DOI:
10.1039/D4SD00165F
(Paper)
Sens. Diagn., 2024,
3, 1562-1570
A fast and highly selective ECL creatinine sensor for diagnosis of chronic kidney disease†
Received
25th May 2024
, Accepted 21st July 2024
First published on 7th August 2024
Abstract
Monitoring of creatinine in human fluid has attracted considerable attention owing to the potential for diagnosis of chronic kidney disease. However, the detection of creatinine has been difficult owing to its electrochemical and optical inertness. In this approach, a highly selective and sensitive electrochemiluminescence (ECL) strategy based on homogeneous carbon quantum dots (CQDs) for the detection of creatinine was introduced. A copper(II) picrate complex was added at the surface of electrode to improve the selectivity of the sensor significantly by the formation of a Janovsky complex. A multi-pulse amperometric technique was applied as a very fast and reliable method for quantitative determination of creatinine. The calibration curve was acquired with a linear range from 1.0 × 10−8 to 1 × 10−5 M with a low detection limit of 8.7 × 10−9 M. The proposed creatinine sensing platform is experimentally very simple and shows high selectivity with a broad linear range of detection. Furthermore, the presented method can determine creatinine in real samples with excellent recoveries.
1. Introduction
Creatinine (Crn) is a mammalian metabolite mainly produced from non-enzymatic hydrolysis of creatine and phosphocreatine during muscle activities.1 As a waste, Crn is continuously removed from the body by the kidneys. The Crn amount in blood and urine is an important health indicator. So, Crn is clinically determined for diagnosis of diseases related to renal, muscular, and cardiovascular dysfunctions.2 Normal concentration levels of creatinine in blood are from 45 to 110 μM, while in urine, typical concentrations range between 3.3 and 27 mM, and in saliva from 8.8 to 26.5 μM. The most common clinical method for determination of this biomarker is the Jaffe method.3 This colorimetric assay based on the reaction between Crn and alkaline picrate is tolerated despite problems like low selectivity and time-consuming sample preparation.4 There are alternative enzymatic detection techniques, but although they are very selective, they are not clinically used because of their high cost.2 Therefore finding new, fast and cost effective analytical methods for detection of Crn is significant and attractive for many researchers.5 So far, different analytical methods such as colorimetric,6–9 chemiluminescence,10 fluorescence,11 chromatography,12 and electrophoresis methods13 have been tried by researchers to find new ways of creatinine determination in real samples. Most of these methods are time consuming and require complicated sample preparation steps. Electrochemical sensors have been developed for sensing of Crn.14,15 Although they have advantages in comparison with other techniques, they usually need expensive enzymes due to the low electrochemical activity of Crn.16
Electrochemiluminescence (ECL) as a developing analytical technique has attracted attention because of advantages like high sensitivity and cheap instrumentation.17 Determination of numerous analytes was possible with ECL techniques in different environments.18,19 Finding novel ECL systems based on new luminophores, construction of new devices, and application of new current/potential profiles are important.20 Homogeneous and heterogeneous (or solid-state) ECL systems are two categories of ECL based on where the luminophore is applied in the electrochemical cell.21,22 When the working electrode is modified with a luminophore (heterogeneous ECL), despite having intense ECL signals, some limitations in repeatability and reproducibility could be observed due to the shortage of the luminophore amount. On the other hand, when the luminophore is applied in the solution (homogeneous ECL), the luminophore is more available in the electrochemical cell, and the electrode can be modified with other materials that provide selectivity to the analyte.23,24 Luminophores with high aqueous solubility, like the well-known ruthenium complexes (e.g. Ru(bpy)32+), are usually employed in homogeneous ECL systems. Carbon quantum dots (CQDs) are also very soluble in water and can be applied in homogeneous ECL systems.
ECL of carbon based quantum dots, especially CQDs, has attracted attention because of some advanced features.25,26 Biological compatibility, cheap precursors, and short/facile synthesis methods are making these quantum dots promising as luminophores in ECL analyses. Nitrogen doped carbon quantum dots (N-CQDs) have recently been used as new luminophores in ECL systems due to their strong ECL signal compared to pristine CQDs.27–29 In some recent studies, N-CQDs synthesized from citric acid and urea by solvothermal methods showed excellent durability with an intense cathodic ECL signal that could be applied for ECL analyses having high potential to replace the expensive Ru(bpy)32+.30,31
The cyclic voltammetry (CV) method is usually applied as the current–potential regime to drive the ECL signal in analytical applications.32 During the application of CV, oxidation and reduction of the luminophore occur in a relatively wide range of potentials. The repeated oxidation–reduction cycles in addition to diffusion could lead to destruction of the luminophore, which limits the stability of ECL signals during the long-term regime of analysis. As voltammograms are not used as analytical signals in ECL sensing, other fast current–potential regimes like pulsed methods could be applied to avoid the mentioned limitations.33 Pulsed techniques have other advantages like sensitivity without responses to capacitive currents, programmable detection and good data management.34 Based on our knowledge, to date only two ECL sensors of Crn have been reported and in both of them, heterogeneous molecularly imprinted ECL (MIT-ECL) systems were used.35,36 Analytical application of pulsed potential methods using homogeneous ECL of CQDs is also a new approach.
