
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceScalable one-pot synthesis of amorphous iron-nickel-boride bifunctional electrocatalysts for enhanced alkaline water electrolysis†
Bennett
Schmitt
 a,
Eva
Murphy
a,
Sinny J.
Trivedi
b,
Qiancheng
Zhang
b,
Brian J.
Rodriguez
b,
Aran
Rafferty
c,
Raman
Bekarevich
d,
Gabor
Ersek
e,
Giuseppe
Portale
e and
M. Veronica
Sofianos
*a
a,
Eva
Murphy
a,
Sinny J.
Trivedi
b,
Qiancheng
Zhang
b,
Brian J.
Rodriguez
b,
Aran
Rafferty
c,
Raman
Bekarevich
d,
Gabor
Ersek
e,
Giuseppe
Portale
e and
M. Veronica
Sofianos
*a
aSchool of Chemical and Bioprocess Engineering, University College Dublin, Belfield, Dublin 4, Ireland. E-mail: mvsofianou@gmail.com
bSchool of Physics, Conway Institute of Biomolecular and Biomedical Research, University College Dublin, Dublin 4, Ireland
cAMBER Research Centre, Naughton Institute, Trinity College Dublin, Dublin 2, D02PN40, Ireland
dAdvanced Microscopy Laboratory, Centre for Research on Adaptive Nanostructures and Nanodevices (CRANN), Trinity College Dublin, Dublin 2, Ireland
ePhysical Chemistry of Polymeric and Nanostructured Materials, Zernike Institute for Advanced Materials, University of Groningen, Nijenborgh 4, Groningen, 9747 AG, The Netherlands
First published on 18th October 2024
Abstract
Green hydrogen is considered an attractive energy vector that can easily replace fossil fuel consumption, meeting global energy demands. Therefore, developing easily scalable, efficient and cost-effective electrocatalysts for water electrolysis is imperative for our transition to a more sustainable energy future. To this end, we demonstrate here a simple and scalable one-pot chemical reduction method for the synthesis of amorphous iron nickel boride nanoparticles with a spherical morphology. The iron to nickel ratio was easily adjusted during synthesis, and how it effects electrocatalytic performance was evaluated. The electrocatalyst powder with the highest concentration of metallic iron, or iron bonded to nickel, exhibited enhanced bifunctional OER and HER electrocatalytic activity, outperforming RuO2, the current state-of-the-art electrocatalyst; reaching industrial current densities, with an OER overpotential of 252 mV at 10 mA cm−2 and 349 mV at 100 mA cm−2. In our view, this work delivers an important method for the scalable synthesis of amorphous bimetallic boride nanoparticles with adjustable electronic structure for achieving enhanced water electrolysis at a minimum cost.
Introduction
Hydrogen is considered an attractive energy vector due to its high energy-to-mass ratio (120 MJ kg−1) that can easily replace fossil fuel consumption whilst meeting global energy demands. When produced from renewable energy, it is referred to as ‘green’ hydrogen. Currently, the main technology used for the production of green hydrogen is water electrolysis, also known as electrochemical water splitting. In this process, electricity is used to split water into hydrogen and oxygen gas. Theoretically, the standard thermodynamic potential of 1.23 V (vs. reversible hydrogen electrode, RHE) is required to split water into H2 and O2 regardless of the electrolyte media. In reality, a potential higher than 1.8 V is needed, as the reactions taking place at both the anode and cathode involve multiple electron transfer steps, resulting in an additional energy requirement to overcome kinetic obstacles and accelerate electron transfer.1–3 This additional energy requirement, also known as overpotential, is the main challenge associated with the production of green hydrogen, and has compromised its commercial viability to date. This is the reason why natural gas, coal or biomass gasification are still the main technologies for producing hydrogen on an industrial scale, accounting for 96% of hydrogen production.4–6 Only the remaining 4% is green hydrogen.7To overcome the kinetic obstacles associated with electrochemical water splitting and to accelerate electron transfer, efficient electrocatalysts are needed. To date, noble metals and their compounds (RuO2 and Pt/C) are the most efficient electrocatalysts for Oxygen (OER) and Hydrogen Evolution Reaction (HER).8–10 The scarcity and high-cost of these materials have limited their commercial application for large scale green hydrogen production. Hence, hydrogen is produced from fossil fuels, in complete contradiction of its green credentials.
A new emerging class of materials that have the potential to replace the current state-of-the-art noble metal based electrocatalysts for both OER and HER, are transition bimetallic compounds.11–14 Integrating the same electrocatalyst at both the anode and cathode of an electrolyser will not only simplify the system, but will reduce the overall production cost.12–14 On the other hand, transition bimetallic phosphides, sulphides, hydroxides, and borides have demonstrated enhanced performance for both OER and HER relative to their monometallic counterparts.15–20 Combining two transition metals and by controlling their relative ratios, one can form a new bimetallic compound endowed with adjustable electronic structure, controllable intermediate adsorption/desorption energy, and a tuneable reaction pathway.18,21,22 Hence, it is anticipated that the intrinsic catalytic activity, durability and stability can be more easily manipulated in bimetallic compounds than their monometallic counterparts. In a comprehensive study, Wang et al. prepared Co–Mo–P nanopillar array catalysts on three-dimensional nickel foam by employing a hydrothermal and phosphorylation method.23 They reported an overpotential of 258 mV for OER and 37 mV for HER at a current density of 10 mA cm−2 in an alkaline electrolyte. Meng et al. reported the electrochemical synthesis of FeNi2S4 with an overpotential of 210 mV for OER and 75 mV for HER at 10 mA cm−2 under alkaline conditions.24 Huang et al. prepared NiFe layered, double hydroxide-based nanosheet arrays using a hydrothermal method, exhibiting 249 mV for alkaline OER and 75 mV for alkaline HER.25 Hong et al. synthesised NixFe1−xB nanoparticles by a facile borothermal reduction method in molten salt.26 Their as-prepared electrocatalyst achieved a current density of 10 mA cm−2 at overpotentials of 282 mV for OER and 63.5 mV for HER, in an alkaline electrolyte.
Inspired by the enormous potential of transition bimetallic compounds, we have devised a simple and scalable one-pot chemical reduction method for the synthesis of iron nickel boride electrocatalyst powders. Their electronic structure was established by the mole ratio of Fe to Ni used during synthesis. How these ratios affect their physicochemical properties was extensively studied using a variety of characterisation techniques. All three electrocatalyst powders exhibited an amorphous crystal structure with average nanoparticle size distributions centred around 22 nm. The iron, nickel and boron atoms were evenly distributed throughout the nanoparticles as demonstrated by elemental mapping. It is worth mentioning that all three electrocatalysts powders exhibited higher OER electrocatalytic activity when compared to RuO2, the current state-of-the-art electrocatalyst. The electrocatalyst powder with the highest concentration of metallic iron, or iron bonded to boron, performed best both in OER and HER, and exhibited bifunctionality. Its recorded OER overpotential was 252 mV at 10 mA cm−2 and 349 mV at 100 mA cm−2, whereas the HER was 486 mV at −10 mA cm−2 and 587 at −100 mA cm−2. This electrocatalyst powder showed good durability in both OER and HER, as demonstrated by chronoamperometry for 10 h testing. In conclusion, this is a fundamental study demonstrating that even narrow changes in the molar ratio between two metals in bimetallic borides can have a dramatic influence in the physicochemical properties of the resulting electrocatalysts, and how these properties further influence OER and HER activity, thus providing valuable knowledge towards the design of bimetallic compounds with specific physicochemical properties for large-scale industrial water splitting applications.
