
Ni single-atom catalysts for highly efficient electrocatalytic CO2 reduction: hierarchical porous carbon as a support and plasma modification†
Qiulin
Ye
a,
Yaqi
Peng
*a,
Dongdong
Wang
a,
Jiabao
Lv
a,
Yaoyue
Yang
 b,
Yue
Liu
b,
Zhifu
Qi
c,
Songqiang
Zhu
c,
Chunliang
Ge
d,
Yan
Yang
d,
Angjian
Wu
*ac and
Shengyong
Lu
*a
b,
Yue
Liu
b,
Zhifu
Qi
c,
Songqiang
Zhu
c,
Chunliang
Ge
d,
Yan
Yang
d,
Angjian
Wu
*ac and
Shengyong
Lu
*a
aState Key Laboratory of Clean Energy Utilization, Zhejiang University, Hangzhou, 310027, P. R. China. E-mail: pengyaqi@zju.edu.cn; wuaj@zju.edu.cn; lushy@zju.edu.cn
bKey Laboratory of Basic Chemistry of the State Ethnic Commission, School of Chemistry and Environment, Southwest Minzu University, Chengdu 610041, Sichuan Province, China
cBaima Lake Laboratory, Hangzhou, 310053, P. R. China
dZhejiang Tiandi Environmental Protection Technology Co., Ltd., Hangzhou 310012, China
First published on 4th December 2023
Abstract
Electrocatalytic CO2 conversion promises a sustainable alternative to producing CO2-based green chemicals and fuels concomitant with the storage of fluctuating renewable energy. Herein, isolated Ni single-atom catalysts supported by nitrogen-doped hierarchical porous carbon (Ni-SACs/NHPC) were designed with large specific surface area (1115.6 m2 g−1) and abundant micropore structures. The synthesized Ni-SACs/NHPC demonstrated excellent activity (CO faradaic efficiency, FECO > 90%) for CO2 electrocatalytic reduction with a broad reaction potential window from −0.74 to −1.24 V vs. reversible hydrogen electrode (RHE), while flow cell design increased the current density while maintaining high CO faradaic efficiency. Additionally, non-thermal Ar-plasma was used to enhance the adhesion at the electrocatalyst–substrate interface, resulting in a significantly high CO partial current density (16.8 mA cm−2) achieved at −1.04 V vs. RHE with CO faradaic efficiency of ∼96%. Density functional theory (DFT) calculations revealed the highly dispersed single-atom Ni could effectively weaken the Ni–C bond energy and avoid the excessive accumulation of *CO to promote electrocatalytic CO2-to-CO conversion.
1. Introduction
The electrochemical CO2 reduction reaction (CO2RR) driven by renewable electricity (e.g. wind energy, solar energy, etc.) has been regarded as a sustainable route to produce green value-added chemicals and fuels while mitigating global CO2 emission.1,2 Nevertheless, high overpotential and low energy efficiency have greatly hindered the commercial development of CO2RR due to the stubborn C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O (∼806 kJ mol−1) activation and simultaneous generation of multiple carbon-based by-products at the approximate potential. Moreover, compared to the kinetically favored hydrogen evolution reaction (HER), the limited solubility of CO2 (0.033 mol L−1) in an aqueous electrolyte further impacts the reaction activity.3,4 Despite substantial endeavors devoted to optimizing electrocatalysts and reaction systems, merely a few C1 products demonstrate promising technology readiness levels and economic feasibility in terms of faradaic efficiency (FE) (>90%) and energy efficiency. The electrocatalytic conversion of CO2 into CO has attracted tremendous attention, given the estimated global CO market of over 2.18 billion dollars.5 However, the current state-of-the-art electrocatalysts still heavily rely on noble metal-based catalysts (e.g., Au and Ag), which are hindered by their high cost and limited availability.6–8
O (∼806 kJ mol−1) activation and simultaneous generation of multiple carbon-based by-products at the approximate potential. Moreover, compared to the kinetically favored hydrogen evolution reaction (HER), the limited solubility of CO2 (0.033 mol L−1) in an aqueous electrolyte further impacts the reaction activity.3,4 Despite substantial endeavors devoted to optimizing electrocatalysts and reaction systems, merely a few C1 products demonstrate promising technology readiness levels and economic feasibility in terms of faradaic efficiency (FE) (>90%) and energy efficiency. The electrocatalytic conversion of CO2 into CO has attracted tremendous attention, given the estimated global CO market of over 2.18 billion dollars.5 However, the current state-of-the-art electrocatalysts still heavily rely on noble metal-based catalysts (e.g., Au and Ag), which are hindered by their high cost and limited availability.6–8
Recently, earth-abundant transition metal-based single-atom catalysts (SACs) such as Ni, Zn and Fe-based catalysts, have been widely developed for the CO2RR towards CO production.9–14 Featured by homogeneous dispersing isolated atoms embedded in matrix substrate, SACs benefit from 100% atomic utilization efficiency and unique electronic structure. Particularly, atomically distributed active sites of SACs with a clear-defined coordination structure provide a promising platform for studying the structure–activity relationship.15–17 Actually, manipulating the essential parameters such as temperature or precursors during SACs preparation allows for the achievement of different coordination structures.18–21 To further boost the efficient electron transfer and mass transportation, it is crucial to optimize the support matrix materials (e.g., metal–organic frameworks, carbon nanotubes, graphene, etc.,) for SACs design.22 As reported, carbon support with the plentiful distribution of mesopores within the range of 7–10 nm facilitates CO2 diffusion, while hierarchical pores contribute to lower reaction barriers.23 Additionally, the carbon matrix with high content of nitrogen could play a role in anchoring the adjacent Ni2+via nitrogen and preventing Ni atoms from agglomeration.24 Therefore, nitrogen-doped hierarchical porous carbon with a large specific surface area and abundant pore structures was proposed as a support material to enhance reaction activity.
