
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceUnveiling the spatial rearrangements of exhausted immobilised multi-enzyme systems through cryo-X-ray fluorescence nanoprobe imaging†
Javier
Santiago-Arcos
 a,
Murielle
Salome
b,
Fernando
López-Gallego
*ac and
Carlos
Sanchez-Cano
*cde
a,
Murielle
Salome
b,
Fernando
López-Gallego
*ac and
Carlos
Sanchez-Cano
*cde
aCenter for Cooperative Research in Biomaterials (CIC biomaGUNE) – Basque Research and Technology Alliance (BRTA), Paseo de Miramón, 182, 20014 Donostia-San Sebastián, Spain. E-mail: flopez@cicbiomagune.es
bThe European Synchrotron, ESRF, 71 Avenue des Martyrs, 38043 Grenoble Cedex 9, CS40220, France
cIkerbasque, Basque Foundation for Science, Plaza Euskadi 5, Bilbao, 48009, Spain
dDonostia International Physics Center, Paseo Manuel de Lardizabal 4, Donostia, 20018, Spain. E-mail: carlos.sanchez@dipc.org
ePolimero eta Material Aurreratuak: Fisika, Kimika eta Teknologia, Kimika Fakultatea, Euskal Herriko Unibertsitatea UPV/EHU, Donostia-San Sebastian, 20018, Spain
First published on 25th November 2024
Abstract
Enzyme immobilisation is of great importance for the fabrication of heterogeneous biocatalysts, as it allows the stabilisation of proteins using a solid support. Moreover, it permits their reuse in continuous and discontinuous reactors. The behaviour of enzymes at the interface with the materials where they are supported is not well understood during operational conditions. Here, we use X-ray fluorescence (XRF) imaging to study the changes in the overall structure of a heterogeneous biocatalyst formed by two unmodified metalloenzymes (a copper-dependent laccase and a zinc-dependent dehydrogenase) upon incubation, either under drastic (high temperature) or operational conditions. Those two enzymes were co-immobilised reversibly (by electrostatic interactions and His-tag metal coordination) to form a cascade reaction that catalyses the NAD+-dependent oxidation of diols coupled to a laccase–mediator for the in situ regeneration of the redox cofactor. Both the protein scaffolds and the metal cofactors undergo rearrangements during operational use or thermal incubation, but they seem to move as a whole unit within the support. Migration inside the support apparently causes only small alterations to the structure of the protein, yet it leads to the exhaustion of the heterogeneous biocatalyst. As such, we show that the use of advanced X-ray spectroscopy with spatial resolution can help obtain a better understanding of the molecular phenomena that occur during the operation of heterogeneous biocatalysts. Overall, this is fundamental to guide the optimisation for more productive and robust bioprocesses based on immobilised enzyme systems.
Introduction
Cell-free multi-enzyme systems can catalyse complex synthetic reaction schemes with exquisite selectivity, yielding industrially relevant chemicals under mild conditions.1,2 However, performing efficient cascade reactions often requires high levels of enzymatic spatial organisation.3,4 Living organisms contain multi-enzyme systems inside their cell organelles and compartments, but controlling the spatial organisation of enzymes is challenging when using cell-free approaches. Nonetheless, it is possible to mimic the cellular compartmentalisation of biosynthetic cascades by confining enzymes into synthetic materials.5–7 Such enzymatic arrays behave as multifunctional heterogeneous biocatalysts that accelerate cell-free biosynthetic pathways; they can also be separated easily from the reactants, once the reaction is completed. Furthermore, enzyme stability and other properties can be improved if the immobilisation protocols are designed carefully.8,9The spatial organisation of individual enzymes within porous materials can be controlled by tuning the immobilisation rate through the chemical interactions responsible for binding the enzymes to the support.10,11 This approach permits embedding the individual components of enzymatic cascades into the same porous carriers with different relative spatial patterns, and allows a certain level of control over the efficiency of the biocatalytic systems. Intraparticle local concentrations and gradients of intermediates seem to be the driving forces that augment the biocatalysts' performance, rather than the proximity of the enzymes forming part of the immobilisation cascade.12 For example, the two-step de-racemisation of alkyl glyceryl ethers requires two enantioselective and complementary NAD(P)-dependent dehydrogenases that are co-immobilised on porous microbeads with a specific spatial organisation.7 While the oxidative dehydrogenase must be located inside the porous material, the reductive dehydrogenase must be immobilised at the surface of the microbeads. This generates a flux of the reaction intermediate alkyl hydroxyketone from the inside to the outside of the beads, minimising the product-related inhibition of the oxidative enzyme and increasing the overall throughput of the cascade.