Experimental
Synthesis of electrocatalyst powders
Three electrocatalyst powders were synthesised by a one-pot chemical reduction method at room temperature under a nitrogen atmosphere. Continuous stirring using a magnetic stirring bar was employed across all synthesis steps. An aqueous solution of sodium hydroxide and sodium borohydride was used as the reducing agent. Specifically, 0.1 M of sodium hydroxide (NaOH, Sigma-Aldrich, ≥98%) was added into a two-neck round bottom flask (250 mL) containing 100 mL of distilled water. Sodium hydroxide was used to prevent the hydrolysis reaction of sodium borohydride when added to water.27 1 M of sodium borohydride (NaBH4, Sigma-Aldrich, ≥98%) was then added to the aqueous solution.For the synthesis of FeNiB-I, 4.5 mmol of iron chloride tetrahydrate (FeCl2·4H2O, Sigma-Aldrich, 99%) and 5 mmol of nickel chloride hexahydrate (NiCl2·6H2O, M & B, 97%) were added together in a two-neck round bottom flask (50 mL) containing 20 mL of distilled water. The mixed metal precursor was then transferred to a syringe and added dropwise to the NaOH/NaBH4 aqueous solution as illustrated in Fig. 1a. Gas hydrogen bubbles were immediately formed and black precipitates were observed. Once the generation of bubbles had ceased, the black precipitates were collected through centrifugation at 6000 rpm, washed twice with distilled water, and finally rinsed with absolute ethanol. The three black powder products were dried overnight in air at 70 °C. The synthesis protocol for samples FeNiB-II and FeNiB-III followed that of FeNiB-I. Specifically, 5 mmol of FeCl2·4H2O and 5 mmol of NiCl2·6H2O was used for the synthesis of FeNiB-II, and 5 mmol of FeCl2·4H2O and 4.5 mmol of NiCl2·6H2O for the synthesis of FeNiB-III, as detailed in Table 1.
 | ||
| Fig. 1 Preparation and characterisation of the catalyst powders. (a) Schematic illustration of the one-pot synthesis, (b) XRD patterns of FeNiB-I, FeNiB-II and FeNiB-III nanoparticles. | ||
| Description of metal salts added to 1 M NaBH4 | Fe/Ni mole ratio | Electrocatalyst powder |
|---|---|---|
| 4.5 mmol FeCl2·4H2O + 5 mmol NiCl2·6H2O | 0.9/1 | FeNiB-I |
| 5 mmol FeCl2·4H2O + 5 mmol NiCl2·6H2O | 1/1 | FeNiB-II |
| 5 mmol FeCl2·4H2O + 4.5 mmol NiCl2·6H2O | 1/0.9 | FeNiB-III |
Physicochemical characterisation of the electrocatalyst powders
Phase observations of the as-prepared electrocatalyst powders was performed by powder X-ray diffraction (XRD) using a Siemens D500 (40 kV, 30 mA) diffractometer from Germany with Cu-Ka radiation (8.04 keV). The powder specimens were scanned in a 2θ range between 20° and 80° with a 0.03° step size, at 1.6 s per step, and rotated at a speed of 30 rpm.Morphological observations were carried out using a FEI Tecnai 20 (Transmission Electron Microscope). For FeNiB-I, additional high-resolution imaging, electron diffraction and elemental mapping was performed using a FEI Titan 80 – 300 kV FEG S/TEM (Scanning/Transmission Electron Microscope) equipped with an ‘Element’ Energy Dispersive X-ray spectroscopy (EDX) detector. Prior to imaging, the powder samples were mixed with ethanol and ultrasonicated for 20 minutes, then drop cast directly onto a 200 M carbon-coated Cu TEM grid (Mason Technology), and left to dry overnight at room temperature.
SAXS experiments were performed using a MINA instrument (University of Groningen). The instrument is equipped with a rotating Cu anode delivering an X-ray beam with wavelength of λ = 0.15413 nm (E = 8 keV). The SAXS patterns were acquired using a Pilatus 300 K Si solid state detector distanced 3000 mm and 240 mm from the sample. The angular range covered by the resultant SAXS patterns was calibrated using the scattering peaks from a Silver Behenate standard powder sample. The 2D patterns were converted by radially averaging around the beam centre into 1D SAXS profiles of I(q) vs. q, where q is the modulus of the scattering vector (q = 4π![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) sinθ/λ), with 2θ being the scattering angle. After 1D transformation, the two profiles were merged to obtain the final SAXS curve, covering a large portion of q-range. Data processing was performed using a MATLAB code. The SAXS profiles were further analysed using the McSAS program28 in order to quantitatively obtain the particle size histogram.
sinθ/λ), with 2θ being the scattering angle. After 1D transformation, the two profiles were merged to obtain the final SAXS curve, covering a large portion of q-range. Data processing was performed using a MATLAB code. The SAXS profiles were further analysed using the McSAS program28 in order to quantitatively obtain the particle size histogram.
Nitrogen gas adsorption analysis was performed at 77 K using a Nova 2400e surface area analyzer (Quantachrome, UK) to determine the specific surface area and micro-mesoporosity of the electrocatalyst powders. Their specific surface area was calculated from the adsorption data at relative pressures between 0.10 and 0.30, by employing the Brunauer–Emmett–Teller (BET) multi-point method.29 The pore size distributions and volumes in the micro- (<2 nm) and meso-range (2–50 nm) were calculated using the Barrett–Joyner–Halenda (BJH) method from the desorption branch of the isotherm.27,30 Prior to analysis, all three samples were outgassed at 150 °C under vacuum for 4 h. Mercury porosimetry was performed on the powder samples using an Autoscan-33 porosimeter (Quantachrome, UK), based on the Washburn equation:
| Pore diameter, D = (−4γcosθ)/P | (1) |
000 psi) required to force penetration of mercury into pores and interparticulate void spaces.
Chemical analysis of the electrocatalyst powders was conducted using a Kratos AXIS Ultra DLD X-ray Photoelectron Spectrometer (XPS) in ultra-high vacuum utilising an Al-Kα X-ray source (1486.7 eV). Casa XPS software was used for data analysis, and calibrated using the surface adventitious C 1s peak at 284.5 eV.
