
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Bis(methylene)-λ5-phosphane anions†
Akihiro
Nomoto
a,
Koh
Sugamata
 *b and
Takahiro
Sasamori
*acd
*b and
Takahiro
Sasamori
*acd
aGraduate School of Science and Technology, University of Tsukuba, 1-1-1 Tennoudai, Tsukuba, Ibaraki 305-8571, Japan. E-mail: sasamori@chem.tsukuba.ac.jp
bDepartment of Chemistry, College of Science, Rikkyo University, 3-34-1 Nishi-Ikebukuro, Toshima-ku, Tokyo 171-8501, Japan. E-mail: sugamata@rikkyo.ac.jp
cDivision of Chemistry, Institute of Pure and Applied Sciences, University of Tsukuba, 1-1-1 Tennoudai, Tsukuba, Ibaraki 305-8571, Japan
dTsukuba Research Center for Energy Materials Sciences (TREMS), University of Tsukuba, 1-1-1 Tennoudai, Tsukuba, Ibaraki 305-8571, Japan
First published on 14th November 2024
Abstract
Bis(methylene)-λ5-phosphane anions, i.e., anionic phosphorus-centered heteroallene-type molecules, were obtained from the desilylation of a bis(silyl)methyl-substituted phosphaalkene. Their molecular structures, which were determined using spectroscopic techniques and single-crystal X-ray diffraction analysis, suggest that the central di-coordinated P atom is engaged in cumulative C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) PC π-bonds with the neighboring C atoms. The π-bond character of the CPC moieties was examined on the basis of the experimental results in combination with theoretical calculations; the results obtained suggest that multiple silyl substitutions at the C atom weaken the CP π-bonding character.
PC π-bonds with the neighboring C atoms. The π-bond character of the CPC moieties was examined on the basis of the experimental results in combination with theoretical calculations; the results obtained suggest that multiple silyl substitutions at the C atom weaken the CP π-bonding character.
Introduction
Bis(methylene)-λ5-phosphanes, i.e., phosphorus-centered heteroallene-type molecules, have attracted much attention as valence isomers of σ3,λ5-phosphiranes owing to their expected unique bonding character due to the cumulative CP π-bonds. In 1982,1 Appel reported the first example of isolable bis(methylene)-λ5-phosphanes (IR, R = C6H11, Ph, and Me2N), whose structural characterization revealed a cumulative CPC π-bond with a tri-coordinated P(V) atom. This σ3,λ5-phosphane, i.e., a phosphorus(V)-centered allene, represented a milestone in main-group-element chemistry as an unprecedented low-coordinated organophosphorus compound alongside West's disilene and Yoshifuji's diphosphene.2,3 The hitherto isolated bis(methylene)-λ5-phosphanes exhibit a distinct bent-allenic structure that is significantly different from the linear structure of all-carbon allenes.4 Bis(methylene)-λ5-phosphanes have so far been obtained by treating the corresponding carbenes or carbenoids with the corresponding phosphaalkenes.5 Furthermore, the P-chloro-substituted analogues of bis(methylene)-λ5-phosphanes (ICl, Fig. 1) can be expected to serve as suitable precursors to a variety of functionalized bis(methylene)-λ5-phosphanes, given that ICl can be easily functionalized at the phosphorus atom via nucleophilic substitution reactions (Fig. 1a).6–11 The diverse reactivity of bis(methylene)-λ5-phosphanes prompted us to focus our attention on the synthesis of a P-anionic bis(methylene)-λ5-phosphane, which is expected to work as a nucleophilic building block for hitherto unknown types of bis(methylene)-λ5-phosphanes.
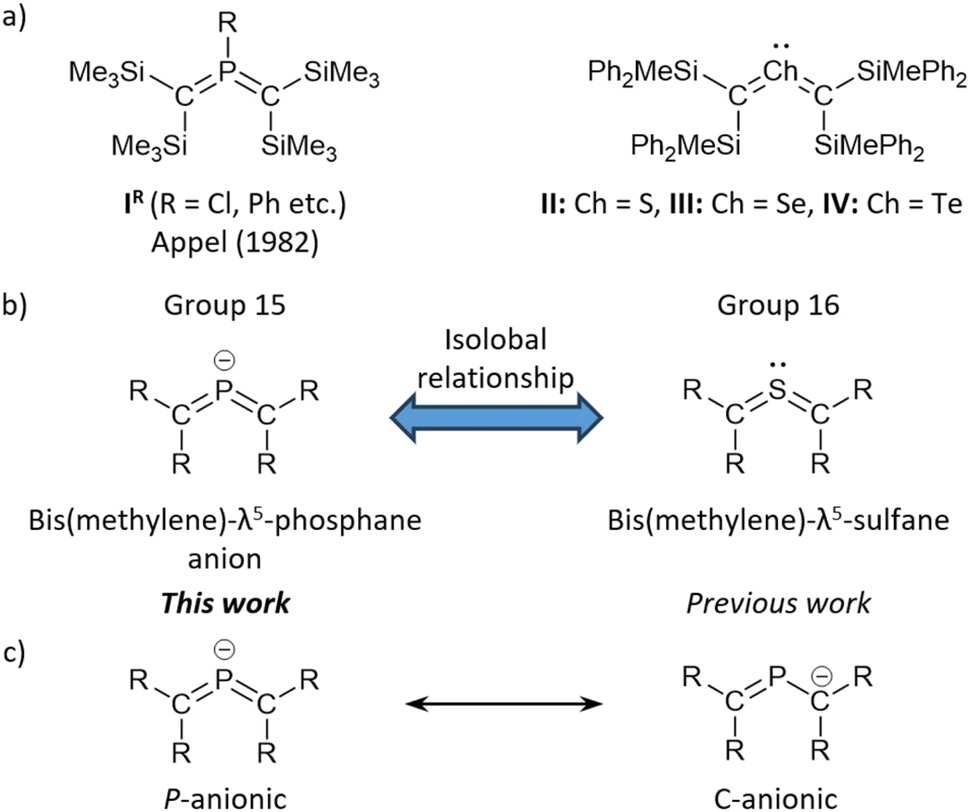 | ||
| Fig. 1 (a) Isolated bis(methylene)-λ5-phosphanes IR and bis(methylene)-λ4-chalcogenanes II–IV. (b) Isolobal relationship between bis(methylene)-λ5-phosphane anion and bis(methylene)-λ4-sulfane. (c) Canonical resonance structures of bis(methylene)-λ5-phosphane anions. | ||