In the present work, we used N-CQDs to determine Crn by ECL. N-CQDs were previously synthesized, characterized by different techniques, and proven as new cathodic ECL luminophores.30,31 Multi-pulsed amperometric methods were used for driving the cathodic ECL. Unlike usual conditions of application of CQDs in ECL systems, synthesized N-CQDs were used for homogeneous ECL. The N-CQDs were dissolved in the electrolyte of the cell together with a S2O82− co-reactant. Homogeneous ECL, with the pulsed amperometry method, allowed us to use dynamic ECL having advantages over traditional static ECL. A glassy carbon electrode (GCE) was modified with copper(II) picrate to improve the selectivity to creatinine. The ECL signals of this system were enhanced linearly by increasing the creatinine concentration. Finally, the sensor was applied to determine four patients urine and blood serum contents of Crn and compared with their clinical analysis results. Scheme 1 briefly summarizes this work.
 |
| | Scheme 1 Schematic of the sensor preparation and application. | |
2. Experimental section
2.1. Reagents and apparatus
Chemicals, reagents, and instruments are reported in the ESI.†
2.2. Synthesis of copper(II) picrate
Cu(Pic)2 was synthesized from copper(II) carbonate (synthesis of copper(II) carbonate is described in the ESI†) and picric acid (PA).37 Briefly, 2.2 mmol of copper(II) carbonate powder was added into 20 ml of PA solution in water (0.2 M) and stirred at 50 °C. Heating and stirring were continued until all of the CO2 bubbles were removed from the solution (about 2 h). Unreacted copper(II) carbonate and PA were removed from the solution by filtration and extraction with dichloromethane, respectively. Finally, yellow-green crystals of copper(II) picrate were obtained by evaporation of the solvent. Then they were dissolved again in water in optimum concentration and stored in a refrigerator (4 °C) to prevent potential explosion danger. It is noteworthy that PA and their dry salts are known to have explosive properties and it is recommended to use them carefully. More details including characterization are provided in the ESI.†
2.3. ECL apparatus
ECL experiments were performed at room temperature using a potentiostat/galvanostat (μStat ECL, DropSens, Spain, controlled with DropView 8400 software) combined with a 25 ml homemade ECL cell placed on a photodiode detector. A three-electrode system consisting of a glassy carbon electrode (GCE) with a diameter of 3 mm (0.07065 cm2) as a working electrode, an Ag/AgCl (saturated KCl) electrode as a reference electrode and a platinum rod as a counter electrode was used for all experiments. A homemade mini stirrer probe was attached to the cell for agitation of the electrolyte. The cell, electrodes, detector and stirrer were placed in a dark box for avoiding entry of external light (Fig. S1†).
2.4. Fabrication of the ECL sensor
A glassy carbon electrode (GCE) was cleaned by polishing with 0.3 μm α-alumina/ethanol slurry to obtain a mirror like surface. After rinsing with deionized water and drying, 10 μL of copper picrate in optimized concentration (1.4 mg mL−1) in 2% Nafion solution was dropped onto the electrode and dried in an oven at 60 °C. The modified electrode was then placed in a dark box for another 10 min and used for experiments.
2.5. Method of ECL
Homogeneous cathodic ECL was applied in this work. N-CQDs as luminophores in a concentration of 0.05 mg mL−1 were added to a PBS buffer solution (pH = 7) containing 0.1 M S2O82− and placed in the cell. An amperometric multi-pulsed method consisting of potential pulses of −1.8 V with a pulse duration of 0.2 s was applied. The resulting ECL and current signals were recorded (Fig. 1A). A mini stirrer was installed in the cell and used for agitating after each addition of analyte during the calibration. The potential of pulses, duration of pulses (pulse times), relaxation time between pulses, concentrations (co-reactant, luminophore, modifier), and the effect of pH were optimized.