Topographical, electrical and electrochemical characterisation of the electrode material
To prepare the electrode material, three separate homogenous inks were formulated containing 5 mg of the electrocatalyst powder, 490 μL of distilled water, 490 μL of absolute ethanol, and 20 μL of 5% Nafion (Sigma Aldrich). The inks were homogenised in an ultrasonic bath for 20 minutes.For the topographical and electrical characterisation of the electrode material, 10 μL of the above ink was drop cast onto an 18 × 18 mm conductive indium tin oxide (ITO) coated glass cover slip (SPI supplies, 06465-AB) using a micropipette and was then dried under an IR lamp. Topography images were obtained using an Atomic Force Microscope (AFM; Asylum Research, MFP-3D) with a Si probe (Nanosensors, NCH) having a nominal resonance frequency of 330 kHz, a tip radius less than 8 nm, and a spring constant of 42 N m−1. Roughness values were averaged from three 5 μm images from representative electrocatalyst powders. Conductive AFM (C-AFM) investigations were carried out using the same AFM equipped with a C-AFM cantilever holder (Asylum Research, ORCA with 2 nA V−1 sensitivity) and a solid Pt AFM probe (Rocky Mountain Nanotechnology, 25PT300B) with a nominal resonance frequency, tip radius, and spring constant of 20 kHz, 15 nm, and 18 N m−1, respectively. The current–voltage (IV) spectroscopic measurements were conducted in contact mode in a ±3.0 V range with a 1 Hz triangle waveform initially ramping up from 0 V and applied from the ORCA holder to the ITO electrode, with the current being measured in the ORCA holder. The measurements were made in at least three locations per sample and 20 IV curves were measured per 20 μm × 20 μm location in a grid array. Before IV measurements, the current was zeroed by applying a small offset. As the force applied to the surface during IV measurements was kept constant, the tip-sample contact areas were the same assuming the mechanical properties of the electrocatalysts powders were the same. The reported resistances of all three electrocatalyst powders were calculated by dividing the corresponding voltage by the maximum observed current; and the mean and standard deviation was determined from ‘n’ number of curves which are 60, 160 (8 locations across two samples), and 60 IV curves from FeNiB-I, FeNiB-II, and FeNiB-III, respectively, under the assumption that other resistances were zero. Maximum current values above 100 pA were used to determine the mean and standard deviation for the FeNiB-I and FeNiB-III electrocatalyst powders, while maximum current values above 20 pA were taken into account for the FeNiB-II sample. The electrocatalyst powders were measured in the sequence of FeNiB-III, FeNiB-II, and FeNiB-I, respectively, with the same probe. Subsequently, FeNiB-III was measured again, with the same probe, and the current value was found to agree with its predecessor; that is, their respective uncertainty limits overlapped. Therefore, the potential risk of contamination could be ruled out as having an effect on the measurements.
For the electrochemical measurements, 10 μL of the as-prepared homogenous ink was drop-casted using a micropipette onto a 5 mm glassy carbon (GC) rotating disk electrode from Metrohm, which served as the working electrode. The drop-casted ink was left to dry under an IR lamp. The surface area of the glassy carbon working electrode was 0.2 cm2, with a total load of electrocatalyst powder equal to 0.05 mg. Due to this significantly small loading mass on the working electrode, no post XRD and XPS characterisation measurements could be performed, since the mass was below the detection limit of the instruments.
All electrochemical measurements were conducted at room temperature (22 °C ± 2 °C) using an Autolab potentiostat (PGSTAT204 with a FRA32M Module, Metrohm) interfaced to a PC with a Nova 2.1.7 software. A leakless silver/silver chloride (AgCl, 3 M KCl, Metrohm) electrode was used as the reference electrode, and a graphite rod as the counter electrode (MW-4131, BASi). An aqueous solution of 1 M potassium hydroxide (KOH, Sigma Aldrich, 90%) equal to pH 14 was used as the electrolyte. RuO2 (99.9%, Sigma Aldrich) and Pt/C (containing 10 wt% of Pt on C, Sigma Aldrich) were used as standards for OER and HER activity. During all electrochemical measurements, the working electrode was rotated at 3000 rpm by a rotation unit (Metrohm) to eliminate bubbles. All measured potentials were converted to the reversible hydrogen electrode (RHE), according to the equation ERHE = EAg/AgCl + 0.197 + 0.059 × pH. Linear sweep voltammetry (LSV) at a scan rate of 5 mV s−1 under an N2 gas (>99.999%, BOC) atmosphere was performed on all electrocatalyst powders to evaluate OER and HER activity. Electrochemical impedance spectroscopy (EIS) was carried out at the open circuit potential (OCP) using an AC perturbation of 10 mV in the frequency range from 100 kHz to 1 Hz. The uncompensated resistance, Rs, was determined from the Nyquist plot (Fig. S1†) and was used to calculate the ohmic drop correction using the equation Ec = Ee − iRs, where Ec is the corrected potential and Ee is the experimental potential (Table S1†).31 The LSV raw data with no iRs correction is presented in the ESI section (Fig. S2 and S3†). The stability performance of the powder samples was studied by performing chronoamperometry for 10 h. Cyclic voltammetry (CV) scans were performed to determine the electrochemical active surface area (ECSA) for each electrocatalyst powder. In detail, CV scans at different scan rates (20, 40, 60, 80, 100 and 120 mV s−1) were carried out in the non-faradaic region (0.84–1.23 V versus RHE). From these scan rate, CV-dependent plots, the difference in anodic and cathodic current densities (ΔJ) at 1.04 V (vs. RHE) was plotted against the corresponding scan rates. The slope obtained by the linear fit is twice the value of the double layer capacitance, Cdl, at the interface of electrolyte and the surface of the electrocatalyst powder.32 A specific capacitance of 40 μF cm−2 was used here for a flat surface at pH 14. The ECSA was calculated according to the equation ECSA = Cdl/(40 μF cm−2) cm2ECSA.
Results and discussion
The iron nickel boride (FeNiB) electrocatalyst powders were synthesised by applying a one-pot NaBH4-mediated reduction process under an inert nitrogen atmosphere, as illustrated in Fig. 1a, at room temperature. This synthesis method can be easily scaled up by simply changing the volume of the pot used to the desired quantities. A proposed reduction reaction mechanism is described in reaction (2), where FeNiB nanoparticles are formed together with sodium chloride (NaCl), boric acid (H3BO3) and hydrogen gas (H2). Both sodium chloride and boric acid are water soluble, and are easily removed by rinsing the collected nanoparticles with water, while any hydrogen gas which forms can escape during nanoparticle collection from the round bottom flask.| FeCl2 + NiCl2 + 4NaBH4 + 9H2O → FeNiB + 4NaCl + 12.5H2 + 3H3BO3 | (2) |
The yield of the above reaction was 97.8%, assuming that no surface oxidation of FeNiB has occurred and that all byproducts have been successfully removed during rinsing.