Unfortunately, during their operational lifespan, biocatalysts can suffer from enzyme inactivation and other processes that cause exhaustion, but such phenomena remain poorly understood,13 especially for supported enzymes (i.e., heterogeneous biocatalysts). For instance, rearrangement of the spatial distribution of immobilised enzymes during operando conditions might alter the overall kinetics of multi-enzyme systems, causing the inactivation of the biocatalyst.14,15 However, the degree of conservation of such spatial patterns under operational exhaustion remains unknown. Thus, understanding this process is fundamental to comprehend the reaction kinetics and deactivation mechanisms of supported biosynthetic enzyme cascades.
The different methods currently used to study these phenomena have been described recently in several reviews.16–18 The approaches based on confocal laser scanning microscopy (CLSM) are particularly useful, but they are limited to enzymes tagged with fluorophores. This limitation precludes the characterisation of immobilised enzymes that are ready for use under operational conditions, since the labelled enzymes may present substantial structural and functional differences compared to unlabelled ones.19,20 Alternatively, FTIR and Raman microscopy can be used to probe enzyme localisation within the support without being labelled, allowing the detection of structural alterations suffered by enzymes during immobilisation or operation.21–25 Unfortunately, protein detection by both methods relies on bands linked to amide bonds found in all enzymes, which have similar wavelenghts to bands linked to other bonds present in most supports that are commonly used to immobilise enzymes. Therefore, individual enzymes from multi-enzymatic systems are hardly distinguished, and discriminating between proteins and supports is not trivial (although machine learning applied to the analysis of Raman hyperspectral imaging may facilitate the process).25–27 In this context, new methodologies are required to characterise simultaneously the rearrangement of multiple enzymes on heterogeneous biocatalysts with good spatial resolution.
X-ray fluorescence (XRF) is a quantitative spectroscopic technique that allows probing electrons located at the inner orbitals of atoms from heavy elements to produce element-specific emissions.28 When combined with spatially resolved scanning approaches, XRF allows simultaneous mapping of different elements within a sample. However, XRF is not very sensitive to light elements, such as those mainly found in biomolecules (i.e., C, O, or N), as they suffer from high self-absorption in most samples.29 Therefore, when studying heterogeneous biocatalysts based on organic supports (e.g., agarose), XRF cannot discriminate between the support and the immobilised enzymes, unless they are metalloenzymes. As such, this technique is very interesting to unravelling the spatial organisation (and possible migration or lixiviation during operational use) of unlabelled metalloenzymes. Remarkably, the element-specific nature of XRF imaging allows us to distinguish between different enzymes in multi-enzymatic systems, if they are carrying different metals. Moreover, by using synchrotron cryo-nanoprobe beamlines, it is possible to obtain such information with spatial resolutions below 20 nm![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 30 and sub-ppm sensitivity31 in conditions close to physiological (or operational) ones, avoiding redistribution of ions and damage to the support material caused by irradiation with X-rays.32–35 However, although XRF has been previously used to study heterogeneous catalysts,33,36–38 its application for applied biocatalysis remains unexplored until this study.
30 and sub-ppm sensitivity31 in conditions close to physiological (or operational) ones, avoiding redistribution of ions and damage to the support material caused by irradiation with X-rays.32–35 However, although XRF has been previously used to study heterogeneous catalysts,33,36–38 its application for applied biocatalysis remains unexplored until this study.
In this work, we use XRF to probe the spatial organisation and rearrangements experienced by immobilised multi-enzyme systems under different inactivation conditions. As a model, we selected the well-established enzyme cascade reaction that yields lactols and lactones from the regioselective oxidation of diols. This cascade involves a Zn-dependent alcohol dehydrogenase for the NAD+-dependent alcohol oxidation, and a Cu-dependent laccase as laccase–mediator system (LMS) for cofactor recycling.39 The two enzymes were co-immobilised and inactivated under high-temperature and operational conditions, and the changes in their enzymatic activity and spatial distribution were evaluated. XRF experiments, supported by CLSM, as well as bulk kinetic, chromatographic and spectroscopic studies, suggest that the exhaustion of the heterogeneous biocatalyst observed after these two inactivation conditions is linked to the spatial rearrangement of the metalloenzymes immobilised on the porous support.