Furthermore, the impact of electrocatalyst–substrate interface on the activity of electrocatalytic reaction should be recognized, although seldom research was done. Ambient non-thermal plasma (NTP) with the advantages of relatively low treatment temperature and rich reactive species, has been widely used for material modification.25 Recent studies have demonstrated plasma treatment with different carrier gases (such as O2, Ar or N2 gas) could alter the surface properties (such as the wettability, defect sites and roughness), leading to enhanced adhesion at the electrocatalyst–substrate interface and overall improvement in reaction performance.26–29 For instance, the utilization of the plasma-enhanced CuCo alloy–substrate interface indicated significant advancements in electrocatalytic reaction performance by optimizing the interface characteristics and promoting the formation of intermediates.30
Herein, we designed atomically dispersed Ni SACs with nitrogen-doped hierarchical porous carbon as the support (Ni-SACs/NHPC) using a facile and scalable step-pyrolysis strategy. The prepared Ni-SACs/NHPC demonstrated excellent activity for electrocatalytic CO2-to-CO conversion within a broad reaction potential window, approaching the highest FE (98.25%) and CO partial current density of 14.97 mA cm−2 at −1.04 V vs. RHE. Additionally, the FECO maintained about 88.2% at 100 mA cm−2 with flow cell. Based on the optimization of the electrocatalyst–substrate interface by Ar plasma, CO partial current density increased to 16.8 mA cm−2 accompanied with high FE. Eventually, in situ electrochemical attenuated total-reflection surface-enhanced infrared adsorption spectroscopy (ATR-SEIRAS) combined with density functional theory (DFT) calculations was used to reveal active sites and reaction mechanism, which revealed CO2 could be electrochemically converted into CO through intermediate *COOH.
2. Experimental
2.1. Materials
The following chemicals were employed without further purification for the synthesis of Ni-SACs/NHPC, including α-cellulose (AR, Aladdin), (NH4)2C2O4 (Shanghai Chemical Reagents, China), KHCO3, Ni(NO3)2·6H2O, α-D-glucose (≥99.5%, RHAWN) and melamine (99%, Aladdin).2.2. Sample preparation
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :3:1, while KHCO3 served as the foaming agent. The mixture was put into a programmed temperature control furnace under N2 atmosphere and calcined at 900 °C for 1 h with a heating rate of 10 °C min−1, followed by dissolved in 500 mL deionized water and magnetically stirred at room temperature for 24 h. Then, the solution was filtered and washed with deionized water until the pH of the filtrate reached 7. Finally, NHPC was obtained after the filtered products being dried at 105 °C in an oven.
:3:1, while KHCO3 served as the foaming agent. The mixture was put into a programmed temperature control furnace under N2 atmosphere and calcined at 900 °C for 1 h with a heating rate of 10 °C min−1, followed by dissolved in 500 mL deionized water and magnetically stirred at room temperature for 24 h. Then, the solution was filtered and washed with deionized water until the pH of the filtrate reached 7. Finally, NHPC was obtained after the filtered products being dried at 105 °C in an oven.
Then, 0.6 g of NHPC, 0.87 g of Ni(NO3)2·6H2O, 12 g of α-glucose and deionized water (50 mL) were mixed under ultrasonic treatment for 1 h. The obtained mixture was centrifuged at 10000 rpm for 5 min and dried at 80 °C overnight, followed by mixing with melamine with a mass ratio of 1:5. Eventually, Ni-SACs/NHPC was obtained after calcination at 800 °C (5 °C min−1) for 2 h under N2 atmosphere.
3. Results and discussion
3.1. Structure of the catalyst
Ni-SACs/NHPC composite was synthesized in Scheme 1 through a step-pyrolysis method using nitrogen-doped hierarchical porous carbon (NHPC) as carbon support. Initially, the NHPC was prepared using a foaming method, resulting in a carbon matrix abundant with defect sites and pore structure. Then, the obtained NHPC was mixed with glucose and melamine, followed by further pyrolyzing at 800 °C for 2 h under N2 atmosphere. This pyrolysis process facilitated the reduction of Ni2+ into metallic Ni while simultaneously incorporating N atoms into NHPC, thus forming Ni-SACs/NHPC. The as-prepared Ni-SACs/NHPC maintained the original hierarchical porous structure with a three-dimensional interconnected pore structure (Fig. 1a and S1†). According to the enlarged high-angle annular dark-field scanning transmission electron microscope (HAADF-STEM) image, homogeneous dispersion of Ni single atom was observed on the self-supported 3D skeleton of NHPC, where the bright dots corresponded to isolated Ni atoms (Fig. 1b). The corresponding energy dispersive spectrometer (EDS) mapping exhibited the homogeneous distribution of Ni, N, C