X-ray diffraction patterns of all three electrocatalyst powders were obtained to investigate their crystalline structure (Fig. 1b). No distinctive diffraction peaks were observed in samples FeNiB-I and FeNiB-II, except for a rather broad peak between 40° and 50°, centred at ∼45°; most probably assigned to the Fe–Ni–B amorphous phase, as observed elsewhere in the literature for amorphous metal borides.8,33–35 On the other hand, the diffraction pattern for the FeNiB-III electrocatalyst powder, with the higher number of Fe moles used during synthesis, exhibited weak diffraction peaks suggestive of the FeNi phase (COD 96-901-0018). This might be due to the FeNiB having a lattice symmetry similar to the P4/mmm (No 123) space group. It seems that when the mole ratio of Fe to Ni is increased to 1/0.9, some kind of crystalline ordering occurs during the chemical reduction process. As frequently reported in the literature, metal borides synthesised by a chemical reduction method using NaBH4 as the reducing agent yield amorphous crystalline structures that need annealing under an inert atmosphere at temperatures higher than 250 °C.36
To further study the nanostructure of the electrocatalyst powders, TEM microscopy and SAXS was performed. As can be observed from the TEM images (Fig. 2a–c), the nanoparticles are spherical in shape but characterised by a large degree of size dispersity. These morphological observations are confirmed by their associated SAXS curves, as reported in Fig. 2d, the general shape of which is characteristic of a collection of spheroidal particles with large size dispersity. It is noteworthy that the SAXS profile for the FeNiB-II electrocatalyst powder that corresponds to a 1:1 mole ratio of Fe to Ni, has a higher intensity at low q-values with respect to FeNiB-I and FeNiB-III. This means that the FeNiB-II has a larger contribution from bigger scattering objects. To quantify the size distribution of the particles, a Monte Carlo regression analysis was performed. See Fig. 2e. It is clear how FeNiB-III and FeNiB-I have a similar average nanoparticle radius with a mean value around 22 nm (21.9 for FeNiB-III and 22.4 for the FeNiB-I), while the FeNiB-II has a mean value of 26.8 nm. Likewise, the size distribution histogram for the FeNiB-II is clearly broadened towards larger radii values with respect to the other two samples.
 | ||
| Fig. 2 Morphological observations and size distribution analysis. TEM micrographs of (a) FeNiB-I, (b) FeNiB-II, (c) FeNiB-III, (d) SAXS patterns of the electrocatalyst powders, and (e) their corresponding volume-weighted particle size distributions, as determined by SAXS. The vertical dashed lines indicate the mean value of the radius distributions. | ||
High resolution TEM imaging and EDX elemental mapping was acquired for FeNiB-I to further understand the morphology and structural properties. In detail, the HRTEM image (Fig. 3a) of FeNiB-I shows no lattice fringes, as is expected from an amorphous structure. This is corroborated by the corresponding SAED pattern, where no diffraction rings are observed. Fig. 3c reveals Fe, Ni and B atoms evenly distributed across the spherical nanoparticles, evidence of a uniform FeNiB phase with no localised occurrences of NiB and/or FeB phases.
 | ||
| Fig. 3 In-depth characterisation of the morphology and elemental composition of the FeNiB-I electrocatalyst. (a) HRTEM image, (b) SAED pattern, (c) High Angle Annular Dark Field (HAADF) image, Energy Dispersive X-ray spectroscopy (EDAX-EDX) elemental mapping of Ni, Fe, B, overlay of Fe/Ni/B and EDX spectrum. | ||
The specific surface area, pore size distribution and pore volumes of the electrocatalyst powders were investigated, with a data summary presented in Table 2. Nitrogen adsorption–desorption isotherms (Fig. 4a–c), show a type II isotherm but with some minimal hysteresis observed between the adsorption and desorption curves at higher relative pressures, associated with the filling and emptying of mesopores by capillary condensation.37 Typical of type-II is the asymptotic increase of adsorbed nitrogen volume observed at relative pressure approaching one, indicating the existence of significant macroporosity. This type of isotherm and hysteresis loop are associated with powders that have a relatively low specific surface area and wedged shaped pores, often corresponding to the voids formed between agglomerated particles. In terms of specific surface area, all three electrocatalyst powders exhibited quite similar values, i.e. 18.8, 25.3 and 25.8 m2 g−1 for FeNiB-I, FeNiB-II and FeNiB-III, respectively. The surface area of FeNiB-I is noticeably lower than that of FeNiB-II and FeNiB-III, and it also has the lowest pore volume (0.054 cm3 g−1). A proportional relationship between surface area and pore volume is observed across the three electrocatalyst powders. The BJH average pore diameter values very dramatically across the sample set, however it must be noted that these values are, in effect, mode averages, representing the highest data point of each graph. The true mean pore diameter, as calculated across the full range of points in each graph, is reported in Column 5 of Table 2. It can be seen that the values for the samples are in close agreement and that they broadly occur around the midpoint in each of the BJH pore size distribution graphs Fig. 4d–f. These pores, or specifically, interparticulate void spaces, form within clusters of nanoparticles. They occur across the entire measurable range (1–50 nm) with the mean values falling firmly in the centre of the mesopore range.
| Electrocatalyst powder | Specific surface area (m2 g−1) | BJH average pore diameter (nm) | BJH total pore volume (cm3 g−1) | BET average pore diameter (nm) | Porosimetry average pore diameter (nm) | Porosimetry surface area (m2 g−1) |
|---|---|---|---|---|---|---|
| FeNiB-I | 18.8 | 1.9 | 0.054 | 11.4 | 12.5 | 18.2 |
| FeNiB-II | 25.3 | 3.6 | 0.065 | 10.1 | 15.3 | 29.8 |
| FeNiB-III | 25.8 | 20.4 | 0.083 | 12.8 | 24.1 | 23.0 |
 | ||
| Fig. 4 Nitrogen adsorption–desorption isotherms for (a) FeNiB-I, (b) FeNiB-II, and (c) FeNiB-III, BJH pore size distributions for (d) FeNiB-I, (e) FeNiB-II, and (f) FeNiB-III, and pore surface area distribution curves, with cumulative surface area overlaid (obtained from Hg porosimetry) for (g) FeNiB-I, (h) FeNiB-II, and (i) FeNiB-III. | ||
Mercury porosimetry was also employed to investigate the porosity and surface area, since the technique has a much larger pore diameter measurement range of 7–8000 nm, approx. Values of surface area measured using porosimetry are 18.2, 29.8, and 23.0 m2 g−1 respectively, for FeNiB-I, FeNiB-II and FeNiB-III, in line with the BET-measured values. Also shown in Table 2 are porosimetry average pore diameter (mode) values, falling in the mesopore range. These most widely occurring values are of the same size scale as the individual nanoparticles themselves, as revealed from TEM and SAXS analysis in Fig. 2 above.