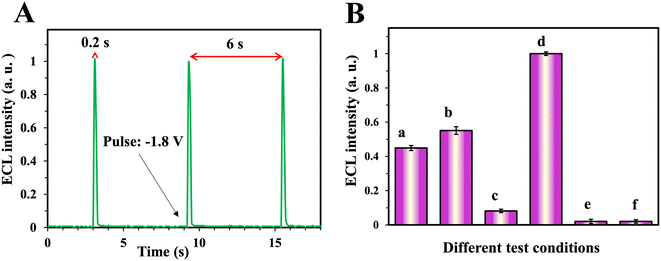 |
| | Fig. 1 (A) ECL pulses, (B) different conditions of ECL tests, a: bare GCE in CQD solution, b: bare GCE in CQD and Crn solution, c: Cu(pic)2/GCE in CQD solution, d: Cu(pic)2/GCE in CQD and Crn solution, e: Cu(pic)2/GCE in PBS solution and f: Cu(pic)2/GCE in Crn and PBS solution. All solutions were 0.1 M K2S2O82−. Error bars are the standard deviation of three measurements. | |
2.6. Preparation of real samples
Human serum was kindly provided by the Iranian Blood Transfusion Organization (IBTO), Ardabil, Iran. Urine and serum samples of four patients diagnosed with kidney disease were obtained from a local hospital in Zanjan, Iran. All experiments were performed in accordance with the guidelines of research and technology of UMA, and approved by the ethics committee at the University of Mohaghegh Ardabili. Informed consent was obtained for human urine samples of this study.
5 ml of methanol was added to 5 ml of each human blood serum sample and sonicated for 10 min. After precipitation of proteins, the samples were centrifuged at 4000 rpm for 15 min to remove the proteins and fats. The obtained clear liquids were then diluted to the final volume of 50 ml in phosphate buffer solution (pH = 7) and used for experiments. To prepare urine samples, 10 ml of patients' urine samples were heated at 80 °C in a water bath for 15 min. After this, the samples were centrifuged at 4000 rpm for 10 min to remove the precipitates and finally the samples were diluted like the serum samples.
3. Results and discussion
3.1. Determination strategy
The Crn measurement method is illustrated in Fig. 1B. Homogeneous ECL of N-CQDs in the case of application of a bare GCE, with moderate signal intensity (column a), was slightly enhanced by the addition of Crn (1 mM) to the solution (column b). As the concentration of added Crn was relatively high, this signal enhancement was not enough for detection. To increase the sensor's selectivity, copper(II) picrate was coated onto the surface of the GCE. The ECL signal of N-CQDs for the copper(II) picrate modified GCE was shallow without Crn (column c). Perhaps interestingly, in the presence of Crn, the ECL signal increased about twice compared to the GCE without coating (column d). This sensitive ECL behavior of the prepared sensor enabled the determination of Crn in a broader range of concentrations. Further investigation showed that the signal of the modified electrode was produced only in the presence of N-CQDs (columns e and f).
3.2. Proposed ECL enhancing mechanism and selectivity
The cathodic ECL mechanism of N-CQDs in the presence of S2O82− is similar to that reported for other CQDs.25,38,39 The mechanism can be described briefly in the following equations:| | | N-CQDs + e− → N-CQDs˙− | (1) |
| | | S2O82− + e− → SO42− + SO4˙− | (2) |
| | | N-CQDs˙− + SO4˙− → N-CQDs* + SO42− | (3) |
As shown in eqn (1) and (2), simultaneous reductions of N-CQDs and persulphate anions on the cathode are the critical steps required to generate ECL in the N-CQD/persulphate system. For describing the Crn determination mechanism, it can be proposed that when the electrode surface is modified with copper(II) picrate, its reduction takes the electrons. Therefore, this modifier hinders the formation of anion radicals (N-CQDs˙− and SO4˙−) leading to a low ECL signal. On the other hand, when Crn is introduced, the formation of a Janovsky complex enhances the electron transfer resulting in an intense ECL signal. This phenomenon allows determining the Crn concentration in the solution with high selectivity. A series of investigations, using FT-IR spectroscopy, were performed to prove the formation of the Janovsky complex from copper(II) picrate and Crn. PA and copper(II) picrate solutions were mixed with Crn solution separately and the resulting complexes were analyzed by FT-IR. As can be seen in Fig. S6,† the FT-IR spectra of the products of copper(II) picrate and Crn are similar to those of the products of PA and Crn (especially areas highlighted with blue and orange colors). This indicates the formation of the complex on the electrode surface during the analysis. Photographs of the solutions, shown in Fig. S7,† confirm the known color of the Janovsky complex.40
3.3. Optimization of analytical conditions
Optimum experimental conditions of Crn measurement with the proposed ECL sensor were determined for analyses with optimal performance. The concentration of N-CQDs (luminophore) in the solution had a direct impact on the ECL intensity. However, as shown in Fig. 2A, excess amounts decreased the ECL intensity. This reducing ECL intensity occurred because of the light absorption properties of the CQDs that in concentrations above 0.05 mg mL−1 (optimum amount) turn the solution darker and hinder the ECL from reaching the detector. Increasing the co-reactant (S2O82−) concentration also increased the ECL intensity until the optimum amount of 0.1 M which is almost the saturation point of this salt in the buffer (Fig. 2B). The amount of Cu(pic)2 as the electrode modifier was optimized by drop casting of solutions with different concentrations of this salt on the electrode and recording the ECL signals of N-CQDs. As Fig. 2C shows, the maximum ECL intensity was obtained at a concentration of 1.42 mg mL−1. The reason probably was that Cu(pic)2 in excess amounts hinders the charge transfer and lowers the ECL signal.