and O over the whole electrocatalyst surface without any noticeable aggregation of Ni metal atoms (Fig. S2†). Further, high-resolution transmission electron microscopy (TEM) images displayed the absence of Ni nanoparticles on the surface of carbon support (Fig. S1†), while lattice-distorted carbon defects were also observed, which could be attributed to the coordination of dispersed Ni SACs with N atoms.32 The X-ray diffraction (XRD) pattern of Ni-SACs/NHPC only possessed typical carbon (002) and (100) facets with the diffraction peaks at 2θ of 24.0° and 44.2°, respectively, without any evident peaks related to Ni crystals, consistent with the previous characterization.33 The mass loading of Ni in the Ni-SACs/NHPC determined by inductively coupled plasma mass spectroscopy (ICP-MS) was about 1.82 wt%. | ||
| Scheme 1 The synthesis process of Ni-SACs/NHPC electrocatalyst. | ||
 | ||
| Fig. 1 (a and b) The aberration-corrected HAADF-STEM images of Ni-SACs/NHPC; (c) XRD pattern of Ni-SACs/NHPC; (d) comparison of nitrogen adsorption–desorption isotherm of NHPC and Ni-SACs/NHPC; (e) pore width distributions of 0.4–5.5 nm calculated by the DFT method; (f) pore width distributions of 3–50 nm calculated by the BJH method. | ||
In addition, N2 adsorption–desorption test was performed to estimate the surface area and porous structure of NHPC and Ni-SACs/NHPC, respectively (Fig. 1d–f and S3†). It was observed that the N2 adsorption–desorption isotherm curves of both Ni-SACs/NHPC and NHPC exhibited the combined characteristics of type I and IV isotherm.34 The specific surface area of Ni-SACs/NHPC was as large as 1115.6 m2 g−1, with rich micropores (Smicro, 1001.3 m2 g−1) and mesopores (Smeso, 114.3 m2 g−1) structure (Table S1†). It was speculated that micropores could provide more active sites and mesopores could promote mass transfer to promote a high CO2RR catalytic activity.35,36
To explore the electronic state and coordination structure of Ni sites at an atomic level, X-ray photoelectron spectroscopy (XPS) combined X-ray absorption spectroscopy was investigated (Fig. S4†). The Ni 2p XPS spectra indicated that the binding energy of Ni 2p3/2 peak was around 855.2 eV, between that of Ni0 (853.4 eV) and Ni2+ (855.6 eV),37,38 which implied that the valence state of Ni was between 0 and 2 (Fig. 2a). The C 1s spectra can be deconvoluted into four typical peaks at 284.75 eV (C–C), 285.00 eV (C–N), 285.84 eV (C–O) and 288.94 eV (CO),39 suggesting that N-species were successfully doped into the carbon matrix (Fig. 2b and Table S2†). By fitting the N 1s peak for Ni-SACs/NHPC, the structure of pyridinic-N (∼398.51 eV), Ni–N (∼399.27 eV), pyrrolic-N (∼400.86 eV), graphitic-N (∼401.75 eV) and oxidized-N (∼404.50 eV) was observed, where the content of Ni–N was 24.46%, which implied the favored Ni–N bonds formed in Ni-SACs/NHPC, consistent with the results of the X-ray absorption fine structure (XAFS) (Fig. 2c and Table S3†).40
 | ||
| Fig. 2 (a) High-resolution XPS Ni 2p spectra of Ni-SACs/NHPC; (b) C 1s spectra and (c) N 1s spectra of Ni-SACs/NHPC; (d) Ni K-edge XANES (e) Ni K-edge Fourier transformed XAFS spectra in the R space of Ni-SACs/NHPC, with Ni foil and NiO as references; (f) the EXAFS fitting in K-space for Ni-SACs/NHPC; the corresponding wavelet transform of (g) NiO; (h) Ni foil; and (i) Ni-SACs/NHPC. | ||
X-ray absorption near edge structure (XANES) and extended X-ray absorption fine structure (EXAFS) of Ni K-edge were performed to further determine the electronic structure and chemical configuration of the Ni-SACs/NHPC. Different from the reference samples (Ni foil and NiO), the Ni adsorption edge of Ni-SACs/NHPC showed similar D4h symmetry to that of nickel phthalein cyanide (NiPc),41 where marked peak A represented 1s to 3d transition, B represented 1s to 4pz transition, C represented 1s to 4px,y transitions and D represented multiple scatter processes (Fig. 2d). The main peak observed from the Fourier transform of the EXAFS spectra for the Ni-SACs/NHPC was 1.86 Å, without the presence of Ni–Ni coordination (typical peak at 2.49 Å) observed in the Ni foil sample. In contrast, the Fourier transform of the EXAFS for the NiO sample displayed the peaks of the Ni–O coordination and the Ni–Ni coordination at 2.11 Å and 2.98 Å, respectively (Fig. 2e). Similarly, the wavelet transforms of Ni-SACs/NHPC exhibited the main intensity maximum at 4.2 Å (Fig. 2i), assigning to the Ni–N coordination, while that of Ni foil sample was at 7.1 Å (Fig. 2h). To further understand the bonding environment of Ni SACs, EXAFS fitting for the R and k space were performed. The average coordination number of Ni–N of Ni-SACs/NHPC was calculated to be 3.9 ± 1.2 and the average bond length was 1.86 ± 0.03 Å (Table S4†), which indicated the formation of Ni–N4 structure in the prepared Ni-SACs/NHPC.