Pore surface area distributions are plotted in Fig. 4g–i, which is the pore surface area with respect to pore diameter. Overlaid are the cumulative surface area curves. From these graphs, the contributions of different pore sizes to the surface area can be observed. For each electrocatalyst powder, the contribution from mesopores is apparent, as was also confirmed using BET analysis. However, there is a much more sizeable and broad contribution attributed to macropores, as large as 400 nm, approx. As mentioned previously, these are not pores within particles, but interparticulate void spaces which exist between agglomerates. The largest void spaces are believed due to the chain-like formations of particles, which join end-to-end, and are most noticeable in Fig. 2(c) above. In terms of the three samples, there are slight variations between the pore surface distributions, with a general shift to smaller void space sizes across the series.
The surface chemical properties of the electrocatalyst powders were studied by X-ray photoelectron spectroscopy (XPS). In the high-resolution spectra of Fe 2p (Fig. 5a–c), the peaks observed at low energy states at ∼707 eV and ∼719 eV originate from metallic Fe or Fe bonded with B,26,38,39 whereas, the four peaks at ∼711 eV, ∼724 eV and at ∼713 eV, ∼728 eV are assigned to Fe2+ and Fe3+, respectively.40 Taking into consideration the area of each deconvoluted peak, the FeNiB-III electrocatalyst powder exhibits the highest concentration of metallic Fe, or Fe bonded with B, when compared to FeNiB-I and FeNiB-II. This is expected since the mole ratio of Fe to Ni used during synthesis was 1:0.9. In relation to the high-resolution spectra of Ni 2p (Fig. 5d–f), the first peak doublets at ∼852 eV and ∼869 eV are assigned to metallic Ni or Ni bonded to B; the second doublet peaks at 855 eV and 873 eV are attributed to Ni3+.34,40 The FeNiB-I electrocatalyst powder exhibited the highest concentration of metallic Ni or Ni bonded to B when considering the area of each deconvoluted peak. This fits well considering the Fe to Ni mole ratio used for FeNiB-I during synthesis was 0.9:1. Lastly, in the high-resolution spectra of B 1 s (Fig. 5g–i), the peak observed at ∼187.9 eV is attributed to metalloid B(0), whereas the peak at ∼191.9 eV is assigned to oxidised boron.41 It is worth mentioning that the 0.9 eV shift at higher binding energies of the metalloid B(0) when compared to pure B (187.0 eV) is due to the fact that B in the FeNiB electrocatalyst powders acts as an electron donor, regulating the electronic properties of the connected Fe and Ni atoms.42
 | ||
| Fig. 5 X-ray photoelectron spectroscopy (XPS) analysis. High-resolution XPS spectra of Fe 2p, Ni 2p, and B 1s of the FeNiB-I (a, d, g), FeNiB-II (b, e, h), and FeNiB-III (c, f, i). | ||
The atomic ratio of Fe to Ni for all three electrocatalysts powders along with their allocated atomic percentage of Fe, and Ni are presented in Table 3, showing good control of Fe to Ni ratio manipulation using our proposed synthesis method. These values were calculated from their XPS survey spectra, without considering the oxygen contribution.
| Electrocatalyst powder | Fe (at%) | Ni (at%) | Fe/Ni atomic ratio |
|---|---|---|---|
| FeNiB-I | 26.23 | 30.43 | 0.86/1 |
| FeNiB-II | 32.51 | 32.29 | 1/1 |
| FeNiB-III | 25.94 | 23.70 | 1/0.91 |
Topographic images are shown in Fig. 6a–c, with average roughness values of 126 ± 25, 254 ± 23, and 67 ± 10 nm for FeNiB-I, FeNiB-II, and FeNiB-III, respectively (Table S2†), noting that AFM is a highly localized measurement that might not capture large area topographic variations. The maximum current was 4.9 ± 1.5 nA for FeNiB-I, 0.043.1 ± 0.02 nA for FeNiB-II, and 8.0 ± 5.6 nA for FeNiB-III in the ±3.0 V range used (Table S3†), showing no clear correlation with roughness. Resistances were 0.65 ± 0.21 GΩ for FeNiB-I, 82 ± 30 GΩ for FeNiB-II, and 0.78 ± 0.93 GΩ for FeNiB-III, (Table S4†), emphasising that FeNiB-II is by far the least conducting of the three electrocatalyst powders. Different thicknesses and homogeneities between samples could account for changes in measured currents. Whilst almost every location showed current above the 100 pA threshold for FeNiB-I and FeNiB-III, less than 22% of the IV curves showed current above a 20 pA threshold for FeNiB-II in the ±3.0 V range, supporting the finding that this sample is considerably less conductive overall. Fig. 6d shows a box plot of calculated resistance values of the samples, as determined from C-AFM IV measurements.
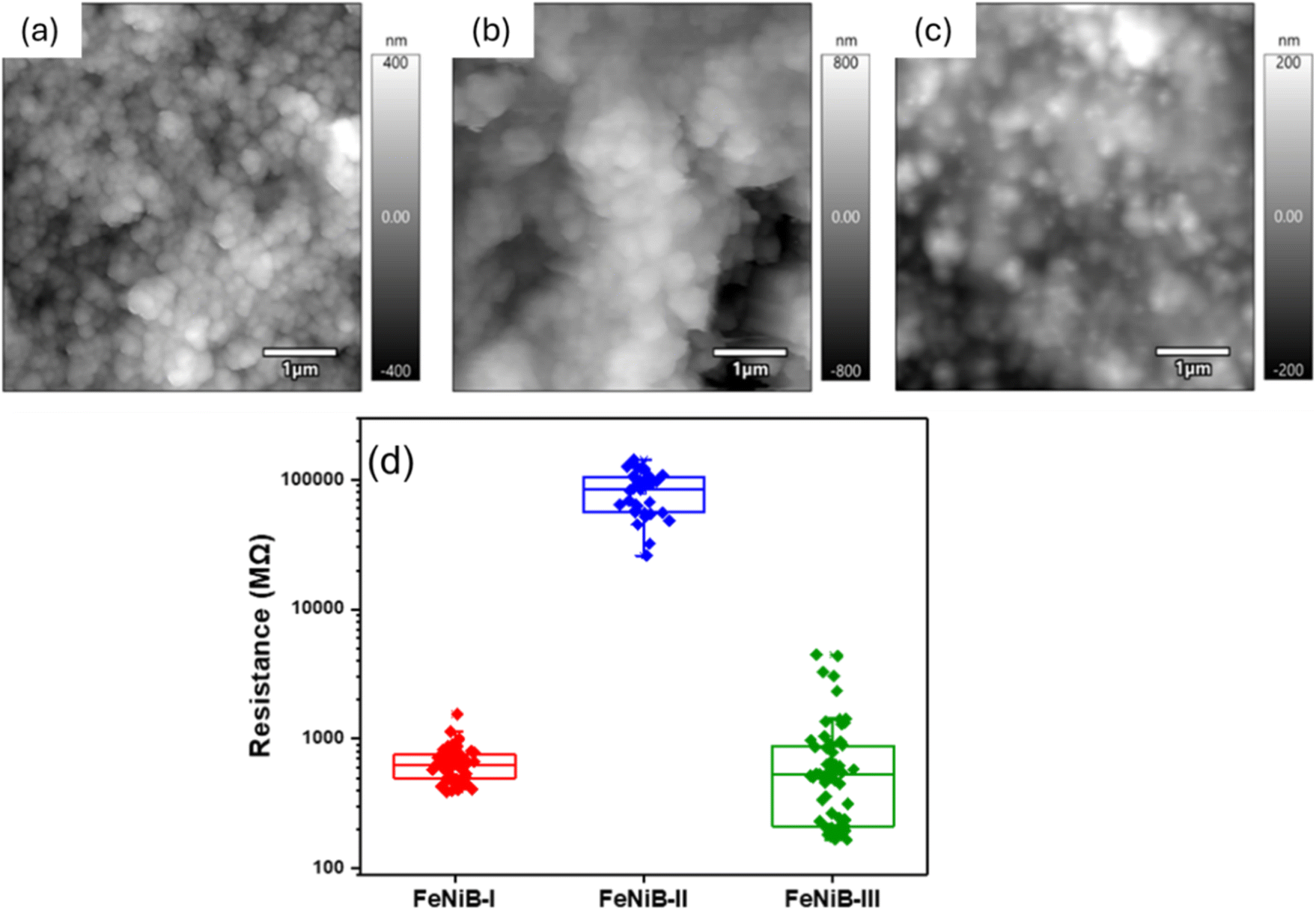 | ||
| Fig. 6 Topographical and electrical characterisation. Representative amplitude modulation mode topography images of (a) FeNiB-I, (b) FeNiB-II, (c) FeNiB-III, and (d) box plot of resistance, as determined from C-AFM IV measurements. | ||