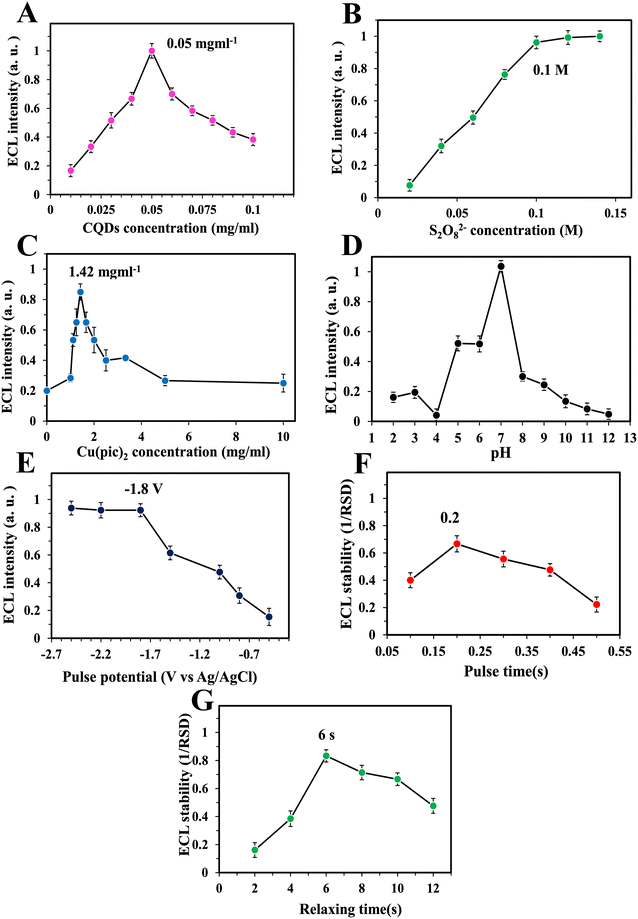 |
| | Fig. 2 Optimization results of different parameters: CQD concentration (A), S2O82− concentration (B), Cu(pic)2 concentration (C), pH (D), pulse potential (E), pulse time (F), and relaxation time (G). Error bars are the standard deviation of three measurements. | |
As is known, the pH of the cell solution in electrochemical measurements is a significant factor. Therefore, phosphate buffers with different pH values were tested as shown in Fig. 2D. The maximum intensity of the ECL was obtained at pH = 7.0. Previous studies have also shown that neutral pH is suitable for having intense ECL signals because of the surface charge of N-CQDs.30,31,41 Three important parameters of multi-pulsed amperometry including pulse potential, pulse time and relaxation time between pulses were optimized. As shown in Fig. 2E, the ECL intensity of the N-CQD/S2O82− system was increased by increasing the applied cathodic potential. The signal increase was observed until −1.8 V and then remained almost constant. This shows that maximal simultaneous electroreduction of the luminophore and co-reactant occurs at this potential resulting in the most intense ECL signal. Therefore, this potential was selected as the potential of pulses for all measurements. The duration of potential application for each pulse (pulse time) was optimized by applying −1.8 V to the working electrode in different periods and signal stability was calculated as 1/RSD for each period. As Fig. 2F shows, the most stable ECL signals were observed in the pulse time of 0.2 s. It is necessary in any pulsed methods to optimize relaxation times (duration with no application of potential between pulses). Therefore, this parameter was optimized in this work. As illustrated in Fig. 2G, the most stable ECL signals with the lowest RSD were observed in the relaxation time of 6 s. It seems that mass transfer of the luminophore and co-reactant to the surface of the electrode needs specific time under these conditions.
