Search PubMed.
- K. Jacobson, P. Liu and B. C. Lagerholm, Cell, 2019, 177, 806–819 Search PubMed.
- T. G. Mason and D. A. Weitz, Phys. Rev. Lett., 1995, 74, 1250–1253 Search PubMed.
- T. G. Mason, Rheol. Acta, 2000, 39, 371–378 Search PubMed.
- T. M. Squires and T. G. Mason, Annu. Rev. Fluid Mech., 2010, 42, 413–438 Search PubMed.
- E. M. Furst and T. M. Squires, Microrheology, Oxford University Press, Oxford, 2017 Search PubMed.
- S. Yamada, D. Wirtz and S. C. Kuo, Biophys. J., 2000, 78, 1736–1747 Search PubMed.
- A. W. Lau, B. D. Hoffman, A. Davies, J. C. Crocker and T. C. Lubensky, Phys. Rev. Lett., 2003, 91, 198101 Search PubMed.
- B. D. Hoffman, G. Massiera, K. M. Van Citters and J. C. Crocker, Proc. Natl. Acad. Sci. U. S. A., 2006, 103, 10259–10264 Search PubMed.
- J. C. Crocker and B. D. Hoffman, Methods Cell Biol., 2007, 83, 141–178 Search PubMed.
- D. Wirtz, Annu. Rev. Biophys., 2009, 38, 301–326 Search PubMed.
- K. M. Schultz and E. M. Furst, Soft Matter, 2012, 8, 6198–6205 Search PubMed.
- A. Rigato, A. Miyagi, S. Scheuring and F. Rico, Nat. Phys., 2017, 13, 771–775 Search PubMed.
- A. C. Title, P. N. Silva, S. Godbersen, L. Hasenöhrl and M. Stoffel, Mol. Metab., 2021, 53, 101267 Search PubMed.
- D. K. Hagman, L. B. Hays, S. D. Parazzoli and V. Poitout, J. Biol. Chem., 2005, 280, 32413–32418 Search PubMed.
- M. B. Hoppa, S. Collins, R. Ramracheya, L. Hodson, S. Amisten, Q. Zhang, P. Johnson, F. M. Ashcroft and P. Rorsman, Cell Metab., 2009, 10, 455–465 Search PubMed.
- E. Fava, J. Dehghany, J. Ouwendijk, A. Müller, A. Niederlein, P. Verkade, M. Meyer-Hermann and M. Solimena, Diabetologia, 2012, 55, 1013–1023 Search PubMed.
- M. T. Valentine, Z. E. Perlman, M. L. Gardel, J. H. Shin, P. Matsudaira, T. J. Mitchison and D. A. Weitz, Biophys. J., 2004, 86, 4004–4014 Search PubMed.
- T. Stylianopoulos, M. Z. Poh, N. Insin, M. G. Bawendi, D. Fukumura, L. L. Munn and R. K. Jain, Biophys. J., 2010, 99, 1342–1349 Search PubMed.
- J. A. McGlynn, N. Wu and K. M. Schultz, J. Appl. Phys., 2020, 127, 201101 Search PubMed.
- L. Blair and E. Dufresne, The MATLAB particle tracking code repository, https://site.physics.georgetown.edu/matlab/ Search PubMed.
- J. Crocker and D. Grier, J. Colloid Interface Sci., 1996, 179, 298–310 Search PubMed.
- R. Parthasarathy, Nat. Methods, 2012, 9, 724–726 Search PubMed.
- K. Luby-Phelps, P. E. Castle, D. L. Taylor and F. Lanni, Proc. Natl. Acad. Sci. U. S. A., 1987, 84, 4910–4913 Search PubMed.
- T. Kalwarczyk, N. Ziebacz, A. Bielejewska, E. Zaboklicka, K. Koynov, J. Szymanski, A. Wilk, A. Patkowski, J. Gapinski, H. J. Butt and R. Hołyst, Nano Lett., 2011, 11, 2157–2163 Search PubMed.