Recently, we have successfully synthesized stable bis(methylene)-λ4-sulfane (II, Fig. 1), which represents the first example of a group-16-element-centered heteroallene, using steric stabilization afforded by silyl groups.12 Moreover, its heavier-element analogues, i.e., bis(methylene)-λ4-selane III and -tellane IV, have been synthesized and fully characterized.13,14 These bis(methylene)-λ4-chalcogenanes, which exhibit pseudo-C2v symmetric coordination geometries with bent allene-type electronic structures, can be interpreted as “2-heteroallenes”,15 characterized by the three-center-four-electron π-bond on the CChC (Ch = S, Se, Te) moiety. Given that bis(methylene)-λ4-chalcogenanes are isolobal to anionic group-15-element-centered 2-heteroallenes (Fig. 1b), we expected that anionic bis(methylene)-λ5-phosphanes could potentially be synthesized using sterically demanding silyl groups as in the cases of II–IV, which may prevent the isomerization to the corresponding phosphiranides.16,17 As described above, bis(methylene)-λ5-phosphane anions should be of great interest not only with respect to their expected unique electronic properties, but also with regard to their potential as precursors to further functionalized bis(methylene)-λ5-phosphanes upon treatment with electrophiles or electron-deficient metals. Moreover, a detailed examination of the intrinsic nature of these species, i.e., whether they should be considered P-anionic bis(methylene)-λ5-phosphanes or C-anionic phosphaalkenes, would be of great importance and interest (Fig. 1c). Here, we present the successful isolation of bis(methylene)-λ5-phosphane anions and their structural characterization.
Results and discussion
To start with, we attempted the synthesis of P-chlorophosphaallene A as a precursor for the bis(methylene)-λ5-phosphane anions according to a previously reported protocol.18 The reaction of bis(silyl)carbenoid RSi2CBrLi (RSi = SiMePh2) with 0.33 equivalents of PCl3 was carried out. Specifically, the treatment of a THF/Et2O solution of the bis(silyl)carbenoid, which was prepared via the lithiation of RSi2CBr2 using t-BuLi at −110 °C, with 0.33 equivalents of PCl3 in Et2O afforded phosphaalkene 1 in 31% yield without the formation of the expected product (A) (Scheme 1). Compound 1, which is air- and moisture-stable, was purified by column chromatography on SiO2 using CH2Cl2/hexane as the eluent. The molecular structure of 1 was characterized by spectroscopic techniques and single-crystal X-ray diffraction (SCXRD) analysis (Fig. 2).19 The 31P NMR chemical shift of 1 (436.5 ppm) is close to those of related phosphaalkenes (ca. 378–439 ppm).20–22 The theoretically estimated value of the chemical shift of 1 (δP = 470 ppm) based on gauge-independent atomic orbital (GIAO) NMR calculations is consistent with the experimental results,23 suggesting π-bond character for the CP bond in solution and a negligible solvent effect. The P1–C1 bond length of 1 (1.675(2) Å) is almost identical to those of the related phosphaalkenes (ca. 1.66 Å)20,21,24 and considerably shorter than the P1–C2 bond of 1 (1.835(2) Å), suggesting a double- and single-bond character for the P1C1 and P1–C2 bonds, respectively. In contrast to the tetrahedral geometry of the C2 atom, the C1 atom features a trigonal planar geometry with a bond-angle sum of 360° around the C1 atom. To elucidate the reaction mechanism, we examined the trapping reactions of the lithiated species generated in situ by the addition of CH3I in the reaction of RSi2CBr2 with t-BuLi.25 The treatment of RSi2CBr2 with 2.9 eq. of t-BuLi followed by the addition of an excess amount of CH3I resulted in the formation of three methylated compounds derived from the intermediates of bis(silyl)carbenoid 2, lithium bis(silyl)methanide 3, and dilithio compound 4 (Scheme S1†). The mechanism for the formation of 1 remains unclear at this stage, even though it seems feasible to assume that 1 is formed via the generation of P-chlorophosphaalkene 5 as a reactive intermediate, followed by its reaction with lithium bis(silyl)methanide 3 together with the elimination of LiCl (Scheme 2).