3.2. Electrochemical reaction activity of CO2RR
The electrocatalytic CO2RR activities of different Ni-based electrocatalysts (Ni-sputter and Ni-SACs/NHPC) were comparatively investigated in a typical sealed H-type cell. The cathode electrolyte consisted of CO2-saturated 0.1 M KHCO3 with continuous purging of CO2 at a gas flow rate of 30 mL min−1, while the anode electrolyte contained 0.1 M KHCO3 using platinum as the counter electrode. Ni-sputter catalysts were prepared by magnetically sputtering, to investigate the size effect of the metal between bulk Ni and Ni SACs. Online gas chromatography (GC) and 1H nuclear magnetic resonance (NMR) were used to quantify the gas-phase and liquid-phase products, respectively. The results indicated that CO and H2 were the only gas-phase products without the occurrence of other carbon-based products in the liquid phase (Fig. S6†). Compared to the utilization of inert N2 as feeding gas, the current density of CO2-saturated electrolytes was magnitude order higher, confirming the successful occurrence of CO2RR over Ni-SACs/NHPC. Interestingly, both Ni-SACs/NHPC and Ni-sputter catalysts exhibited increasing current densities with potential, but the selectivity of the products differed significantly (Fig. S7†). For Ni-sputter electrocatalysts, only H2 was generated, with increasing the potential exacerbating the HER. In contrast, nearly 100% FE of CO was detected using Ni-SACs/NHPC electrocatalysts, indicating that reducing the Ni size from bulk-scale to single-atom scale dramatically switched the product selectivity from H2 to CO (Fig. 3b and c). Furthermore, the long-term stability of the CO2RR using Ni-SACs/NHPC was evaluated at −0.94 V vs. RHE, revealing a slight decrease in total current density after 18 h of continuous operation while maintaining high selectivity towards CO production (Fig. 3d). Besides, the performance of previously reported works based on Ni-based single-atom catalysts for electrocatalytic CO2-to-CO conversion in H-type cell were also compared, indicated in Table S5,† which revealed prepared Ni SACs/NHPC for CO generation exhibiting superior activity. | ||
| Fig. 3 (a) The linear sweep voltammetric (LSV) curves of Ni-SACs/NHPC in CO2-saturated 0.1 M KHCO3 electrolyte at a scan rate of 50 mV s−1 and N2-saturated 0.1 M KHCO3 electrolyte at a scan rate of 5 mV s−1. (b) The effect of potential on the product FEs over different electrocatalysts (Ni-sputter and Ni-SACs/NHPC); (c) the effect of potential on the variation of CO partial current densities and H2 partial current densities over different electrocatalysts (Ni-sputter and Ni-SACs/NHPC); (d) the stability test for Ni-SACs/NHPC at −0.94 V vs. RHE. | ||
To further evaluate the activity of the catalyst, gas diffusion electrodes with Ni-SACs/NPHC coated were tested in a flow cell system using chronopotentiometry, while the detailed set was described in ESI.† It was observed that the FECO firstly increased and then decreased with the increase of set current density. In the flow cell, the gas diffusion was greatly improved compared to the conventional H-cell. The maximum value of FECO approached about 88.2% at 100 mA cm−2. However, when the current density further increased from 150 mA cm−2 to 200 mA cm−2, the FECO significantly decreased. This decline might be attributed to water flooding of the cathode electrode during reactions, leading to an intensified hydrogen evolution reaction and inhibiting the occurrence of CO2RR (Fig. S8†).
3.3. NTP modification on the electrocatalyst–substrate interface for CO2RR