- M. Guo, A. J. Ehrlicher, M. H. Jensen, M. Renz, J. R. Moore, R. D. Goldman, J. Lippincott-Schwartz, F. C. Mackintosh and D. A. Weitz, Cell, 2014, 158, 822–832 Search PubMed.
- K. Nishizawa, M. Bremerich, H. Ayade, C. F. Schmidt, T. Ariga and D. Mizuno, Sci. Adv., 2017, 3, e1700318 Search PubMed.
- G. R. Kneller, J. Chem. Phys., 2011, 134, 224106 Search PubMed.
- K. Goswami and K. L. Sebastian, J. Stat. Mech.: Theory Exp., 2019, 083501 Search PubMed.
- F. Höfling and T. Franosch, Rep. Prog. Phys., 2013, 76, 046602 Search PubMed.
- K. M. Bracey, K. H. Ho, D. Yampolsky, G. Gu, I. Kaverina and W. R. Holmes, Biophys. J., 2020, 118, 193–206 Search PubMed.
- R. Metzler, J. H. Jeon, A. G. Cherstvy and E. Barkai, Phys. Chem. Chem. Phys., 2014, 16, 24128–24164 Search PubMed.
- A. B. Verkhovsky, T. M. Svitkina and G. G. Borisy, J. Cell Biol., 1995, 131, 989–1002 Search PubMed.
- K. M. Van Citters, B. D. Hoffman, G. Massiera and J. C. Crocker, Biophys. J., 2006, 91, 3946–3956 Search PubMed.
- P. G. De Gennes, J. Chem. Phys., 1971, 55, 572–579 Search PubMed.
- P. G. de Gennes, Scaling Concepts in Polymer Physics, Cornell University Press, Ithaca, New York, 1979 Search PubMed.
- P. G. de Gennes and L. Leger, Annu. Rev. Phys. Chem., 1982, 33, 49–61 Search PubMed.
- M. A. Kalwat, S. M. Yoder, Z. Wang and D. C. Thurmond, Biochem. Pharmacol., 2013, 85, 808–816 Search PubMed.
- K. M. Bracey, G. Gu and I. Kaverina, Front. Cell Dev. Biol., 2022, 10, 1–10 Search PubMed.
- P. Hoboth, A. Müller, A. Ivanova, H. Mziaut, J. Dehghany, A. Sönmez, M. Lachnit, M. Meyer-Hermann, Y. Kalaidzidis and M. Solimena, Proc. Natl. Acad. Sci. U. S. A., 2015, 112, E667–E676 Search PubMed.
- K. P. Trogden, J. Lee, K. M. Bracey, K. H. Ho, H. McKinney, X. Zhu, G. Arpag, T. G. Folland, A. B. Osipovich, M. A. Magnuson, M. Zanic, G. Gu, W. R. Holmes and I. Kaverina, eLife, 2021, 10, e59912 Search PubMed.
- A. Varadi, T. Tsuboi and G. A. Rutter, Mol. Biol. Cell, 2005, 16, 2670–2680 Search PubMed.
- C. Arous and P. A. Halban, Am. J. Physiol.: Endocrinol. Metab., 2015, 309, E611–E620 Search PubMed.
- R. Veluthakal and D. C. Thurmond, Cells, 2021, 10, 1503 Search PubMed.
- B. R. Parry, I. V. Surovtsev, M. T. Cabeen, C. S. O'Hern, E. R. Dufresne and C. Jacobs-Wagner, Cell, 2014, 156, 183–194 Search PubMed.
- R. P. Joyner, J. H. Tang, J. Helenius, E. Dultz, C. Brune, L. J. Holt, S. Huet, D. J. Müller and K. Weis, eLife, 2016, 5, 1–26 Search PubMed.
- J. P. Rich, G. H. McKinley and P. S. Doyle, J. Rheol., 2011, 55, 273–299 Search PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3sm01141k |
| ‡ Present address: Department of Materials Science and Engineering, Stanford University, 94305 Stanford, CA, USA. |
| This journal is © The Royal Society of Chemistry 2024 |