 | ||
| Scheme 1 The synthesis of phosphaalkene 1. | ||
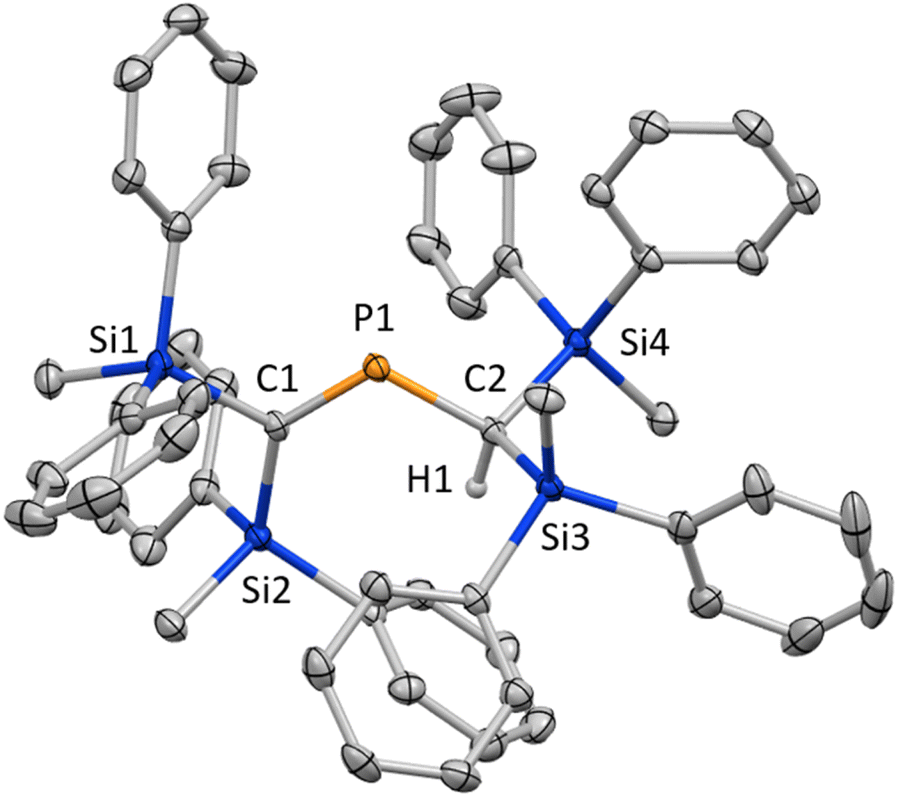 | ||
| Fig. 2 Molecular structure of 1 in the crystalline state with thermal ellipsoids at 50% probability; all hydrogen atoms except for H1 are omitted for clarity. Selected bond lengths [Å] and angles [°]: C1–P1 1.675(2), P1–C2 1.835(2), and C1–P1–C2 112.63(9).19 | ||
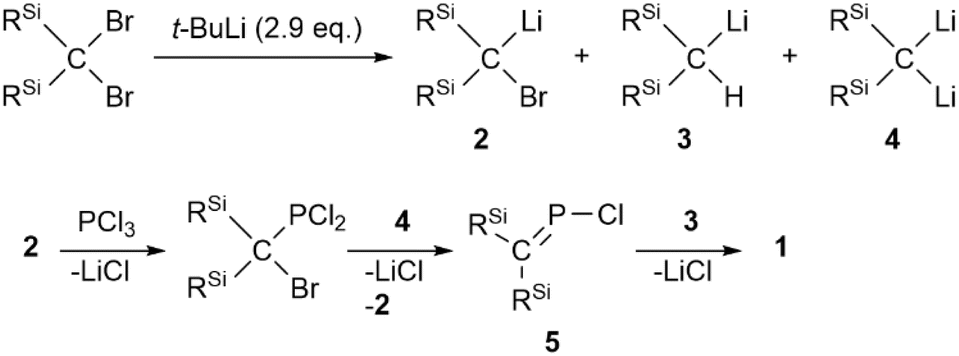 | ||
| Scheme 2 Plausible reaction mechanism for the formation of phosphaalkene 1. | ||
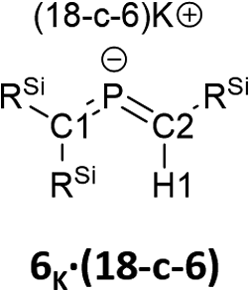
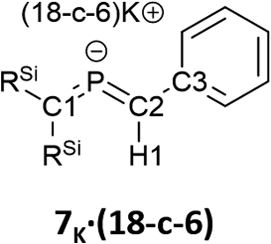
Subsequently, we attempted the deprotonation of 1 in the expectation of the formation of bis(methylene)-λ5-phosphane anion 8K·(18-C-6)via an E2-elimination using potassium hexamethyldisilazide (KHMDS) in the presence of 18-crown-6 in toluene. However, unexpectedly, desilylated compounds 6K·(18-c-6) (30%) and 7K·(18-c-6) (2%) were obtained as crystalline compounds (Scheme 3; entry 1). In the case of using a cryptand instead of 18-crown-6, only 6K·(cryptand) was formed (entry 2). Using LiHMDS or NaHMDS as a base also furnished 6M·(ligand) exclusively (entries 3 and 4). In contrast, when KOt-Bu in combination with 18-crown-6 was used instead of KHMDS/18-crown-6, a complicated mixture was obtained.