Results and discussion
A well-established enzyme cascade that is used to perform consecutive double oxidation of diols into lactols and lactones, while concomitantly regenerating NAD+/NADH cofactors by an LMS,39,40 was co-immobilised on agarose microbeads with 50 μm diameter. These beads were functionalised with cobalt-chelates and primary amino groups (AG-A/Co2+; see ESI† for more details). Such microbeads act as heterofunctional supports that allow the co-immobilisation of several enzymes on the same surface through different chemistries.41–44 This type of heterofunctional support has been exploited for region-directed immobilisation and rigidification of enzymes. Lastly, they have proven to be very useful in co-immobilising multi-enzyme systems.45,46The His-tagged Zn-dependent alcohol dehydrogenase from Bacillus stearothermophilus (BsADH)47 and the Cu-dependent laccase from Aspergillus sp. (AsLAC)48 were bound to the agarose beads through a sequential process (Fig. 1A, and S1†). First, the AsLAC (isoelectric point ≈ 4) was immobilised on AG-A/Co2+ at pH 5 for 2 h through the electrostatic interaction between acidic residues at the surface of the enzyme and the positively charged amine groups from the support. Then, BsADH was bound at pH 7 for a shorter time (15 min) through the coordination of the imidazoles from the His-tag with cobalt chelates anchored to the bead. Table 1 shows that both enzymes were immobilised quantitatively.
 | ||
| Fig. 1 (A) A schematic of the co-immobilisation chemistries of BsADH and AsLAC on agarose beads that were functionalised with Co-chelates and primary amine groups (AG-A/Co2+). (B) Phase contrast imaging of a single bead of AG-A/Co2+ bearing BsADH and AsLAC. (C) Quantitative XRF maps of Zn, Cu, and K of the same AG-A/Co2+ bead. (D) Quantitative XRF maps of Zn and Cu normalised to K content/pixel of the same AG-A/Co2+ bead. Pixel intensity represents metal density according to the colour scale. The scale bars are 10 μm. | ||
| Enzyme | Enzyme load mg g−1 | Ψ (%) | Recovered activityb U g−1/(%) |
|---|---|---|---|
| a Immobilisation yield, Ψ = (immobilised activity/offered activity) × 100. b % Relative recovered activity is defined as the coefficient between the specific activity of the immobilised enzyme and the specific activity of the soluble one × 100. | |||
| BsADH | 0.99 | 99 | 0.86/(26) |
| AsLAC | 0.99 | 99 | 0.03/(8) |
However, the activity recovered (U g−1) from BsADH was an order of magnitude higher than that from the immobilised AsLAC. The smaller mass activity of AsLAC is due to the 46-fold lower specific activity of the free enzyme and to a 2.5 times larger reduction in the recovered activity upon immobilisation compared to BsADH. Similar percentages of recovered activity have been found previously for the individual loading of each enzyme on other porous carriers through similar immobilisation chemistries.6,43,49,50
The spatial organisation of the BsADH and AsLAC after being immobilised on the agarose microbeads was characterised by unveiling the distribution of their Zn and Cu cofactors (respectively) under cryogenic conditions with XRF at beamline ID16A (ESRF). Beads loaded with the 2-enzyme system were deposited on silicon nitride windows and plunge-frozen in liquid ethane. Fig. 1B and C show phase contrast and XRF maps of single porous agarose microbeads (AG-A/Co2+) with co-immobilised BsADH and AsLAC. These images were acquired using an X-ray beam set to 17 keV and focused to 48.6 × 41.6 nm2 (horizontal × vertical) with an incident flux at the sample of 1.55 × 1011 phs/s. Coarse scans were done using 400 × 400 nm2 steps (100 ms dwell time) to detect areas of interest, and fine mapping of at least 3 beads was performed using a step size of 100 × 100 nm2 (50 ms dwell time). Co, K, Cu, and Zn XRF K-emissions were visible in all the biocatalysts analysed (Fig. S2†). Fig. 1C and S3† show the elemental maps of Zn and Cu corresponding to the spatial localisation of BsADH and AsLAC (respectively) within 2D projections of the beads. Moreover, K elemental maps from the beads were also obtained in the same way as the reference element (Fig. 1C). A small amount of K was present in the water (type 2 laboratory grade) used to prepare the loading and running buffers employed in this study. Thus, it is evenly distributed throughout the porous volume of the agarose support and can be used as an internal standard to estimate the volume of the bead contained in each pixel. As such, maps showing the quantity of Cu and Zn, relative to the volume of the pixel studied, were generated by normalising the maps for each element with K maps obtained for the same beads. Volume-normalised maps give a better idea of the enzyme's penetration across the support surface. These maps revealed that BsADH is mainly located in clusters at the outer surface of the porous microbeads, while AsLAC is homogeneously distributed through them (Fig. 1D and S4†). This spatial distribution matches the one revealed by more conventional methods based on CLSM using fluorophore-labelled enzymes (Fig. S5†). Note that this analysis was performed in a population of n > 10 beads and showed a low standard deviation on the mean radius at which each enzyme is infiltrated within the different beads. Enzyme localization presents a very homogeneous distribution in all analysed beads, indicating low heterogenicity across the samples.