The OER performances of the electrocatalyst powders were compared against commercial RuO2 and are presented in Fig. 7. All three electrocatalyst powders outperformed RuO2 in OER activity (Fig. 7a), reaching very high current densities equal to 1200 mA cm−2, comparable to industrial current densities values, thus demonstrating great potential for commercial water electrolysis applications.43 FeNiB-III exhibited the best OER activity, with the smallest Tafel slope equal to 44.2 mV dec−1 (Fig. 7b), and 252 mV overpotential at 10 mA cm−2 and 349 mV at 100 mA cm−2 (Fig. 7c). This enhanced activity may be attributed to its surface chemistry, where metallic Fe or Fe bonded to B may favour the absorption of OH* during the initial stages of OER. The next best performing sample was FeNiB-I, with a 84.8 mV dec−1 Tafel slope (Fig. 7b), and an overpotential equal to 287 mV at 10 mA cm−2 and 341 mV at 100 mA cm−2 (Fig. 7c). FeNiB-II under-performed relative to the other two samples, but still exhibited better OER activity in comparison to commercial RuO2. A slight curve is noticed in the polarisation curve of RuO2 which may be due to the surface formation and release of bubbles during water electrolysis or a surface reconstruction process during OER. In detail, the Tafel slope for FeNiB-II was 65.3 mV dec−1, exhibiting an overpotential equal to 307 mV at 10 mA cm−2 and 422 mV at 100 mV cm−2 (Fig. 7b and c), whereas, the Tafel slope for RuO2 was 86.9 mV dec−1, with 309 mV of an overpotential at 10 mA cm−2 and 449 mV at 100 mA cm−2 (Fig. 7b and c). Since the FeNiB-III was the best performing OER sample, it was chosen to undergo chronoamperometry testing at 1.8 V vs. RHE at a high current density (∼600 mA cm−2) in order to evaluate its durability. Its current density remained stable over time with an increase of only 0.7% during the 10 h stability test.
 | ||
| Fig. 7 OER performance of the electrocatalyst powders. (a) Polarization curves, (b) Tafel slopes, (c) overpotentials at 10 and 100 mA cm−2, (d) chronoamperometric curve of FeNiB-III at 1.8 V (vs. RHE) for 10 h. | ||
The proposed OER mechanisms in alkaline media are as follows:44
(1) Initially OH* forms on the electrocatalyst's surface, and then O* and *OOH intermediate species are formed in subsequent steps.
| OH− + * → OH* + e− | (3) |
| OH* + OH− → O* + H2O + e− | (4) |
| O* + OH− → *OOH + e− | (5) |
| *OOH + OH− → * + O2 + H2O + e− | (6) |
(2) Two adsorbed oxygen atoms couple to produce an oxygen molecule.
| O* + O* → O2 | (7) |
Fig. 8 presents the HER performances of the electrocatalyst powders compared to commercial PtC. It is interesting to note the same trend in HER electrocatalytic activity among the electrocatalyst powders as observed in OER, demonstrating true bifunctionality. PtC demonstrated the best HER activity, with a Tafel slope equal to 154.86 mV dec−1, and 129 mV of overpotential at −10 mA cm−2, and 489 at −100 mA cm−2. It must be pointed out, however, that FeNiB-III surpassed PtC in terms of HER activity at current densities higher than −300 mA cm−2, demonstrating the great potential of these electrocatalysts for commercial water electrolysis applications. FeNiB-III was the best performing of the electrocatalyst powders, exhibiting a Tafel slope equal to 89.6 mV dec−1, and 486 mV overpotential at −10 mA cm−2 and 587 at −100 mA cm−2. This improved activity may be attributed to the intrinsic properties of FeNiB-III, since it demonstrated higher conductivity according to C-AFM, and hence e− are able to reach the electrocatalyst's surface easier in comparison to the other two samples and promote the Volmer reaction step of HER. The second best performing sample was FeNiB-I, showing a Tafel slope of 208.5 mV dec−1, an overpotential equal to 463 mV at −10 mA cm−2 and 614 at −100 mA cm−2. FeNiB-II demonstrated the poorest HER activity but exhibited the smallest Tafel slope overall, including PtC, and was equal to 82.6 mV dec−1. Its overpotential at −10 mA cm−2 was 562 mV and at −100 mA cm−2 was 653 mV. The best performing electrocatalyst powder, FeNiB-III, was selected for chronoamperometry testing in order to test its durability at −0.64 V vs. RHE at a high current density value (∼−200 mA cm−2). Over a 10 h period, the current density remained relatively stable.
 | ||
| Fig. 8 HER performance of the electrocatalyst powders. (a) Polarization curves, (b) Tafel slopes, (c) overpotentials at 10 and 100 mA cm−2, (d) chronoamperometric curve of FeNiB-I at −0.64 V (vs. RHE) for 10 h. | ||
The proposed HER mechanisms in alkaline media are described in the following steps:45
(1) Molecular H2O, couples with an electron, resulting in an adsorbed hydrogen intermediate H* on the catalyst.