 | ||
| Scheme 3 The synthesis of bis(methylene)-λ5-phosphane anions 6M·(ligand) and 7M·(ligand). | ||
Accordingly, it can be concluded that it is crucial to use the HMDS anion for the generation of 6M·(ligand), and that the accompanied potassium cation causes the simultaneous formation of 7K·(ligand). The formation of 6M·(ligand) should most likely be interpreted in terms of a favorable nucleophilic attack of the HMDS anion on the electrophilic Si atom, even though the details of the mechanism for the formation of 7K·(ligand) remain unclear at this stage.26 As shown in Scheme 4, theoretical calculations suggested that the formation of 6 together with amino silane (Me3Si)2NSiMePh2 in the reaction of 1 with the HMDS anion should be thermodynamically more favorable (ΔEZERO = −20.3 kcal mol−1) compared to the reaction of phosphaalkene 1 with (Me3Si)2N− to give tetrasilyl bis(met hylene)-λ5-phosphane anion 8 and (Me3Si)2NH (ΔEZERO = −13.8 kcal mol−1). To further investigate the reaction mechanism for the desilylation reaction, we performed theoretical calculations on the potential energy surface of both a deprotonation reaction and a desilylation reaction of the phosphaalkene (SM) using model compounds as shown in Fig. S55.† The reaction barrier for the deprotonation product viaTS1 is smaller than that of the desilylation product viaTS2, while the product of the desilylation reaction (Pr2) is significantly stable compared to that of the deprotonation reaction (Pr1). As a result, it was found that the formation of bis(methylene)-λ5-phosphane anion 6 is a thermodynamically favored reaction.
 | ||
| Scheme 4 Comparison of ΔEZERO between the desilylation and deprotonation reactions of 1. | ||
Bis(methylene)-λ5-phosphane anions 6K·(18-c-6) and 7K·(18-c-6) exhibit moderate thermal stability in the solid state (6K, m.p.: 93 °C (decomp); 7K, m.p.: 53 °C (decomp.)) and high thermal stability in solution, i.e., the NMR spectra of 6K·(18-c-6) and 7K·(18-c-6) in C6D6 remained unchanged after 24 h at 80 °C.
The characterization of 6K·(18-c-6) and 7K·(18-c-6) was accomplished by multinuclear NMR and UV-vis spectroscopy, mass spectrometry, and SCXRD analysis.19 In the molecular structures of 6K·(18-c-6) and 7K·(18-c-6) (Fig. 3), both the protons on C2 of 6K·(18-c-6) and 7K·(18-c-6) were located based on the residual Q-peaks, which represent residual electron density peaks in the differential electron-density map, in the LSQ process (see the ESI†). In the crystal structure of 6K·(18-c-6), the potassium cation is coordinated by an 18-crown-6 ether and two phenyl rings, which results in the formation of an infinite chain structure in the solid state (Fig. 3a and S42†). In contrast, in 7K·(18-c-6), the potassium cation is coordinated by an 18-crown-6 ether and one phenyl ring connected to the allene moiety, resulting in a monomeric structure. It should also be noted here that the phenyl ring is co-planar to the CPC moiety, probably due to π-conjugation, which is indicative of the considerable π-bond character of both CP bonds. The central CPC moiety in 6K·(18-c-6) is bent (C1–P1–C2: 112.23(5)°) with almost identical CP bond lengths (C1–P1: 1.723(1) Å; P1–C2: 1.694(1) Å). In the case of 7K·(18-c-6), the allene moiety also shows a bent allene-type structure (C1–P1–C2: 112.6(1)°; C1–P1: 1.717(2) Å; P1–C2: 1.690(3) Å). The C–P bonds are considerably shorter than typical C–P single bonds (e.g., 1.835(2) Å in 1) but slightly longer than typical CP double bonds (e.g., 1.675(2) Å in 1). The fact that the C1 = P1 bond (1.723(1) Å) is slightly longer than the C2 = P1 bond (1.694(1) Å) should most likely be rationalized in terms of the predominant contribution of the resonance structure of bis(methylene)-λ5-phosphane anion 6 bearing the CPC allenic π-bonds along with the partial contributions of 2-phosphapropenyl anion 6A rather than 6B (Fig. 4), wherein the anion charge is partially localized on the C1 atom probably due to the considerable α-effect of the two adjacent silyl groups. The C1–P1 bonds (1.716(3) Å and 1.717(2) Å) in 7K·(18-c-6) are slightly longer than the P1–C2 bonds (1.694(3) Å and 1.690(3) Å) and those of 6K·(18-c-6). These structural features are similar to those of a previously reported bis(methylene)-λ4-sulfane.12 In contrast, as shown in Fig. 5, the dihedral angles (φ) between the two terminal carbon planes of the allene moieties in 6K·(18-c-6) (8.4°) and 7K·(18-c-6) (4.5°/3.4°) are very small, suggesting an almost coplanar geometry, which is different from that of the reported bis(methylene)-λ4-sulfane (51.9°). Theoretical calculations indicated that the dihedral angles between the terminal carbon planes of the allene moieties (φ) tend to increase with increasing steric demand of the substituents on the terminal carbons (Fig. S46†). Thus, it can be concluded that a bis(methylene)-λ5-phosphane anion should exhibit an intrinsically coplanar geometry. Moreover, the C1–P1–C2 bond angles of 6K·(18-c-6) (112.23(5)°) and 7K·(18-c-6) (112.6(1)°) are almost the same as that of 1 (112.6(1)°), but significantly narrower than those of the hitherto reported bis(methylene)-λ5-phosphanes (127–137°),7 indicating high s-character for the lone pair on the phosphorus atoms of 6K·(18-c-6) and 7K·(18-c-6) as well as high p-character of R–P(C)2 σ-/π-bonds.