In previous studies, we demonstrated that the spatial organisation of an enzyme on a porous support depends on the competition between the kinetics of immobilisation and diffusion.10 Therefore, the differences in the spatial organisation of BsADH and AsLAC rely on the specific kinetics governing their immobilisation processes. The immobilisation of BsADH on AG-A/Co2+ is extremely fast (100% immobilisation in less than 15 min), which agrees with its spatial localisation at the outer surface of the beads as revealed by XRF mapping. Cobalt XRF maps (Fig. S6†) also show high-density areas with this element at the outer surface of the porous support. Such spatial overlapping observed in Co and Zn maps supports the selective interaction between the His-tag of the protein and the Co chelates, which drives the immobilisation of BsADH. Instead, quantitative immobilisation of AsLAC on AG-A/Co2+ needs longer times (2 h) under the conditions studied. In this case, the binding of AsLAC to the support seems to be directed by electrostatic interactions with amino groups of the bead because similar kinetics were observed when the enzyme was immobilised on AG-A supports (agarose beads functionalized only with amine groups; Fig. S7†). Our volume-normalised XRF maps reveal that slower binding kinetics lead to a uniform distribution of AsLAC within the porous particles, which is independent of the spatial localization of Co. This confirms that the immobilisation of AsLAC does not require the interaction between the enzyme and the metal chelates located in the bead. Moreover, the distribution of AsLAC in the biocatalyst might also help explain the low mass activity of the enzyme after immobilisation on AG-A/Co2+. Oxygen availability may be limited for the laccase as it must diffuse across the internal porous structure of the support to reach AsLAC. A similar reduction in activity was observed for other oxygen-dependent enzymes (like NADH oxidases), when they were immobilised on the inner surface of porous materials.51,52
To probe any possible migration or spatial rearrangement of the BsADH and/or AsLAC that were co-immobilised on AG-A/Co2+ during activity exhaustion, we subjected the biocatalysts to either thermal inactivation or operando conditions. Next, the enzyme activity and intrinsic protein fluorescence were monitored as a function of the inactivation time. Finally, the inactive samples were analysed by XRF-mapping (at least 3 beads per sample). Fig. 2A shows the thermal inactivation kinetics at 70 °C. In the first 15 min of incubation, we observed a sharp decay in the activity of each enzyme, but more than 60% of their initial activity was maintained for at least 2 hours. When the intrinsic protein fluorescence was monitored, we observed rather small changes over time in the ratio between the fluorescence intensity at 350 and 330 (I350/I330) nm, and no variation in the maximum emission wavelength (λmax = 330) was seen. This result indicates that the immobilised enzymes undergo limited unfolding during the thermal incubation (Fig. 2B and S8†). The mild loss of enzyme activity and the small changes in protein-related fluorescence suggest that the high temperatures applied do not induce large conformational changes or trigger extensive enzyme leaching in both of the enzymes. Despite the small changes in activity and protein fluorescence, XRF elemental maps (Fig. 2C, S9 and S10†) show a clear change in the spatial distribution of Zn upon thermal shock as areas with high concentrations of Zn corresponding to clusters of BsADH disappeared after thermal inactivation, resulting in a more uniform distribution of Zn across the bead (Fig. 2C and S5†). Similar changes are observed in the distribution of Co within the beads (Fig. S6†). This insight suggests the redistribution or lixiviation within the porous support of either the Co coordinated to the chelating groups or the His-BsADH bound to the Co-chelates at the surface of the support. On the contrary, Cu XRF elemental maps (Fig. 2C and S9†) showed a less noticeable spatial rearrangement of Cu inside the beads, suggesting that AsLAC maintains its uniform spatial distribution before and after thermal shock as no metal clusters were observed. These results, for both BsADH and AsLAC, were also confirmed by CLSM (Fig. S11†). Unfortunately, XRF and CLSM imaging could not be done in an ideal correlative manner to understand better the distribution of both metal cofactors and protein scaffolds within the same bead. BsADH and AsLAC are not fluorescent, and labelling them with a fluorophore affects their enzymatic activity (hampering their use for operational studies). Correlative analysis of the beads with label-free FTIR or Raman-based microscopic techniques was also considered. However, such techniques cannot provide information on the individual distribution of BsADH and AsLAC within a sample, while the extra manipulation and irradiation could damage the beads before the XRF experiments.