To get insight into the effect of NTP treatment on electrocatalytic CO2RR activity, different injected powers of NTP were investigated in terms of FECO and CO partial current density (Fig. 4a and b). In general, a slight change of FECO was observed using carbon substrate with and without NTP treatment. Both the Ni-SACs/NHPC50 and Ni-SACs/NHPC100 exhibited high FECO with a relatively broad potential window. Ni-SACs/NHPC100 demonstrated higher CO partial current density than Ni-SACs/NHPC50 and Ni-SACs/NHPC regardless of the potential variation and reached its peak value of 25 mA cm−2 at −1.24 V vs. RHE, which increased by 16.4% compared to that of Ni-SACs/NHPC, implying a favored charge exchange at the Ar-plasma modified electrocatalyst–substrate interface. By further increase of plasma power (150 W), higher CO partial current density was observed at the expense of the decrease of FE. Furthermore, electrochemical characteristics including electrochemical active surface area (ECSA), Tafel slopes and electrochemical impedance tests were compared (Fig. 4c–e). The ECSA was calculated based on the double-layer capacitance (Cdl), which indicated the Cdl of Ni-SACs/NHPC100 (2.398 mF cm−2) was higher than the Cdl of Ni-SACs/NHPC (1.92 mF cm−2) and Ni-SACs/NHPC50 (1.132 mF cm−2), respectively (Fig. S9†). According to Tafel slopes, much lower values of Ni-SACs/NHPC50 (361 mV dec−1) and Ni-SACs/NHPC100 (388 mV dec−1) were observed compared to the counterparts without Ar plasma treatment (420 mV dec−1), which confirmed the favored reaction kinetic via substrate modification. Similarly, the smallest semicircle diameter was observed using Ni-SACs/NHPC100 electrocatalyst based on the Nyquist plots, which meant that Ni-SACs/NHPC100 possessed the lowest charge transfer resistance (Rct) and fastest charge transfer process. And Ni-SACs/NHPC100 maintained long-term stability at −1.04 V vs. RHE with relatively high FE.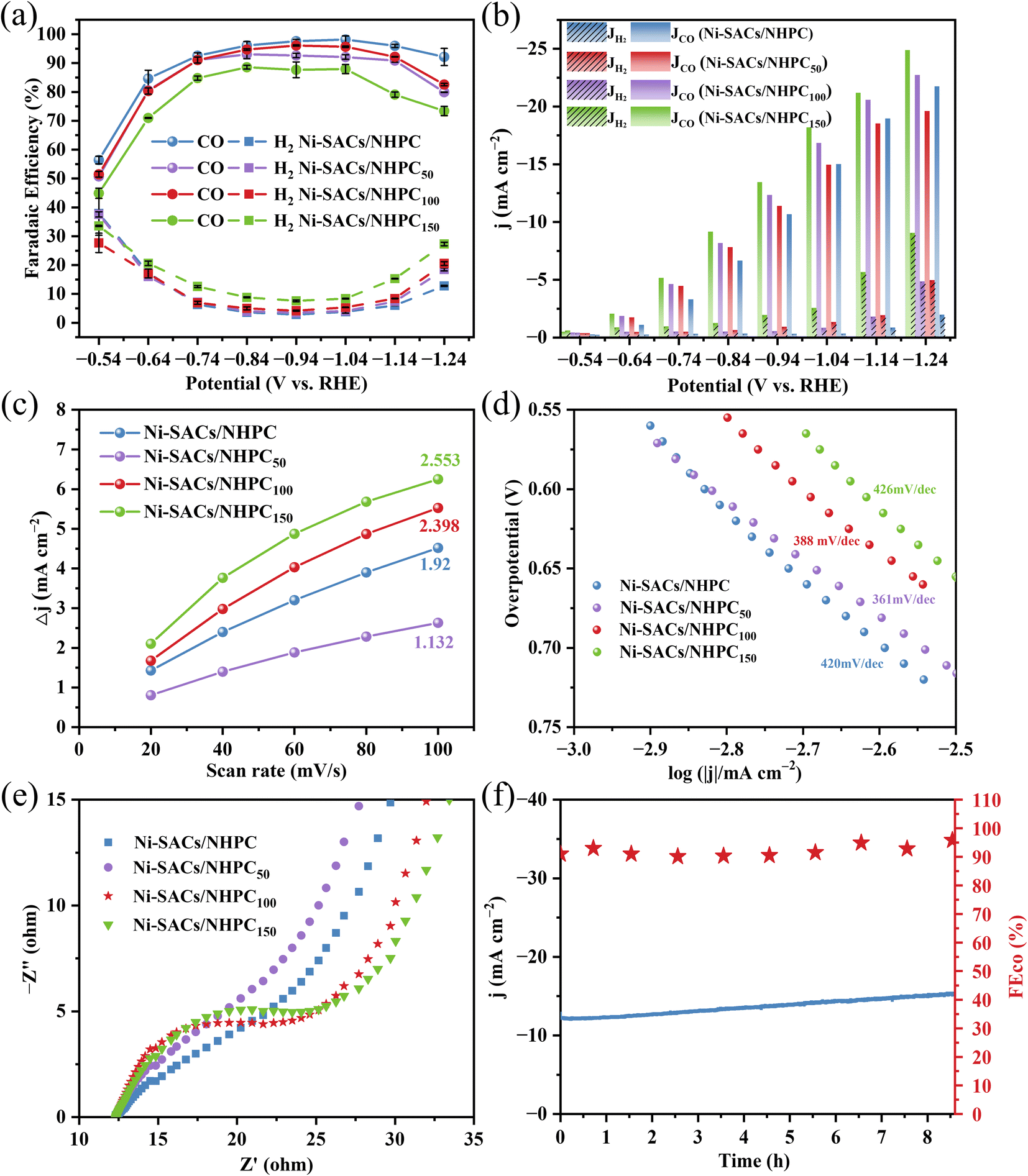 | ||
| Fig. 4 Ar-plasma modification on Ni-SACs/NHPC electrocatalyst. The effect of ICP power (50 W, 100 W and 150 W) on (a) FE and (b) CO partial current densities with the potential varying from −0.54 to −1.24 V vs. RHE; (c) electrochemically active surface areas (ECSAs) tests based on the double-layer capacitance of the different electrocatalysts. (d) Tafel plots and (e) Nyquist plots of Ni-SACs/NHPC using different ICP-treated carbon substrates. (f) The stability test for Ni-SACs/NHPC100 at −1.04 V vs. RHE. | ||
Then, the atomic force microscopy (AFM) and contact angle test were applied to evaluate the surface characteristics of the carbon substrate by NTP modification. The plasma-treated carbon substrate demonstrated abundant fine spines, distinct from the flat and smooth surface without plasma treatment (Fig. S10†). Based on the calculation of average surface roughness (Ra), the Ra of Ni-SACs/NHPC100 was 4.6 nm, which was much larger than the pristine carbon substrate with Ra of about 0.854 nm. As a result, the contact angle of the carbon substrate decreased from 153.8° to 125.7° after Ar-plasma treatment. It was speculated that the plasma treatment would change the wettability of the pristine carbon paper, while the decrease of the hydrophobicity might promote higher CO partial current density.
3.4. DFT calculation and in situ ATR-SEIRAS characterization
To explore the mechanism of CO2RR and identify the key intermediates, in situ attenuated total reflection surface enhanced infrared reflection absorption spectroscopy (ATR-SEIRAS) was performed under a potential range from −0.3 V to −1.25 V vs. RHE (Fig. 5a). The band centered at 2342 cm−1 was assigned to the fast desorption of CO2 and the bands that appeared between 2033 cm−1 and 1920 cm−1 were attributed to *CO.42,43 Peaks at 1760 cm−1 and 1230 cm−1 were considered to be CO stretching of the intermediate *COOH and HCO3−, respectively.44–46 The signal of intermediates *COOH and *CO began to appear at ∼−0.45 V to −0.55 V vs. RHE, which indicated the initiation of CO2RR. Negatively shifting the potential to −0.65 V vs. RHE, the appearance of the CO2 band might be related to the strong adsorption of CO2 under the electric field and lead to the accumulation of CO2 on the surface. Furthermore, the band intensity for integrated *CO achieved its maximum value at −0.95 V vs. RHE, consistent with the electrochemical evaluation.