| H2O + e− + * → H* + OH−, Volmer reaction | (8) |
(2) The absorbed hydrogen intermediate H* combines then with molecular H2O and an electron to produce a hydrogen molecule.
| H* + H2O + e− → H2 + OH−, Heyrovsky reaction | (9) |
(3) Two adsorbed hydrogen atoms couple to produce a hydrogen molecule.
| H* + H* → H2, Tafel reaction | (10) |
The electrochemical surface (ECSA) was used to evaluate the structure–activity relationship of the FeNiB electrocatalyst powders in relation to OER and HER activity. A higher ECSA value indicates a higher number of active sites which can play a role in enhancing electrocatalytic activity. Fig. S4† shows the CV curves of the electrocatalyst powders at 20, 40, 60, 80, 100 and 120 mV s−1 scan rates, with FeNiB-I and FeNiB-III exhibiting very similar values of Cdl (Table S4†), equal to 132.46 and 127.86 μF cm−2, respectively. Whereas, the Cdl value for FeNiB-II was 94.26 μF cm−2. The ECSA for FeNiB-I was 3.32 cm2, for FeNiB-II was 2.36 cm2 and for FeNiB-III was 3.20 cm2. The OER and HER kinetics were further evaluated by EIS (Fig. S3†), in particular by comparing the obtained charge transfer resistance values of the electrocatalyst powders (Table S5†). The FeNiB-III exhibited the smallest charge transfer resistance value, and the highest OER and HER activity, in comparison with FeNiB-I and FeNiB-II. It interesting to observe that both FeNiB-I and FeNiB-III have very similar physicochemical properties in relation to specific surface area, particle size, ECSA and Cdl, with the only differences being that FeNiB-III exhibited a lower roughness, higher conductivity, and lower charge transfer resistance, which may be linked to the higher concentration of Fe0/Fe–B derived by the 1:0.9 Fe to Ni mole ratio used during synthesis. This illustrates the importance of having an adjustable electronic structure when it comes to electrocatalytic activity, independent maybe of the physicochemical properties. Adjustable electronic structure means controllable intermediate adsorption/desorption energy, and a tuneable reaction pathway, resulting in higher electrocatalytic activity. It is evident in the FeNiB electrocatalyst powders that a higher concentration of Fe0/Fe–B leads to better catalytic activity in both OER and HER, in comparison to the Ni0/Ni–B concentration.
Conclusions
Bimetallic iron nickel boride nanoparticles were successfully synthesised using a simple and up-scalable one-pot chemical reduction method. The ratio between iron and nickel was easily adjusted during synthesis, with both iron and nickel atoms proven to be evenly distributed across the nanoparticles. The nanoparticulate samples were shown to be amorphous, non-porous, and spherical, with well-matched particle size distributions. The presence of meso- and macro-porosity was attributed to interparticulate void spaces between the particles. The electrocatalyst powder with the highest concentration of metallic iron or iron bonded to boron exhibited higher conductivity and electrocatalytic activity both in OER and HER, reaching high current densities, especially for OER, on a par with industrial current densities. This improved activity may be attributed to the fact that surfaces with higher concentration of metallic iron or iron bonded to boron can favour the absorption of OH* species responsible for the first reaction steps of OER, whereas at the same time their higher conductive surface allow more e− to contribute towards the initial reaction steps in HER. Overall, this study demonstrates a practical and up-scalable method for synthesising amorphous bimetallic boride nanoparticles with controllable electronic structure for improved water electrolysis performance. Narrow changes in the molar ratio of Fe to Ni outside the 1/1 molar ratio, can have dramatic effects towards the activity of bimetallic boride electrocatalysts. We believe this work can prompt new strategies and ways of thinking in terms of engineering bimetallic boride nanoparticles (having adjustable electronic structures), with scope to scale-up for larger industrial applications.Data availability
The majority of the data supporting this article have been included as part of the ESI.† Additional data that support the findings of this work are available from the corresponding author upon reasonable request.Author contributions
The manuscript was written through contributions of all authors. All authors have given approval to the final version of the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
MVS acknowledges the financial support from the UCD Ad Astra fellowship programme, and the Royal Chemical Society for the Research Enablement Grant #72049. SJT, QZ and BJR acknowledge the financial support of Science Foundation Ireland (SFI) under the US-Ireland R&D Partnership Programme Grant Number SFI/21/US/3765. HR-TEM and EDX analysis were performed at the SFI-funded CRANN Advanced Microscopy Laboratory (AML).References
- Y. Liu, P. Vijayakumar, Q. Liu, T. Sakthivel, F. Chen and Z. Dai, Nano-Micro Lett., 2022, 14, 43 CrossRef CAS PubMed.
- K. Zhou, J. Huang, D. Xiang, A. Deng, J. Du and H. Liu, J. Energy Chem., 2024, 94, 340–356 CrossRef CAS.
- X. Li, L. Zhao, J. Yu, X. Liu, X. Zhang, H. Liu and W. Zhou, Nano-Micro Lett., 2020, 12, 1–29 CrossRef CAS PubMed.
- S. Jiang, H. Suo, T. Zhang, C. Liao, Y. Wang, Q. Zhao and W. Lai, Catalysts, 2022, 12, 123 CrossRef CAS.
- S. Fukuzumi, Y.-M. Lee and W. Nam, ChemSusChem, 2017, 10, 4264–4276 CrossRef CAS PubMed.
- J. M. Ogden, Annu. Rev. Energy, 1999, 24, 227–279 CrossRef.
- S. Y. Tee, K. Y. Win, W. S. Teo, L.-D. Koh, S. Liu, C. P. Teng and M.-Y. Han, Adv. Sci., 2017, 4, 1600337 CrossRef.
- B. Fu, V. Tzitzios, Q. Zhang, B. Rodriguez, M. Pissas and M. V. Sofianos, Nanomaterials, 2023, 13, 300 CrossRef CAS.
- F. A. Ghafar, D. Etherton, S. Liu, C. E. Buckley, N. J. English, D. S. Silvester and M. V. Sofianos, J. Electrochem. Soc., 2022, 169, 096507 CrossRef.
- S. Aralekallu, K. Sannegowda Lokesh and V. Singh, Fuel, 2024, 357, 129753 CrossRef CAS.
- M.-I. Jamesh, Y. Kuang and X. Sun, ChemCatChem, 2019, 11, 1550–1575 CrossRef CAS.
- M. Yang, T. Wei, J. He, Q. Liu, L. Feng, H. Li, J. Luo and X. Liu, Nano Res., 2024, 17, 1209–1216 CrossRef CAS.
- Z. Han, D. Zhang, H. Wang, G. Zheng, M. Liu and Y. He, Acta Phys. Sin., 2024, 40(9), 2307034 Search PubMed.
- T. Hou, T. Wei, Y. Wu, L. Zhang, J. Ding, Q. Liu, L. Feng and X. Liu X, J. Colloid Interface Sci., 2024, 15, 834–840 CrossRef.