 | ||
| Fig. 2 (A) Time-course of activity decay of BsADH and AsLAC that were co-immobilised on AG-A/Co2+ during incubation at 70 °C. (B) Fluorescence intensity at the maximum emission wavelength of the two co-immobilised enzymes at different thermal inactivation times. (C) Quantitative XRF maps of Zn and Cu normalised to K content/pixel of one single bead of AG-A/Co2+ bearing BsADH and AsLAC before (0 h, 70 °C) and after thermal shock (2 h, 70 °C). Pixel intensity represents metal density according to the colour scale. The scale bars are 10 μm. | ||
The operational stability of the biocatalyst was also studied by following the selective oxidation of 1,5-pentanediol (catalysed by BsADH) coupled to the in situ NAD+ regeneration (driven by AsLAC and the electron mediator acetosyringone) (Fig. 3A). In our experiments, a fresh heterogeneous biocatalyst achieved 37 ± 8% diol conversion and a lactol yield of 20 ± 2% (as analysed by GC-FID Fig. S12†). However, after the biocatalyst was used for consecutive batch reaction cycles, the diol conversion steadily decayed, reaching conversions below 10% after the third consecutive operational cycle (Fig. 3B). Additionally, the system was able to carry out the second oxidation of the lactol to yield the lactone (2.8 ± 0.1%) only during the first two operational cycles (Fig. S12†). Again, XRF imaging of beads after the first operational cycle showed only minor changes in Cu distribution compared to the fresh sample. However, the Zn clusters found in fresh microbeads almost disappeared after 1 operational run (Fig. 3C, S13 and S14†). This result agrees with the elemental distribution of Cu and Zn found in the beads after 2 hours of thermal inactivation (Fig. 2C and S9†).
 | ||
| Fig. 3 (A) Reaction scheme for the regioselective oxidation of diols catalysed by BsADH coupled to an AsLAC as the LMS (using acetosyringone as the mediator). (B) Conversion of diol after consecutive reaction cycles using BsADH and AsLAC that were co-immobilised on AG-A/Co2+ (20 mM 1,5-pentanediol, 1 mM NAD+, 1 mM acetosyringone in 5 mM of phosphate buffer at pH 7 and 30 °C). (C) Quantitative XRF maps of Zn and Cu normalised to K content/pixel of one single bead of AG-A/Co2+ bearing BsADH and AsLAC before (fresh) and after the 1st reaction cycle (cycle 1). Pixel intensity represents metal density according to the colour scale. The scale bars are 10 μm. | ||
Intensity histograms obtained from the same XRF maps (Fig. 4) confirm that both thermal inactivation and operational use of the heterogeneous biocatalyst induce significant changes in the spatial distribution of Zn. Histograms obtained from biocatalyst beads after their first operational cycle or thermal inactivation show a large reduction in their tails (related to areas with high concentrations of Zn), compared to fresh systems. This is due to the disappearance of BsADH clusters that were observed both after thermal shock and operational use. Histograms also indicate small changes in the distribution of Cu, with a small displacement in their maximum toward higher concentrations of Cu. However, a reduction in the overall area with higher concentrations of the metal was observed. This quantitative analysis supports that the metal cofactor of BsADH (i.e., Zn) was spatially reorganised within the biocatalyst beads to a greater extent than the metal cofactor of AsLAC (i.e., Cu) during the exhaustion processes. Notably, spatial rearrangements of Zn and Cu inside the beads may be explained through different mechanisms. They include the metal escape from the enzyme active sites and release from the protein scaffold, the metal exchange between the enzyme active sites and the metal chelates in the support, the lixiviation of the holoenzymes (both metal and protein scaffolds) from the support to the bulk, and/or the spatial redistribution of holoenzymes. Any of these causes or a combination of them could explain the exhaustion of the heterogeneous biocatalysts observed during either thermal incubation or operational use.