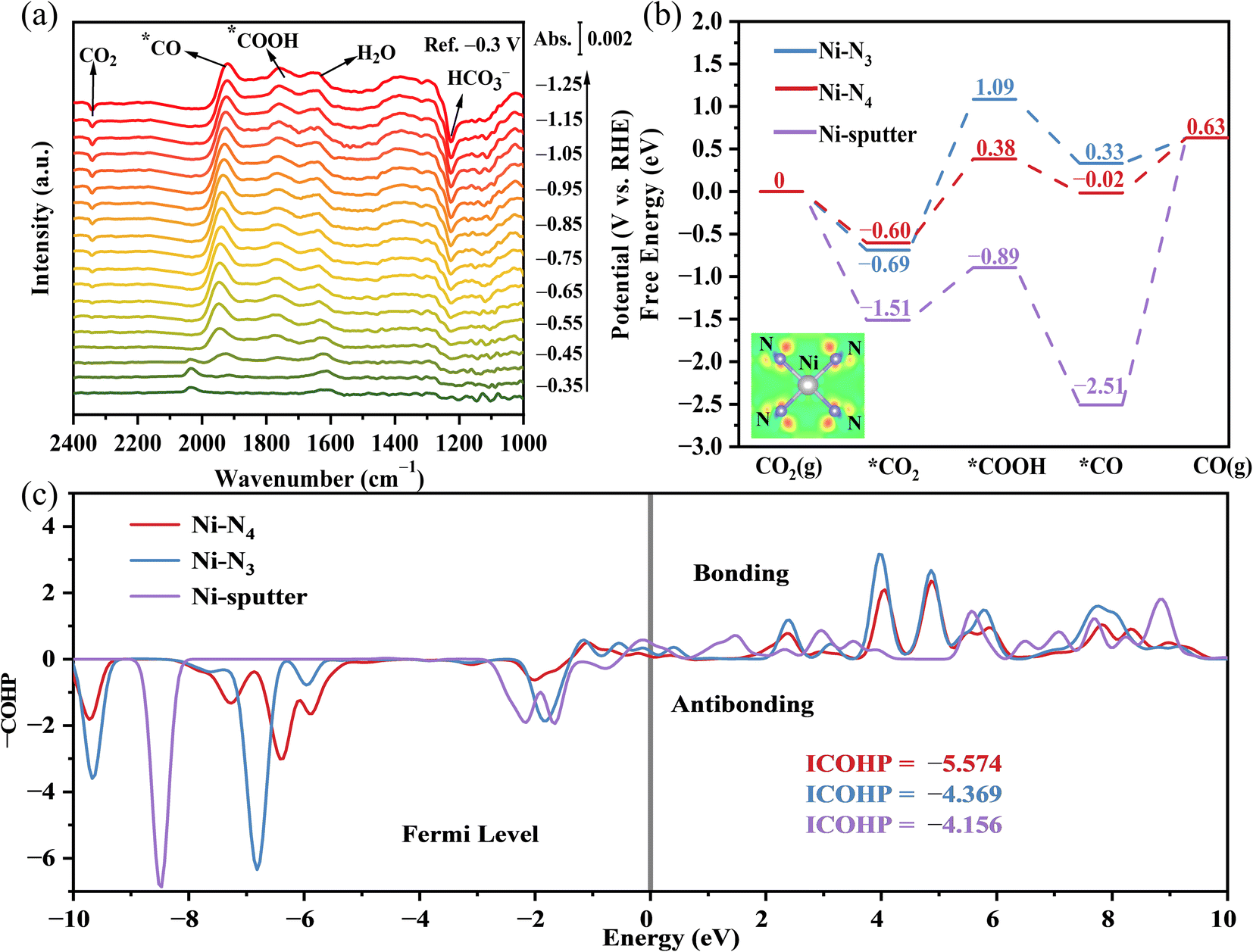 | ||
| Fig. 5 (a) In situ ATR-SEIRAS results of Ni-SACs/NHPC in 0.1 M KHCO3 electrolyte at different potentials; (b) reaction free-energy diagram for electrocatalytic CO2RR to CO on different catalysts. The inset images demonstrated the cross-section of charge density difference on Ni–N4 single atom configuration. The gray atoms represented Ni atoms and the purple atoms represented N atoms. The blue and red areas represented electron dissipation and electron accumulation. (c) COHP of Ni–C bond of *CO, the negative value indicates a bonding orbital. | ||
DFT calculations were performed to reveal the thermodynamic characteristics during CO2RR. The minimal rate-determining step (RDS) energy barrier of the N4-coordinated Ni SACs (0.60 eV) demonstrated the origin of the excellent catalytic activity of Ni-SACs/NHPC than Ni–N3 (1.09 eV) and Ni-sputter (2.51 eV), consistent with the accumulation band of *COOH in ATR-SEIRAS spectra. In contrast, higher CO2 adsorption (−1.51 eV than −0.69 eV of Ni–N3 and −0.60 eV of Ni–N4, respectively) and lower CO desorption (+2.51 eV than −0.33 eV of Ni–N3 and +0.02 eV of Ni–N4, respectively) resulted into the fast accumulation of adsorbed CO2 and intermediates over the catalyst surface, thereby hindering the total electrocatalytic reaction rate.
To explore the catalytic activity improvement by adjusting the local electronic environment of Ni SACs under N4 coordination, the differential charge density of Ni atoms coordinated to N4 was simulated as shown in the inset image of Fig. 5b. The electron dissipation around Ni and the electron accumulation around N demonstrated the construction of unsaturated electron-losing Ni active center, which modulated the ability of Ni to adsorb CO2. On the other hand, the occupation of active sites by CO poisoning also inhibited the catalyst activity, and Crystal Orbital Hamilton Population (COHP) calculations were performed to judge the resistance to CO poisoning by quantitatively analyzing the *CO adsorption on different catalyst surfaces (Fig. 5c). Since CO tends to form hollow adsorption by Ni cluster (three Ni–C bonds), strong Ni–C bonding energy (−4.156 × 3 = −12.468) was difficult to avoid CO poisoning. The highly dispersed single-atom Ni weakened the Ni–C bond energy, thereby avoiding the excessive accumulation of *CO and promoting the catalyst performance. Moreover, the comparison of Ni–N4 (−4.369) and Ni–N3 (−5.574) also highlights the ability of the Ni–N4 catalyst to release *CO after CO2 reduction.