3.4. Crn analysis results
Different amounts of Crn were gradually injected into the cell containing the luminophore and co-reactant under optimum conditions, and ECL pulses were recorded to obtain the calibration curve. As shown in Fig. 3A, ECL signals were gradually increased with the increase of the Crn concentration. On the other hand, as Fig. 3B illustrates, there was no relationship between currents derived from pulses and the Crn concentration. A linear relationship between the intensity of ECL signals and the logarithm of Crn concentrations was observed (Fig. 3C). The linear range of this measurement was obtained to be between 1 × 10−8 and 1 × 10−5 mol L−1 based on the equation of ECL = 0.2838![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) log[Crn] + 2.4233 and R2 = 0.9843. This linearity is very compatible with the Crn levels in human (women and men) blood serum and urine which are 4.5 × 10−5 to 1.1 × 10−4 and 3.3 × 10−3 to 2.7 × 10−2 mol L−1, respectively.42,43 This shows that the new sensor could be applied for real sample analysis in clinical ranges even after several times of dilutions. Calculations using calibration data resulted in a detection limit (LOD) of 8.7 × 10−9 mol L−1 and a quantification limit (LOQ) of 1.7 × 10−8 mol L−1 (S/N = 3). Table 1 shows a short review of reported Crn detection methods from the performance point of view. Compared with other recent studies, this reveals that the new sensor has a broad linear range with a very low detection limit.
log[Crn] + 2.4233 and R2 = 0.9843. This linearity is very compatible with the Crn levels in human (women and men) blood serum and urine which are 4.5 × 10−5 to 1.1 × 10−4 and 3.3 × 10−3 to 2.7 × 10−2 mol L−1, respectively.42,43 This shows that the new sensor could be applied for real sample analysis in clinical ranges even after several times of dilutions. Calculations using calibration data resulted in a detection limit (LOD) of 8.7 × 10−9 mol L−1 and a quantification limit (LOQ) of 1.7 × 10−8 mol L−1 (S/N = 3). Table 1 shows a short review of reported Crn detection methods from the performance point of view. Compared with other recent studies, this reveals that the new sensor has a broad linear range with a very low detection limit.
 |
| | Fig. 3 (A) ECL intensity of the optimized sensor in different concentrations of Crn: (a) 1 × 10−8, (b) 5 × 10−8, (c) 1 × 10−7, (d) 5 × 10−7, (e) 1 × 10−6, (f) 5 × 10−6 and (g) 1 × 10−5 mol L−1. (B) Currents derived from pulses in different concentration of Crn. (C) Calibration curve. (D) Effect of real samples on the calibration curve. Error bars are the standard deviation of three measurements. | |
Table 1 Performance parameters for different methods for Crn determination compared to this work
| Method |
Real sample |
Linear range (M) |
LOD (M) |
Reference |
| Voltammetric |
Artificial urine |
10−3−5 × 10−3 |
5 × 10−4 |
44
|
| Capillary electrophoresis |
Human serum and urine |
1 × 10−4–3 × 10−2 |
3.5 × 10−5 |
13
|
| Colorimetric |
Human serum and urine |
3 × 10−5–5 × 10−4 |
2.2 × 10−5 |
6
|
| Amperometric |
Not reported |
5 × 10−6–1 × 10−4 |
1 × 10−7 |
45
|
| Amperometric |
Human serum |
3 × 10−6−1 × 10−3 |
1 × 10−7 |
14
|
| Voltammetric |
Human serum |
6 × 10−7–1 × 10−6 |
7.4 × 10−7 |
46
|
| Enzyme-less electrochemical |
Human urine |
1 × 10−5−2 × 10−4 |
7.2 × 10−7 |
47
|
| Amperometric |
Human serum and urine |
5 × 10−7−1.5 × 10−4 |
7.7 × 10−8 |
48
|
| MIT-ECL |
Human serum and urine |
5 × 10−9–1 × 10−3 |
5 × 10−10 |
35
|
| Fluorescence |
Not reported |
1 × 10−4–1 × 10−3 |
3.2 × 10−5 |
11
|
| Voltammetric |
Human serum |
5 × 10−8–4 × 10−5 |
1.13 × 10−8 |
49
|
| Chemiluminescence |
Human urine |
1 × 10−7–3 × 10−5 |
7.2 × 10−8 |
10
|
| Colorimetric |
Human urine |
1 × 10−4–2 × 10−2 |
8 × 10−5 |
50
|
| Homogeneous ECL |
Human serum and urine |
1 × 10−8–1 × 10−5 |
8.7 × 10−9 |
This work |
3.5. Measurement of Crn in real samples
The effect of matrix sample on sensor performance was studied by the addition of constant amounts of prepared real samples to the ECL cell using different concentrations of Crn and recording the ECL signals. Data are shown in Fig. 3D. Linear ranges were identical and curves (and equations) were very similar to those for the calibration. These results show that the calibration equation could be applied for fast determination of Crn in real samples even without using the time-consuming standard addition method.