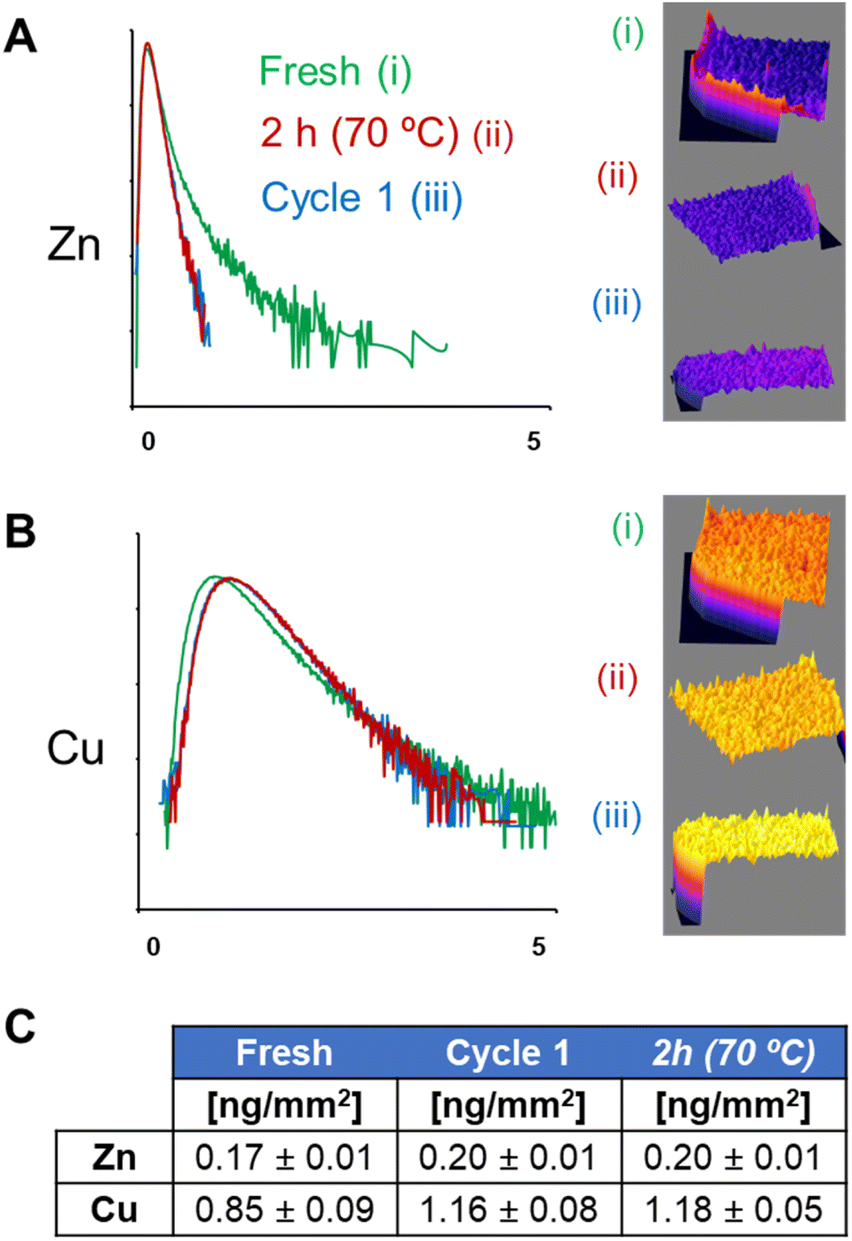 | ||
| Fig. 4 Normalised log-scale histograms (relative counts vs. metal density in ng ng−1 K−1) were obtained from XRF maps of Zn (A) and Cu (B) of (i) fresh, (ii) thermal inactivated (2 h, 70 °C), or (iii) 1st reaction cycle (cycle 1) of the AG-A/Co2+ beads bearing BsADH and AsLAC. (C) Average pixel intensity (in ng mm−2) of Zn and Cu on the XRF maps of (i) fresh, (ii) thermal inactivated (2 h, 70 °C), or (iii) 1st reaction cycle (cycle 1) of the AG-A/Co2+ beads bearing BsADH and AsLAC. | ||
Yet, both the CLSM (Fig. S11†) and XRF elemental mapping of different beads (Fig. 2C and 3C) show similar changes in the spatial distribution of enzymes and metals (respectively). This makes a massive escape of Zn and Cu from the protein scaffold unlikely, suggesting that both components of the enzymes behave in the same way during operational use or inactivation.
Moreover, the quantification from the acquired XRF maps (Fig. 4C) indicates that the average concentrations of Cu and Zn inside particles are mostly conserved (considering the semi-quantitative nature of the technique) during the exhaustion processes (both thermal and operational). This confirms that there is no significant leaching of metals from either the holoenzymes or the agarose particles. Instead, BsADH and AsLAC seem to migrate differently within the beads, during both thermal inactivation and operational conditions. According to previous studies,14 such protein migration is possible because BsADH is immobilised reversibly on the agarose beads through the His-tag, and AsLAC is also bound to the carrier through weak electrostatic interactions. Therefore, we suggest that both holoenzymes undergo macromolecular crawling through the surface of the porous support. Such events normally cause structural distortions on the immobilised enzymes, leading to their partial inactivation.14,15 In the case of Cu-dependent AsLAC, these spatial rearrangements are less pronounced than those observed for Zn-dependent BsADH. Equally, AsLAC suffers minimal functional impairment, losing only 5% of the initial activity after the first operational run, compared to a 50% activity loss for BsADH. These changes suggest a direct relation between the extent of the migration of an enzyme within the particle, the amount of structural distortions that they suffer, and their inactivation during operational use (Fig. S15†). Thus, explaining the large levels of rearrangement and inactivation of BsADH as compared to the more moderate changes observed for AsLAC. Therefore, the lower operational stability of BsADH seems to limit the robustness of our biocatalyst.