4. Conclusions
In summary, isolated Ni SACs dispersed on hierarchically porous N-doped carbon were successfully synthesized. Due to a large specific surface area of carbon support with abundant microscopes and mesopores, Ni-SACs/NHPC demonstrated outstanding activity and stability for CO2 electrochemical reduction, with the highest CO partial current density (14.97 mA cm−2) and FECO of 98.25% at −1.04 V vs. RHE in a H-type cell while the FECO reached about 88.2% at 100 mA cm−2 in a flow cell. In addition, Ni-SACs/NHPC with NTP-treated carbon substrate exhibited high partial CO current density, which was attributed to improved charge transfer caused by surface roughness and wettability change. According to ATR-SEIRAS spectra and DFT calculations, the cumulative *COOH signal combined with the highest energy barrier of *CO2 to *COOH revealed the formation of *COOH was the rate-determining step during CO2RR. Local electron and orbital analysis demonstrated that the highly dispersed Ni–N4 promoted the evolution of intermediates and accelerated the dissociation of target product CO, which contributed to the excellent performance of Ni-SACs/NHPC.Author contributions
Q. Y. carried out the experiments, conducted the synthesis of catalysts and performed characterizations. A. W. conceived the project. Q. Y., A. W. and Y. P. wrote the manuscript. D. W. performed characterizations. J. L. calculated the DFT results. Y. Y. and Y. L. carried out the in situ ATR-SEIRAS experiments. Z. Q., S. Z., C. G., Y. Y. and S. L. analyzed the experimental results and revised the manuscript. All authors discussed the results and commented on the manuscript.Conflicts of interest
The authors declare no competing interests.Acknowledgements
This work was supported by the Pioneer R&D Program of Zhejiang Province (2022C03040), “the Fundamental Research Funds for the Central Universities” (226-2022-00159; 2022LHJH01-03; 2022ZFJH004; 2022QZJH14) and the Ecological Civilization Project, Zhejiang University.References
- Y. Zhu, X. Yang, C. Peng, C. Priest, Y. Mei and G. Wu, Small, 2021, 17(16), 2005148 CrossRef CAS.
- X. Lv, Q. Liu, J. Wang, X. Wu, X. Li, Y. Yang, J. Yan, A. Wu and H. B. Wu, Appl. Catal., B, 2023, 324, 122272 CrossRef CAS.
- H. B. Yang, S.-F. Hung, S. Liu, K. Yuan, S. Miao, L. Zhang, X. Huang, H.-Y Wang, W. Cai, R. Chen, J. Gao, X. Yang, W. Chen, Y. Huang, H. M. Chen, C. M. Li, T. Zhang and B. Liu, Nat. Energy, 2018, 3(2), 140–147 CrossRef CAS.
- M. Konig, S. H. Lin, J. Vaes, D. Pant and E. Klemm, Faraday Discuss., 2021, 230, 360–374 RSC.
- T. T. Zheng, K. Jiang and H. T. Wang, Adv. Mater., 2018, 30(48), 1802066 CrossRef.
- T. N. Huan, P. Prakash, P. Simon, G. Rousse, E. Gravel, E. Doris and M. Fontecave, ChemSusChem, 2016, 9(17), 2317–2320 CrossRef CAS.
- M. Ma, B. J. Trzesniewski, J. Xie and W. A. Smith, Angew. Chem., Int. Ed., 2016, 55(33), 9748–9752 CrossRef CAS.
- H. Lee, S. K. Kim and S. H. Ahn, J. Ind. Eng. Chem., 2017, 54, 218–225 CrossRef CAS.
- P. Su, K. Iwase, S. Nakanishi, K. Hashimoto and K. Kamiya, Small, 2016, 12(44), 6083–6089 CrossRef CAS.
- Z. Chen, X. Zhang, W. Liu, M. Jiao, K. Mou, X. Zhang and L. Liu, Energy Environ. Sci., 2021, 14(4), 2349–2356 RSC.
- C. H. Zhang, S. Z. Yang, J. J. Wu, M. J. Liu, S. Yazdi, M. Q. Ren, J. W. Sha, J. Zhong, K. Q. Nie, A. S. Jalilov, Z. Y. Li, H. M. Li, B. I. Yakobson, Q. Wu, E. L. Ringe, H. Xu, P. M. Ajayan and J. M. Tour, Adv. Energy Mater., 2018, 8(19), 1703487 CrossRef.
- H. N. Zhang, J. Li, S. B. Xi, Y. H. Du, X. Hai, J. Y. Wang, H. M. Xu, G. Wu, J. Zhang, J. Lu and J. Z. Wang, Angew. Chem., Int. Ed., 2019, 58(42), 14871–14876 CrossRef CAS.
- W. Yuan, Y. Ma, H. Wu and L. Cheng, J. Energy Chem., 2022, 65, 254–279 CrossRef CAS.
- J. Li, J. Liu and B. Yang, J. Energy Chem., 2021, 53, 20–25 CrossRef CAS.
- Q. Zhang and J. Guan, Adv. Funct. Mater., 2020, 30(31), 2000768 CrossRef CAS.
- W. Ju, A. Bagger, G. P. Hao, A. S. Varela, I. Sinev, V. Bon, B. Roldan Cuenya, S. Kaskel, J. Rossmeisl and P. Strasser, Nat. Commun., 2017, 8, 944 CrossRef.
- C. Jia, S. Li, Y. Zhao, R. K. Hocking, W. Ren, X. Chen, Z. Su, W. Yang, Y. Wang, S. Zheng, F. Pan and C. Zhao, Adv. Funct. Mater., 2021, 31(51), 2107072 CrossRef CAS.
- X. Yang, J. Cheng, X. Yang, Y. Xu, W. Sun, N. Liu and J. Liu, ACS Sustain. Chem. Eng., 2021, 9(18), 6438–6445 CrossRef CAS.
- G. Xiao, R. Lu, J. Liu, X. Liao, Z. Wang and Y. Zhao, Nano Res., 2022, 15(4), 3073–3081 CrossRef CAS.
- Y. Zhang, L. Jiao, W. Yang, C. Xie and H.-L Jiang, Angew. Chem., Int. Ed., 2021, 60(14), 7607–7611 CrossRef CAS PubMed.
- Q. Fan, P. Hou, C. Choi, T.-S Wu, S. Hong, F. Li, Y.-L Soo, P. Kang, Y. Jung and Z. Sun, Adv. Energy Mater., 2020, 10(5), 1903068 CrossRef CAS.