3.6. Selectivity, stability, and reproducibility of the sensor
To evaluate the sensor's selectivity, different interfering compounds and ions that could be present in real samples (uric acid, glucose, urea, ascorbic acid, ammonium, oxalate, phosphate, etc.) were tested. As illustrated in Fig. 4A, no interfering effects of these compounds were observed at a concentration of 1 mM. For more selectivity studies, these compounds at the same concentration were also tested with the sensor in the presence of Crn at 10 μM concentration (100 times lower than the interferences) and the results showed that no ECL signal enhancement or quenching happened (Fig. 4B), indicating the good selectivity of the sensor to Crn. The sensor stability was investigated by recording the ECL signals during the given time and the results are shown in Fig. 4C. The intensity of the signals is almost identical to the RSD of 2.24% revealing the stable properties of the new sensor. For further investigations of possible structural changes in the sensor during determination of Crn, cyclic voltammetry (CV) was performed before and after the stability test. As shown in Fig. S7,† the intensity of two reversible redox peaks of the [Fe(CN)6]3−/4− system slightly decreased using a modified electrode (curve b) compared with a bare GCE (curve a). On the other hand, there were no recognizable redox current changes in the case of the used sensor (curve c), indicating that the electrochemical performance of the sensor was almost stable after use. Ten measurements of Crn (10 μM) were performed using ten freshly prepared sensors in ten days to study the reproducibility. As Fig. 4D shows, all measurement results were very close to each other (RSD = 3.19%), implying that the sensor can be prepared and used several times successfully.
 |
| | Fig. 4 (A) Selectivity of the new sensor in the presence of 1 × 10−3 mol L−1 different contaminants: K+, Na+, chloride, phosphate, uric acid, ascorbic acid, glucose, oxalate, ammonium, urea, citrate and 1 × 10−5 mol L−1 Crn. (B) Selectivity in the mixture of 1 × 10−3 mol L−1 contaminants with 1 × 10−5 mol L−1 Crn. (C) Stability and (D) reproducibility. Error bars are the standard deviation of three measurements. | |
3.7. Patient samples analyses
For testing the efficiency of the sensor for real sample analysis, serum and urine samples of four patients (suffering from kidney diseases) with known contents of Crn (all were at abnormal levels) were taken from a hospital and analyzed with the new sensor. For each determination, three potential pulses were applied to the sensor and thus each measurement took less than a minute. The calibration equation was used for calculation of Crn concentrations. As shown in Table 2, the results are very close to those of the clinical colorimetric analyses with acceptable accuracy.
Table 2 Determination of Crn in serum and urine of four patients
| Patients |
Samples |
Clinical results (M) |
Sensor's results (M) |
Accuracya (%) |
|
Accuracies are based on the difference between clinical and sensor's results and are equal to (sensor's result − clinical result)/clinical result × 100. All results are the average of three measurements.
|
| 1 |
Serum |
3.96 × 10−4 |
3.86 × 10−4 |
−2.55 |
| Urine |
3.15 × 10−3 |
3.20 × 10−3 |
1.85 |
| 2 |
Serum |
4.48 × 10−4 |
4.69 × 10−4 |
−2.24 |
| Urine |
2.80 × 10−3 |
2.82 × 10−3 |
0.58 |
| 3 |
Serum |
2.50 × 10−4 |
2.54 × 10−4 |
1.71 |
| Urine |
3.50 × 10−3 |
3.42 × 10−3 |
−2.15 |
| 4 |
Serum |
2.96 × 10−4 |
3.02 × 10−4 |
2.10 |
| Urine |
2.85 × 10−3 |
2.91 × 10−3 |
2.09 |
4. Conclusions
New homogeneous ECL based on an N-CQD/S2O82− system was used to selectively determine Crn. Pulsed amperometry was used as a new, fast, and precise current–potential regime in this work. Copper(II) picrate was synthesized and used as an electrode modifier to make the sensor highly selective to Crn. The combination of the pulse method and homogeneous luminophore application allowed having a dynamic ECL determination that made the analysis of samples faster in comparison with other methods. From the real sample analysis point of view, the proposed ECL sensor shows accurate results in determining the urine and serum Crn contents of four patients diagnosed with kidney disease. In addition to having potential for diagnosis of chronic kidney disease, this study could also open new ways of luminophore and method applications in ECL sensors.
Data availability
The data supporting this article have been included as part of the ESI.†
Conflicts of interest
There are no conflict to declare.
Acknowledgements
The authors gratefully acknowledge the support of this work by the University of Mohaghegh Ardabili research council, Ardabil, Iran. The authors thank Professor Frank Marken for his valuable comments to improve the manuscript.
References
- M. Wyss and R. Kaddurah-Daouk, Physiol. Rev., 2000, 80, 1107–1213 CrossRef CAS PubMed.