The interpretation of the results obtained in these experiments considers the presence of certain heterogeneity between beads. As such, single-particle experiments using CLSM or similar microscopy techniques typically analyse ∼10 beads. Studying that many samples with XRF under cryogenic conditions is rather difficult due to long acquisition times and limited access time. We are aware of this limitation and the possible consequences that it can have on the significance of the data obtained.53 Regardless, a minimum of 3 beads were imaged at the highest resolution used per sample (4× fresh, 3× thermal inactivated, and 3× after 1st reaction cycle). Moreover, our experiments using CLSM on analogue biocatalysts, prepared by immobilizing fluorescently labeled BsADH and AsLAC enzymes, were conducted on much larger populations and revealed low variability among the beads. Notably, beads of the same type consistently exhibited the same trends in enzyme localisation according to both the XRF and CLSM experiments. Therefore, the low particle variability (heterogenicity) in the samples studied here supports the conclusions drawn.
Conclusions
We have used XRF imaging to probe the spatial organisation of a heterogeneous biocatalyst that was composed of two unmodified metalloenzymes (binding Zn or Cu) immobilised on porous agarose-based carriers. The elemental maps obtained revealed the distinctive distribution of each type of metalloenzymes within the particles, and indicated that an internal localisation of oxygen-dependent enzymes (such as AsLAC) within the beads limits the catalytic efficiency of the heterogeneous biocatalyst. Moreover, XRF imaging of particles that were used to oxidise diols to lactols and lactones provided important insights into the spatial rearrangements experienced by the immobilised enzymes during operational runs, helping us to understand the inactivation mechanisms responsible for the exhaustion of our heterogeneous biocatalysts. Our experiments suggest the presence of different extents of rearrangement in the spatial localisation of each of the enzymes during operation. Despite this spatial redistribution, both enzymes remained bound to their metal cofactor with the protein scaffold and remained immobilised within the particle. Such reorganisations were apparently allowed by the weak interactions between the enzymes and the support, which facilitated intraparticle enzyme migration through macromolecular crawling on the bead surface. Furthermore, the process ultimately caused structural distortions in the proteins, leading to a loss of enzymatic activity and exhaustion of the biocatalyst.Overall, our results show the potential of XRF imaging with suitable spatial and temporal resolution to characterise both pre- and post-operational heterogeneous biocatalysts. Similar analytical methods have already been used to understand the performance of heterogeneous catalysts before, during, and after their operation,33,36–38,54 but they were not previously applied for the functional characterization of heterogeneous biocatalysts. Remarkably, the information obtained from such experiments can provide insights into phenomena that were previously elusive to traditional ensemble measurements, and is of great value to optimise fabrication of new biocatalysts. Therefore, further use of XRF and other X-ray spectroscopic methods should help develop more robust and efficient heterogeneous biocatalysts for industrial purposes.
Data availability
The XRF data presented in this study are openly available via the European Synchrotron Radiation Facility at https://doi.org/10.15151/ESRF-ES-449124298. The rest of the data are contained within the article.Author contributions
JSA, FLG, and CSC conceived the project and designed experiments. JSA performed enzymatic experiments. JSA, MS, and CSC performed XRF experiments. All authors contributed to the writing and editing of the manuscript and agreed on the final version.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The XRF experiments were performed on beamline ID16A (proposal MA-4866) at the European Synchrotron Radiation Facility (ESRF), Grenoble, France. This work was performed under the support of the Severo Ochoa Centres and Maria de Maeztu Units of Excellence Program of the Spanish State Research Agency – Grant No. CEX2018-000867-S (DIPC) and MDM-2017-0720 (CIC biomaGUNE). The authors thank the Spanish State Research Agency (grant PID2020-118176RJ-I100 to CSC, grant RTI2018-094398-B-I00 to FLG, the FPI fellowship PRE2019-090835 to JSA) and the European Research Council (ERC-Co-METACELL-818089) grant to FLG, and the Gipuzkoa Foru Aldundia (Gipuzkoa Fellows program; grant number 2019-FELL-000018-01/62/2019 to CSC) for financial support. The Table of Contents and Fig. 1A were created in https://www.biorender.com/.References
- M. Teshima, V. P. Willers and V. Sieber, Curr. Opin. Biotechnol., 2023, 79, 102868 CrossRef CAS PubMed.
- S. Burgener, S. Luo, R. McLean, T. E. Miller and T. J. Erb, Nat. Catal., 2020, 3, 186–192 CrossRef.
- L. J. Sweetlove and A. R. Fernie, Annu. Rev. Plant Biol., 2013, 64, 723–746 CrossRef CAS PubMed.
- D. L. Schmitt and S. An, Biochemistry, 2017, 56, 3184–3196 CrossRef CAS PubMed.
- W. J. Altenburg, N. A. Yewdall, D. F. M. Vervoort, M. H. M. E. van Stevendaal, A. F. Mason and J. C. M. van Hest, Nat. Commun., 2020, 11, 6282 CrossRef CAS PubMed.