- S. Chen, T. Liu, S. O. Olanrele, Z. Lian, C. Si, Z. Chen and B. Li, J. Energy Chem., 2021, 54, 143–150 CrossRef CAS.
- B. Mohanty, S. Basu and B. K. Jena, J. Energy Chem., 2022, 70, 444–471 CrossRef CAS.
- K. Mou, Z. Chen, X. Zhang, M. Jiao, X. Zhang, X. Ge, W. Zhang and L. Liu, Small, 2019, 15(49), 1903668 CrossRef CAS.
- A. J. Wu, J. Yang, B. Xu, X. Y. Wu, Y. H. Wang, X. J. Lv, Y. C. Ma, A. N. Xu, J. G. Zheng, Q. H. Tan, Y. Q. Peng, Z. F. Qi, H. F. Qi, J. F. Li, Y. L. Wang, J. Harding, X. Tu, A. Q. Wang, J. H. Yan and X. D. Li, Appl. Catal., B, 2021, 299, 120667 CrossRef CAS.
- K. Jiang, Y. F. Huang, G. S. Zeng, F. M. Toma, W. A. Goddard and A. T. Bell, ACS Energy Lett., 2020, 5(4), 1206–1214 CrossRef CAS.
- E. Haye, N. Job, Y. Wang, S. Penninckx, V. Stergiopoulos, N. Tumanov, M. Cardinal, Y. Busby, J.-F. Colomer, B.-L. Su, J.-J. Pireaux and L. Houssiau, J. Colloid Interface Sci., 2020, 570, 312–321 CrossRef CAS.
- K. K. Samanta, M. Jassal and A. K. Agrawal, Surf. Coat. Technol., 2009, 203(10), 1336–1342 CrossRef CAS.
- Y. Liang, Y. Han, J.-s. Li, J. Wang, D. Liu and Q. Fan, J. Energy Chem., 2022, 70, 643–655 CrossRef CAS.
- A. Wu, Y. Zhou, J. Lv, D. Zhang, Y. Peng, Q. Ye, P. Fu, W. Wang, X. Lin, S. Liu, M. Xu, Z. Qi, S. Zhu, W. Zhu, J. Yan, X. Tu and X. Li, ACS Sustain. Chem. Eng., 2022, 10(44), 14539–14548 CrossRef CAS.
- S. Y. Lu, X. L. Huang, M. H. Tang, Y. Q. Peng, S. C. Wang and C. P. Makwarimba, Environ. Pollut., 2021, 284, 117113 CrossRef CAS PubMed.
- X. Yang, J. Cheng, B. Z. Fang, X. X. Xuan, N. Liu, X. Yang and J. H. Zhou, Nanoscale, 2020, 12(35), 18437–18445 RSC.
- M. Song, B. Jin, R. Xiao, L. Yang, Y. Wu, Z. Zhong and Y. Huang, Biomass Bioenergy, 2013, 48, 250–256 CrossRef CAS.
- M. Naderi, Progress in Filtration and Separation, Academic Press, Oxford, 2015 Search PubMed.
- W. Xiong, H. Li, H. Wang, J. Yi, H. You, S. Zhang, Y. Hou, M. Cao, T. Zhang and R. Cao, Small, 2020, 16(41), 2003943 CrossRef CAS.
- J. Yang, Z. Qiu, C. Zhao, W. Wei, W. Chen, Z. Li, Y. Qu, J. Dong, J. Luo, Z. Li and Y. Wu, Angew. Chem., Int. Ed., 2018, 57(43), 14095–14100 CrossRef CAS.
- S. Uhlenbrock, C. Scharfschwerdt, M. Neumann, G. Illing and H. J. Freund, J. Phys.: Condens.Matter, 1992, 4(40), 7973–7978 CrossRef CAS.
- A. M. Tarditi, N. Barroso, A. E. Galetti, L. A. Arrua, L. Cornaglia and M. C. Abello, Surf. Interface Anal., 2014, 46(8), 521–529 CrossRef CAS.
- X. N. Chen, X. H. Wang and D. Fang, Fullerenes, Nanotubes Carbon Nanostruct., 2020, 28(12), 1048–1058 CrossRef CAS.
- L. H. Liu, S. Liu, L. Li, H. F. Qi, H. B. Yang, Y. Q. Huang, Z. D. Wei, L. Li, J. M. Xu and B. Liu, J. Mater. Chem. A, 2020, 8(13), 6190–6195 RSC.
- Z. W. Cai, P. Du, W. H. Liang, H. Zhang, P. Wu, C. X. Cai and Z. J. Yan, J. Mater. Chem. A, 2020, 8(30), 15012–15022 RSC.
- X. Qin, S. Zhu, F. Xiao, L. Zhang and M. Shao, ACS Energy Lett., 2019, 4(7), 1778–1783 CrossRef CAS.
- H. B. Zhang, J. Wei, J. C. Dong, G. G. Liu, L. Shi, P. F. An, G. X. Zhao, J. T. Kong, X. J. Wang, X. G. Meng, J. Zhang and J. H. Ye, Angew. Chem., Int. Ed., 2016, 55(46), 14308–14312 Search PubMed.
- Y. Hou, Y.-L. Liang, P.-C. Shi, Y.-B Huang and R. Cao, Appl. Catal., B, 2020, 271, 118929 CrossRef CAS.
- C. Cai, B. Liu, K. Liu, P. Li, J. Fu, Y. Wang, W. Li, C. Tian, Y. Kang and A. Stefancu, Angew. Chem., Int. Ed., 2022, 61, e202212640 CrossRef CAS.
- S. W. Han, T. H. Ha, C. H. Kim and K. Kim, Langmuir, 1998, 14(21), 6113–6120 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3se00858d |
| This journal is © The Royal Society of Chemistry 2024 |