-
N. Turner, N. Lameire, D. J. Goldsmith, C. G. Winearls, J. Himmelfarb and G. Remuzzi, Oxford textbook of clinical nephrology, Oxford University Press Oxford, Oxford, 4th edn, 2015 Search PubMed.
-
A. Jain, R. Jain and S. Jain, in Basic Techniques in Biochemistry, Microbiology and Molecular Biology: Principles and Techniques, ed. A. Jain, R. Jain and S. Jain, Springer US, New York, NY, 2020, pp. 201–203, DOI:10.1007/978-1-4939-9861-6_46.
- R. K. Rakesh Kumar, M. O. Shaikh and C.-H. Chuang, Anal. Chim. Acta, 2021, 1183, 338748 CrossRef CAS.
- E. P. Randviir and C. E. Banks, Sens. Actuators, B, 2013, 183, 239–252 CrossRef CAS.
- S. Shariati and G. Khayatian, Microfluid. Nanofluid., 2022, 26, 30 CrossRef CAS.
- U. Sivasankaran, T. C. Jos and K. Girish Kumar, Anal. Biochem., 2018, 544, 1–6 CrossRef CAS.
- J. Guan, Y. Xiong, M. Wang, Q. Liu and X. Chen, Sens. Actuators, B, 2024, 399, 134842 CrossRef CAS.
- I. Lewińska, M. Speichert, M. Granica and Ł. Tymecki, Sens. Actuators, B, 2021, 340, 129915 CrossRef.
- S. Hanif, P. John, W. Gao, M. Saqib, L. Qi and G. Xu, Biosens. Bioelectron., 2016, 75, 347–351 CrossRef CAS.
- H. D. Duong and J. I. Rhee, Sensors, 2017, 17, 2570 CrossRef PubMed.
- D. Tsikas, A. Wolf, A. Mitschke, F.-M. Gutzki, W. Will and M. Bader, J. Chromatogr., B, 2010, 878, 2582–2592 CrossRef CAS.
- O. B. de Oliveira Moreira, J. C. Queiroz de Souza, J. M. Beraldo Candido, M. P. do Nascimento, P. R. Chellini, L. M. de Lemos and M. A. L. de Oliveira, Talanta, 2023, 258, 124465 CrossRef CAS PubMed.
- V. Serafín, P. Hernández, L. Agüí, P. Yáñez-Sedeño and J. M. Pingarrón, Electrochim. Acta, 2013, 97, 175–183 CrossRef.
- K. Ngamchuea, C. Moonla, A. Watwiangkham, S. Wannapaiboon and S. Suthirakun, Electrochim. Acta, 2022, 428, 140951 CrossRef CAS.
- U. Lad, S. Khokhar and G. M. Kale, Anal. Chem., 2008, 80, 7910–7917 CrossRef CAS.
- W. Miao, Chem. Rev., 2008, 108, 2506–2553 CrossRef CAS PubMed.
- L. Li, Y. Chen and J.-J. Zhu, Anal. Chem., 2017, 89, 358–371 CrossRef CAS.
- X. Ying, L. Zhou, W. Fu, Y. Wang and B. Su, Sens. Diagn., 2023, 2, 480–491 RSC.
- S. O'Connor, L. Dennany and E. O'Reilly, Bioelectrochemistry, 2023, 149, 108286 CrossRef.
- M. Sornambigai, L. Bouffier, N. Sojic and S. S. Kumar, Anal. Bioanal. Chem., 2023, 415(24), 5875 CrossRef CAS PubMed.
- C. Venkateswara Raju, G. Kalaiyarasan, S. Paramasivam, J. Joseph and S. Senthil Kumar, Electrochim. Acta, 2020, 331, 135391 CrossRef CAS.
- H. Nasrollahpour, B. Khalilzadeh, A. Naseri, M. Sillanpää and C. H. Chia, Anal. Chem., 2022, 94, 349–365 CrossRef CAS.
- X. Li, Y. Huang, J. Chen, S. Zhuo, Z. Lin and J. Chen, Bioelectrochemistry, 2022, 147, 108189 CrossRef CAS PubMed.
-
M. Amiri, H. Afshary and Y. Sefid-Sefidehkhan, in Electrochemistry: Volume 17, ed. C. Banks, The Royal Society of Chemistry, 2023, 10.1039/BK9781839169366-00304.
- S. Das, B. Saha, M. Tiwari and D. K. Tiwari, Sens. Diagn., 2023, 2, 268–289 RSC.
- Y. Han, Y. Jia, Y. Du, Y. Li, X. Ren, H. Ma, D. Wu, X. Kuang, D. Fan and Q. Wei, Anal. Chem., 2023, 95, 6655–6663 CrossRef CAS.
- F. Niu, Y. Xu, J. Liu, Z. Song, M. Liu and J. Liu, Electrochim. Acta, 2017, 236, 239–251 CrossRef CAS.