- J. Luo, J.-H. Im, M. T. Mayer, M. Schreier, M. K. Nazeeruddin, N.-G. Park, S. D. Tilley, H. J. Fan and M. Grätzel, Science, 2014, 345, 1593–1596 CrossRef CAS PubMed.
- S. Cobo, J. Heidkamp, P.-A. Jacques, J. Fize, V. Fourmond, L. Guetaz, B. Jousselme, V. Ivanova, H. Dau and S. Palacin, Nat. Mater., 2012, 11, 802–807 CrossRef CAS PubMed.
- H. Wang, H.-W. Lee, Y. Deng, Z. Lu, P.-C. Hsu, Y. Liu, D. Lin and Y. Cui, Nat. Commun., 2015, 6, 7261 CrossRef CAS.
- S. Sanati, A. Morsali and H. García, Energy Environ. Sci., 2022, 15, 3119–3151 RSC.
- M. Streckova, O. Petrus, A. Guboova, R. Orinakova, V. Girman, C. Bera, M. Batkova, M. Balaz, J. Shepa and J. Dusza, J. Alloys Compd., 2022, 923, 166472 CrossRef CAS.
- P. Zhai, Y. Zhang, Y. Wu, J. Gao, B. Zhang, S. Cao, Y. Zhang, Z. Li, L. Sun and J. Hou, Nat. Commun., 2020, 11, 5462 CrossRef CAS PubMed.
- X. Zheng, X. Han, Y. Cao, Y. Zhang, D. Nordlund, J. Wang, S. Chou, H. Liu, L. Li, C. Zhong, Y. Deng and W. Hu, Adv. Mater., 2020, 32, 2000607 CrossRef CAS.
- Y. Xiong, Y. Yang, F. J. DiSalvo and H. D. Abruña, ACS Nano, 2020, 14, 13069–13080 CrossRef CAS PubMed.
- Q. Wang, Y. Song, X. Liu, H. Liang, S. Li, S. Wang, Y. Sun and Y. Zhang, Ionics, 2024, 30, 1523–1530 CrossRef CAS.
- L. Meng, H. Xuan, J. Wang, X. Liang, Y. Li, J. Yang and P. Han, Int. J. Hydrogen Energy, 2024, 51, 271–280 CrossRef CAS.
- Y. Huang, X. Liu, X. Li, X. Guo, T. Zhou, H. Feng, S. Li, Y. Zhu, J. Zhu and P. K. Shen, Sustainable Mater. Technol., 2022, 34, e00508 CrossRef CAS.
- W. Hong, S. Sun, Y. Kong, Y. Hu and G. Chen, J. Mater. Chem. A, 2020, 8, 7360–7367 RSC.
- V. G. Minkina, S. I. Shabunya, V. I. Kalinin, V. V. Martynenko and A. L. Smirnova, Int. J. Hydrogen Energy, 2012, 37, 3313–3318 CrossRef CAS.
- I. Bressler, B. R. Pauw and A. F. Thunemann, J. Appl. Crystallogr., 2015, 48, 962–969 CrossRef CAS.
- S. Brunauer, P. H. Emmett and E. Teller, J. Am. Chem. Soc., 1938, 60, 309–319 CrossRef CAS.
- E. P. Barrett, L. G. Joyner and P. P. Halenda, J. Am. Chem. Soc., 1951, 73, 373–380 CrossRef CAS.
- S. Ma, R. Farla, K. Bao, A. Tayal, Y. Zhao, Q. Tao, X. Yang, T. Ma, P. Zhu and T. Cui, Nanoscale, 2021, 13, 18570–18577 RSC.
- A. Chunduri, S. Gupta, O. Bapat, A. Bhide, R. Fernandes, M. K. Patel, V. Bambole, A. Miotello and N. Patel, Appl. Catal., B, 2019, 259, 118051 CrossRef CAS.
- F. A. Ghafar, D. Etherton, S. Liu, C. Buckley, N. English, D. Silvester-Dean and M. V. Sofianos, J. Electrochem. Soc., 2022, 169, 096507 CrossRef.
- X. Lin, V. Tzitzios, Q. Zhang, B. J. Rodriguez, A. Rafferty, R. Bekarevich, M. Pissas and M. V. Sofianos, Sustainable Energy Fuels, 2024, 13(2), 300 Search PubMed.
- N. Wang, A. Xu, P. Ou, S.-F. Hung, A. Ozden, Y.-R. Lu, J. Abed, Z. Wang, Y. Yan, M.-J. Sun, Y. Xia, M. Han, J. Han, K. Yao, F.-Y. Wu, P.-H. Chen, A. Vomiero, A. Seifitokaldani, X. Sun, D. Sinton, Y. Liu, E. H. Sargent and H. Liang, Nat. Commun., 2021, 12, 6089 CrossRef CAS PubMed.
- B. Ganem and J. O. Osby, Chem. Rev., 1986, 86, 763–780 CrossRef CAS.
- M. Lorenz, C. Paganini, G. Storti and M. Morbidelli, Materials, 2019, 12, 1580 CrossRef CAS.
- M. Arivu, J. Masud, S. Umapathi and M. Nath, Electrochem. Commun., 2018, 86, 121–125 CrossRef CAS.
- Z. Chen, R. Zheng, H. Zou, R. Wang, C. Huang, W. Dai, W. Wei, L. Duan, B.-J. Ni and H. Chen, Chem. Eng. J., 2023, 465, 142684 CrossRef CAS.
- J. Wang, S. Qing, X. Tong, K. Zhang, G. Luo, J. Ding and L. Xu, Appl. Surf. Sci., 2023, 640, 158330 CrossRef CAS.
- K. Zhang, G. Zhang, J. Qu and H. Liu, Small, 2018, 14, 1802760 CrossRef PubMed.
- J. Masa, S. Piontek, P. Wilde, H. Antoni, T. Eckhard, Y.-T. Chen, M. Muhler, U.-P. Apfel and W. Schuhmann, Adv. Energy Mater., 2019, 9, 1900796 CrossRef.
- J. K. Lee, C. Lee, K. F. Fahy, B. Zhao, J. M. LaManna, E. Baltic, D. L. Jacobson, D. S. Hussey and A. Bazylak, Cell Rep. Phys. Sci., 2020, 1, 100147 CrossRef CAS.
- X. Xie, L. Du, L. Yan, S. Park, Y. Qiu, J. Sokolowski, W. Wang and Y. Shao, Adv. Funct. Mater., 2022, 32, 2110036 CrossRef CAS.
- J. Wang, X. Yue, Y. Yang, S. Sirisomboonchai, P. Wang, X. Ma, A. Abudula and G. Guan, J. Alloys Compd., 2020, 819, 153346 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Additional materials characterizations and electrochemical measurements. See DOI: https://doi.org/10.1039/d4se01073f |
| This journal is © The Royal Society of Chemistry 2024 |