 | ||
| Fig. 3 (a) Molecular structure of 6K·(18-c-6) in the crystalline state with thermal ellipsoids at 50% probability; all hydrogen atoms except for H1 and 18-crown-6 are omitted for clarity. Selected bond lengths [Å] and angles [°]: C1–P1 1.723(1), P1–C2 1.694(1), and C1–P1–C2 112.23(5). (b) Two independent molecules (7K-A and 7K-B) were found in the unit cell. Molecular structure of one of the two crystallographically independent molecules in the unit cell of 7K·(18-c-6) in the crystalline state with thermal ellipsoids at 50% probability; all hydrogen atoms except for H1 and 18-crown-6 are omitted for clarity. Selected bond lengths [Å] and angles [°] [7K-A]: C1–P1 1.717(2), P1–C2 1.690(3), C2–C3 1.449(3), C1–P1–C2 112.6(1) [7K-B], P2–C47 1.716(3), C48–P2 1.694(3), and C48–P2–C47 112.5(1).19 | ||
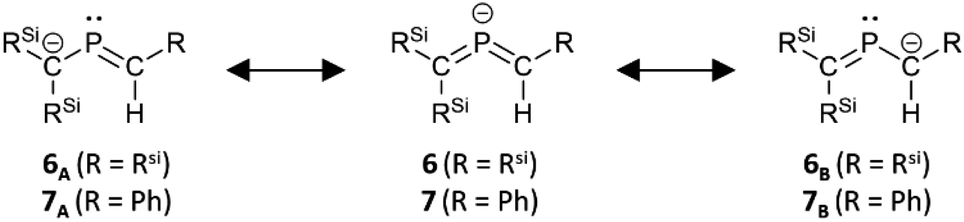 | ||
| Fig. 4 Canonical resonance structures of bis(methylene)-λ5-phosphane anions 6 and 7. | ||
 | ||
| Fig. 5 Comparison of structural parameters between bis(methylene)-λ5-phosphane anions and bis(methylene)-λ4-sulfanes. | ||
The structural optimization of bis(methylene)-λ5-phosphane anions 6 and 7 using DFT calculations23 was able to closely reproduce the experimentally observed structures (Fig. S43 and S44†). Natural-bond-orbital (NBO) calculations on the optimized structure of 6 showed one lone pair at the P atom (HOMO−1), two C–P σ-bonds, and a 3-center-4-electron π-bond on the C–P–C moiety as the LUMO+10 (anti-bonding), HOMO (π*(PC)), and HOMO−2 (π(PC)) (Fig. 6).23 On the other hand, the 3-center-4-electron π-bond in 7 is composed of the LUMO (anti-bonding), HOMO (non-bonding), and HOMO−2 (bonding), whereby the HOMO–LUMO gap is narrowed by the π-conjugation with the attached phenyl group. The estimated bond orders of the C–P bonds in 6 and 7, based on their Wiberg bond indices (WBIs) of 1.48 (P1–C2) and 1.24 (C1–P1) for 6, as well as 1.45 (P1–C2) and 1.24 (C1–P1) for 7, are slightly smaller than that of the CP double bond in 1 (1.66) and larger than the value for the C–P single bond in 1 (0.91), indicating π-bonding character for the C–P bonds in 6 and 7. The calculated natural population analysis (NPA) charge on the phosphorus atom was +0.8 for 6 and +0.7 for 7, while the charges on the adjacent carbon atoms were −1.7 on C1 for 6 and 7 and −1.4 on C2 for 6 and −0.7 for 7. The NPA charge distribution on 6 and 7 was similar to those of bis(methylene)-λ4-sulfanes, which have an isoelectronic relationship with the bis(methylene)-λ5-phosphane anions (Table 1). Considering the aforementioned results in their entirety, it should be concluded that the overall structure of bis(methylene)-λ5-phosphane anion 6 is characterized by not only 3-center-4-electron π-bonds in the CPC allene bonding but also a partial contribution of resonance structure 6A rather than 6B.