- H. Wang, A. Abdussalam and G. Xu, Bioelectrochemistry, 2022, 148, 108249 CrossRef CAS PubMed.
- H. Afshary, M. Amiri, A. Bezaatpour and M. Wark, J. Electrochem. Soc., 2022, 169, 026523 CrossRef CAS.
- H. Afshary, M. Amiri, F. Marken, N. B. McKeown and M. Amiri, Anal. Bioanal. Chem., 2023, 415, 2727–2736 CrossRef CAS PubMed.
-
A. J. Bard and A. J. Bard, Electrogenerated chemiluminescence, Marcel Dekker, New York, 2004 Search PubMed.
- W. Zhu, Q. Wang, H. Ma, X. Lv, D. Wu, X. Sun, B. Du and Q. Wei, Sci. Rep., 2016, 6, 24599 CrossRef CAS.
-
E. Núñez-Bajo and M. T. F. Abedul, in Laboratory Methods in Dynamic Electroanalysis, ed. M. T. F. Abedul, Elsevier, 2020, pp. 329–338, DOI:10.1016/B978-0-12-815932-3.00032-2.
- B. Babamiri, A. Salimi, R. Hallaj and M. Hasanzadeh, Biosens. Bioelectron., 2018, 107, 272–279 CrossRef CAS PubMed.
- N. Cao, F. Zhao and B. Zeng, Sens. Actuators, B, 2020, 306, 127591 CrossRef CAS.
- T. M. Makoto Matsukawa, K.-i. Miyamoto, M. Iida, M. Yoshida, H. Morinaga and S. Fujiwara, Sci. Technol. Energ. Mater., 2003, 64, 175–181 Search PubMed.
- X. Qin, C. Zhang, Z. Whitworth, Z. Zhan, K. Chu, P. Hu, S. Jahanghiri, J. Zhou, J. Chen, Q. Zhang and Z. Ding, Advanced Sensor and Energy Materials, 2023, 100062, DOI:10.1016/j.asems.2023.100062.
- T. Han, T. Yan, Y. Li, W. Cao, X. Pang, Q. Huang and Q. Wei, Carbon, 2015, 91, 144–152 CrossRef CAS.
- S. Chaiyo, K. Kalcher, A. Apilux, O. Chailapakul and W. Siangproh, Analyst, 2018, 143, 5453–5460 RSC.
- Y. Li, R. Li, Z. Zhu, J. Liu, P. Pan, Y. Qi and Z. Yang, Microchem. J., 2022, 183, 108073 CrossRef CAS.
- C. L. Gonzalez-Gallardo, N. Arjona, L. Álvarez-Contreras and M. Guerra-Balcázar, RSC Adv., 2022, 12, 30785–30802 RSC.
- M. J. Pedrozo-Peñafiel, T. Lópes, L. M. Gutiérrez-Beleño, M. E. H. M. Da Costa, D. G. Larrudé and R. Q. Aucelio, J. Electroanal. Chem., 2020, 878, 114561 CrossRef.
- A. Nene, C. Phanthong, W. Surareungchai and M. Somasundrum, J. Solid State Electrochem., 2023, 27, 2869–2875 CrossRef CAS.
- S. Caliskan, E. Yildirim, D. A. Anakok and S. Cete, J. Solid State Electrochem., 2022, 26, 549–557 CrossRef CAS.
- M. Jamil, B. Fatima, D. Hussain, T. A. Chohan, S. Majeed, M. Imran, A. A. Khan, S. Manzoor, R. Nawaz, M. N. Ashiq and M. Najam-ul-Haq, Bioelectrochemistry, 2021, 140, 107815 CrossRef CAS.
- R. K. R. Kumar, A. Kumar, M. O. Shaikh, C.-Y. Liao and C.-H. Chuang, Sens. Actuators, B, 2024, 399, 134787 CrossRef CAS.
- N. Nontawong, M. Amatatongchai, P. Jarujamrus, D. Nacapricha and P. A. Lieberzeit, Sens. Actuators, B, 2021, 334, 129636 CrossRef CAS.
- P. Ming, Y. Niu, Y. Liu, J. Wang, H. Lai, Q. Zhou and H. Zhai, Langmuir, 2023, 39, 13656–13667 CrossRef CAS PubMed.
- Y. He, X. Zhang and H. Yu, Microchim. Acta, 2015, 182, 2037–2043 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Reagents, instruments, some of the syntheses and optimization parts. See DOI: https://doi.org/10.1039/d4sd00165f |
|
| This journal is © The Royal Society of Chemistry 2024 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a  a and
Mandana
Amiri
a and
Mandana
Amiri