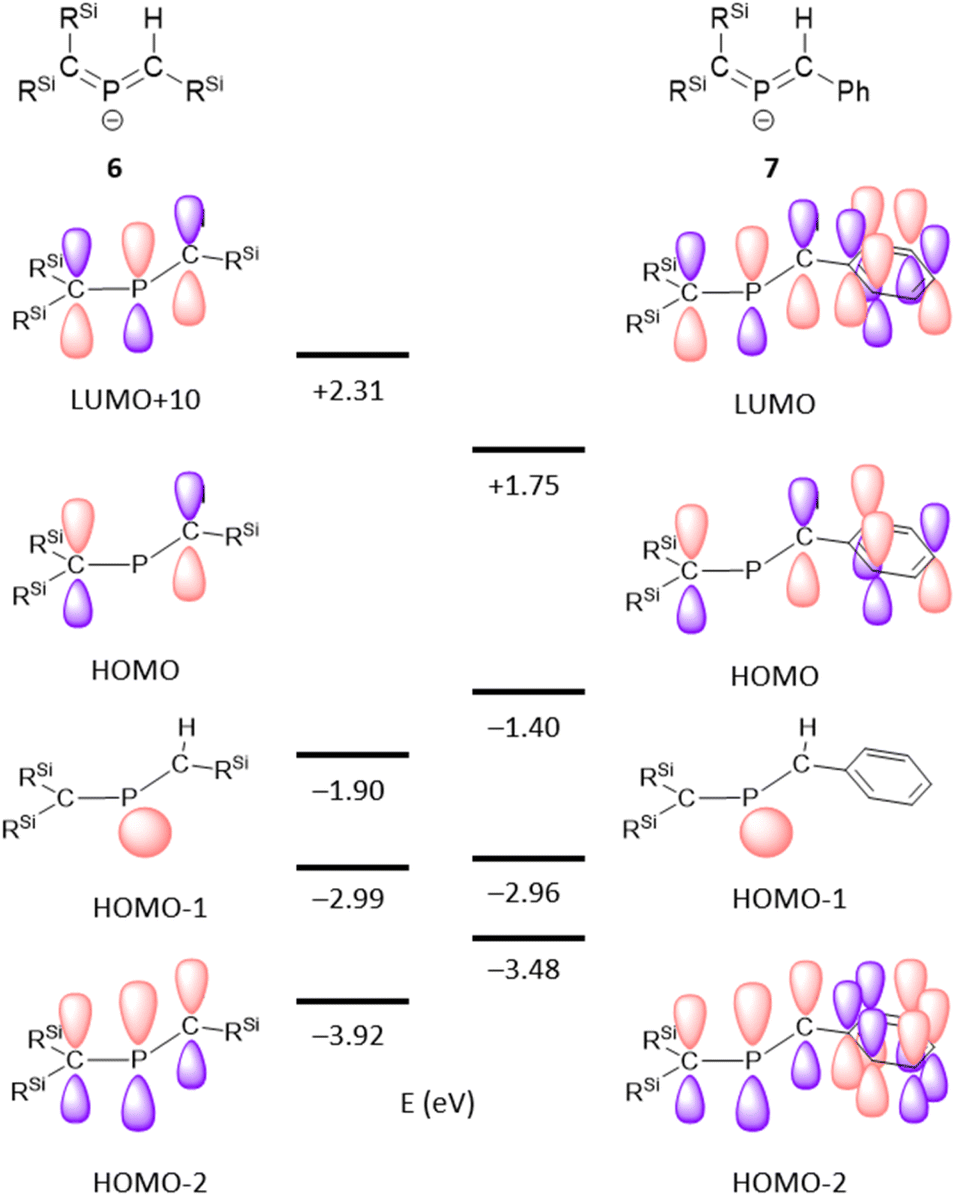 | ||
| Fig. 6 Kohn–Sham orbitals of 6 (left) and 7 (right), calculated at the B3PW91-D3(bj)/6-311G(3d) level. | ||
|
|
|
|
|
|
|---|---|---|---|---|
| a Unidentified. b Could not be identified due to overlapping. c 2 J P−H in Hz. | ||||
| δ 31P | 339.0 | 306.8 | 326.3, 334.1 | 256.6 |
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) |
||||
| δ 13 C | ||||
| C1 | 75.0 | 72.4 | —a, 141.2 | 139.0 |
| (1JC−P in Hz) | (82) | (74) | (—a, 67) | (54) |
| C3 | 108.1 | 128.0 | 177.8, 173.2 | 175.9 |
| (1JC−P in Hz) | (68) | (44) | (53, 66) | (35) |
| C3 | — | 121.2 | — | 140.2 |
| (1JC−P in Hz) | — | (19) | — | (14) |
|
||||
| δ 1 H | ||||
| H1 | 6.08 | —b | 7.89, 7.77 | 8.21 |
| (2JH−P in Hz) | (16.8) | (14.7)c | (25, 18) | (26) |

The 31P NMR spectra of 6K·(18-c-6) in o-difluorobenzene and 7K·(18-c-6) in benzene-d6 at room temperature showed at 339.0 ppm for 6K·(18-c-6) and at 306.8 ppm for 7K·(18-c-6), which were significantly low-field shifted compared to that of potassium diphenylphosphide, Ph2PK (−10.0 ppm), but slightly high-field shifted compared to that of 1 (δ = 436.5). These chemical shifts thus fall within the reported range for bis(methylene)-λ5-phosphanes (120–347 ppm).9 The 31P NMR chemical shifts estimated by the GIAO calculations for 6 (350 ppm) and 7 (317 ppm) are consistent with the experimental results (for details, see the ESI†).23
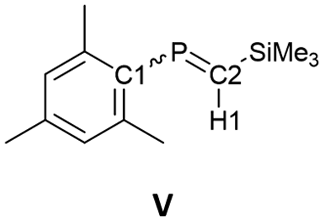
The 13C NMR spectrum of 6K·(18-c-6) in o-difluorobenzene at 333 K showed two signals at 75.0 ppm (C1) and 108.1 ppm (C2). These values fall within the reported range for bis(methylene)-λ5-phosphanes (31.6–122.3 ppm).9 Furthermore, the phosphorus-carbon coupling constants (1JC−P = 82 Hz for C1 and 68 Hz for C2) and 7K·(18-c-6) (1JC−P = 74 Hz for C1 and 44 Hz for C2) are larger than those of previously reported bis(methylene)-λ5-phosphanes (25.6–74.7 Hz), phosphaalkenes27V and (E)-VI (1JC−P = 34.5–78.1 Hz for >CP– species), and those of the C–P single bond in V and (E)-VI (Table 1). This result indicates that the C–P bonds in 6K·(18-c-6) and 7K·(18-c-6) exhibit multiple-bond character with high p-character.
The 1H NMR spectrum of 6K·(18-c-6) showed that the proton on C2 (6.08 ppm) is significantly high-field shifted compared to that in 1 (3.72 ppm), indicating an increase in the effect of magnetic anisotropy due to the π-electrons of 6K·(18-c-6). The proton on C2 in 7K·(18-c-6) could not be observed due to significant overlap with the phenyl protons. The P–H coupling constant in 6·(18-c-6) (2JH−P = 16.8 Hz) is larger than that in (Me3Si)2CHPCl2 (14.3 Hz) but smaller than those of phosphaalkenes V and (E)-VI (18–26 Hz; Table 1).
The UV-vis spectra of 6K·(18-c-6) in benzene and 7K·(18-c-6) in toluene at room temperature exhibited characteristic adsorptions at λmax = 378 nm (ε = 7.8 × 103 L mol−1 cm−1) and 474 nm (ε = 1.1 × 104 L mol−1 cm−1), respectively. Time-dependent DFT calculations for 6 and 7 showed excitation energies of λ = 408 nm and 470 nm (Fig. S13 and S19†), respectively, for the HOMO–LUMO electron transitions (π–π*), indicating a bathochromic shift due to the π-conjugation of the phenyl group in 7K·(18-c-6). Taking these experimental and theoretical investigations into account, it can be concluded that both 6 and 7 contain two CP π-bonds, i.e., they should behave in solution as bent allene-type compounds with cumulative CP π bonds.
Finaly, reactions of 6K·(18-c-6) and 7K·(18-c-6) with t-Bu3PHBF4 as a protonating reagent were performed to investigate the nucleophilicity/basicity of the allene moieties as indicated by the canonical structures shown in Fig. 4. Both reactions proceeded selectively to produce the corresponding proton adducts 9 and 10 (Scheme 5). Although the anionic charges of 6 and 7 can be expected to be predominantly located on their C1 atoms as resonance structures 6A and 7A, the protonation occurred at their C2 atoms, suggesting that the negative charges on the C1 atoms are significantly stabilized by the double-silyl-α-effect, and thus the C2 atoms should be more basic than the C1 atoms due to their reactive CP π-bond character. In addition, the theoretical calculations of the products indicated that 9 is thermodynamically more stable than 9′ by ΔEZero = 3.4 kcal mol−1 (Fig. S47†), which would support the selective formation of the C2-protonated products 9 and 10.
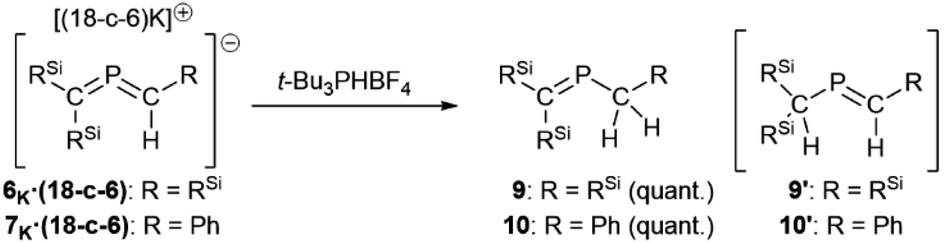 | ||
| Scheme 5 Protonation of 6K and 7K with t-Bu3PHBF4. | ||
Conclusions
In summary, the first isolable bis(methylene)-λ5-phosphane anions (6K·(18-c-6) and 7K·(18-c-6)) were synthesized by the desilylation of the corresponding phosphaalkene with KHMDS. Spectroscopic and single-crystal X-ray diffraction analyses in combination with theoretical calculations revealed that 6 and 7 show a bent and planar allene structure with two cumulative PC π-bonds in the CPC allene moiety forming a so-called three-center-four-electron π-bond. Further investigations into the reactivity of 6 and 7 are currently in progress in our laboratories and the results will be disclosed elsewhere in due course.
Data availability
The data supporting this article have been included as part of the ESI.†Author contributions
The project was designed and conducted by K. S. and T. S. Experimental work such as synthesis and characterization was carried out by A. N. All authors contributed to writing the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was financially supported by MEXT JSPS KAKENHI grants 24K08388, 23H01943, 22K18332, and 21KK0094, by the Collaborative Research Program of the Institute for Chemical Research at Kyoto University (2023–13), a project subsidized by the New Energy and Industrial Technology Development Organization (NEDO), and by the JST CREST grant JPMJCR19R4. We would also like to thank Mr Toshiaki Noda for the expert manufacturing of custom-tailored glassware. We acknowledge the generous assistance of SPring-8 with X-ray diffraction measurements under proposal numbers BL02B1: 2023A1539, 2023A1771, 2023A1785, 2023A1794, 2023A1859, 2023A1925, 2023B1675, 2023B1806, 2023B1878, and 2024A1857. Computational time was generously provided by the Supercomputer Laboratory at the Institute for Chemical Research (Kyoto University). Computations were also partially carried out using resources of the Research Center for Computational Science, Okazaki, Japan (projects: 24-IMS-C377/24-IMS-C397).Notes and references
- R. Appel, J. Peters and A. Westerhaus, Angew. Chem., Int. Ed. Engl., 1982, 21, 80–81 CrossRef.
- R. West, M. J. Fink and J. Michl, Science, 1981, 214, 1343–1344 CrossRef CAS PubMed.
- M. Yoshifuji, I. Shima, N. Inamoto, K. Hirotsu and T. Higuchi, J. Am. Chem. Soc., 1981, 103, 4587–4589 CrossRef CAS.
- D. R. Taylor, Chem. Rev., 1967, 67, 317–359 CrossRef CAS.
- R. Appel, E. Gaitzsch, K.-H. Dunker and F. Knoch, Chem. Ber., 1986, 119, 535–542 CrossRef CAS.
- R. Appel, E. Gaitzsch and F. Knoch, Angew. Chem., Int. Ed. Engl., 1985, 24, 589–590 CrossRef.
- R. Appel, E. Gaitzsch, K.-H. Dunker and F. Knoch, Chem. Ber., 1986, 119, 535–542 CrossRef CAS.
- R. Appel, P. Schulte and F. Knoch, Phosphorus, Sulfur Silicon Relat. Elem., 1988, 36, 147–151 CrossRef CAS.
- P. Becker, H. Brombach, G. David, M. Leuer, H.-J. Metternich and E. Niecke, Chem. Ber., 1992, 125, 771–782 CrossRef CAS.
- H. J. Metternich and E. Niecke, Angew. Chem., Int. Ed. Engl., 1991, 30, 312–313 CrossRef.
- H.-J. Metternich, E. Niecke, J. F. Nixon, R. Bartsch, P. B. Hitchcock and M. F. Meidine, Chem. Ber., 1991, 124, 1973–1976 CrossRef CAS.
- K. Sugamata, D. Hashizume, Y. Suzuki, T. Sasamori and S. Ishii, Chem.–Eur. J., 2018, 24, 6922–6926 CrossRef CAS PubMed.
- K. Sugamata, Y. Urao and M. Minoura, Chem. Commun., 2019, 55, 8254–8257 RSC.
- K. Sugamata, T. Asakawa, Y. Urao and M. Minoura, Inorg. Chem., 2022, 61, 17641–17645 CrossRef CAS PubMed.
- K. Sugamata and T. Sasamori, Dalton Trans., 2023, 52, 9882–9892 RSC.
- E. Niecke, W. W. Schoeller and D.-A. Wildbredt, Angew. Chem., Int. Ed. Engl., 1981, 20, 131–132 CrossRef.
- E. Niecke, M. Leuer, D.-A. Wildbredt and W. W. Schoeller, J. Chem. Soc., Chem. Commun., 1983, 1171–1172 RSC.
- R. Appel and A. Westerhaus, Tetrahedron Lett., 1982, 23, 2017–2018 CrossRef CAS.
- T. Sasamori and CSD Communication, Experimental Crystal Structure Determination, 2024 Search PubMed; Compound 1, DOI:10.5517/ccdc.csd.cc2l0zg2 (a) Compound 6K, (18-c-6), DOI:10.5517/ccdc.csd.cc2l0zh3; (b) Compound 7K, (18-c-6), DOI:10.5517/ccdc.csd.cc2l0zj4.
- T. Sasamori, J. M. Villalba Franco, J.-D. Guo, K. Sugamata, S. Nagase, R. Streubel and N. Tokitoh, Eur. J. Inorg. Chem., 2016, 2016, 678–684 CrossRef CAS.
- K. Sugamata, T. Sasamori and N. Tokitoh, Chem. Lett., 2014, 43, 95–96 CrossRef CAS.
- K. Issleib, H. Schmidt and C. Wirkner, Z. Anorg. Allg. Chem., 1981, 473, 85–90 CrossRef CAS.
- (a) Calculated at the B3PW91-D3(bj)/6-311G(3d)//B3PW91-D3(bj)/6-311G(3d) level using the Gaussian 16 program.; (b) NBO calculations were performed using the NBO7 program, see: E. D. Glendening, C. R. Landis and G. Weinhold, J. Comput. Chem., 2019, 40, 2234–2241 CrossRef CAS PubMed.
- A. H. Cowley, R. A. Jones, J. G. Lasch, N. C. Norman, C. A. Stewart, A. L. Stuart, J. L. Atwood, W. E. Hunter and H. M. Zhang, J. Am. Chem. Soc., 1984, 106, 7015–7020 CrossRef CAS.
- In each reaction leading to the formation of the corresponding bis(methylene)-λ5-phosphane anions 6M, the products exhibited extremely low solubility to organic solvent, making it challenging to analyze the product ratios due to the large amounts of insoluble solids in the crude products. Therefore, in all cases, the yields were determined using NMR spectroscopy after purification. The starting material 1 was detected only in entry 3, while in the other reactions, 1 was completely consumed, and no 7M was produced except for entry 1.
- Similar phenyl-group shift from the silicon atom to the carbon atom has been reported, see: (a) I. Fleming, S. K. Patel and C. J. Urch, J. Chem. Soc., Perkin Trans. 1, 1989, 115–124 RSC; (b) J. J. Eisch, Ind. Eng. Chem. Prod. Res. Dev., 1975, 14, 11–21 CrossRef CAS.
- R. Appel, J. Menzel, F. Knoch and P. Volz, Z. Anorg. Allg. Chem., 1986, 534, 100–108 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4sc07246d |
| This journal is © The Royal Society of Chemistry 2024 |