- J. Santiago-Arcos, S. Velasco-Lozano, E. Diamanti, A. I. Benítez-Mateos, D. Grajales-Hernández, F. Paradisi and F. López-Gallego, ACS Sustain. Chem. Eng., 2024, 12, 9474–9489 CrossRef CAS PubMed.
- D. A. Grajales-Hernández, E. Diamanti, R. Moro, S. Velasco-Lozano, E. Pires and F. López-Gallego, ACS Catal., 2023, 13, 15620–15632 CrossRef.
- U. Guzik, K. Hupert-Kocurek and D. Wojcieszyńska, Molecules, 2014, 19, 8995–9018 CrossRef PubMed.
- J. M. Bolivar, J. M. Woodley and R. Fernandez-Lafuente, Chem. Soc. Rev., 2022, 51, 6251–6290 RSC.
- J. M. Bolivar, A. Hidalgo, L. Sanchez-Ruiloba, J. Berenguer, J. M. Guisan and F. Lopez-Gallego, J. Biotechnol., 2011, 155, 412–420 CrossRef CAS PubMed.
- F. Lopez-Gallego, Methods Enzymol., 2019, 617, 385–411 CAS.
- G. A. Ellis, W. P. Klein, G. Lasarte-Aragonés, M. Thakur, S. A. Walper and I. L. Medintz, ACS Catal., 2019, 9, 10812–10869 CrossRef CAS.
- A. J. Martín, S. Mitchell, C. Mondelli, S. Jaydev and J. Pérez-Ramírez, Nat. Catal., 2022, 5, 854–866 CrossRef.
- E. Diamanti, S. Arana-Peña, P. Ramos-Cabrer, N. Comino, D. Carballares, R. Fernandez-Lafuente and F. López-Gallego, Adv. Mater. Interfaces, 2022, 9, 2200263 CrossRef CAS.
- A. F. Chaparro Sosa, R. M. Bednar, R. A. Mehl, D. K. Schwartz and J. L. Kaar, J. Am. Chem. Soc., 2021, 143, 7154–7163 CrossRef CAS PubMed.
- J. M. Bolivar, I. Eisl and B. Nidetzky, Catal. Today, 2016, 259, 66–80 CrossRef CAS.
- J. M. Bolivar and B. Nidetzky, Molecules, 2019, 24, 3460 CrossRef CAS PubMed.
- E. Diamanti and F. López-Gallego, Angew. Chem., Int. Ed., 2024, 63, e202319248 CrossRef CAS PubMed.
- Z. Huang, G. Li, C. Zhang and X.-H. Xing, Enzyme Microb. Technol., 2016, 83, 1–6 CrossRef CAS PubMed.
- W. Wriggers, S. Chakravarty and P. A. Jennings, Pept. Sci., 2006, 80, 736–746 CrossRef PubMed.
- Y. Mei, L. Miller, W. Gao and R. A. Gross, Biomacromolecules, 2003, 4, 70–74 CrossRef CAS PubMed.
- J. P. Smith, M. Liu, M. L. Lauro, M. Balasubramanian, J. H. Forstater, S. T. Grosser, Z. E. X. Dance, T. A. Rhodes, X. Bu and K. S. Booksh, Analyst, 2020, 145, 7571–7581 RSC.
- O. Pauli, A. Ecker, A. Cruz-Izquierdo, A. Basso and S. Serban, Catalysts, 2022, 12, 989 CrossRef CAS.
- I.-I. Rădoi, D. E. Bedolla, L. Vaccari, A. Todea, F. Zappaterra, A. Volkov and L. Gardossi, Catal. Sci. Technol., 2023, 13, 4955–4967 RSC.
- H. Wei and J. P. Smith, ACS Cent. Sci., 2023, 9, 1913–1926 CrossRef CAS PubMed.
- N. M. Ralbovsky and J. P. Smith, Biocatalysis Pharm. Res., 2023, 40, 1479–1490 CrossRef CAS PubMed.
- H. Wei and J. P. Smith, Mol. Pharmaceutics, 2024, 21, 5565–5576 CrossRef CAS PubMed.
- M. J. Pushie, I. J. Pickering, M. Korbas, M. J. Hackett and G. N. George, Chem. Rev., 2014, 114, 8499–8541 CrossRef CAS PubMed.
- P. Kikongi, J. Salvas and R. Gosselin, X-Ray Spectrom., 2017, 46, 347–355 CrossRef CAS.
- J. C. da Silva, A. Pacureanu, Y. Yang, S. Bohic, C. Morawe, R. Barrett and P. Cloetens, Optica, 2017, 4, 492 CrossRef CAS.
- J. Decelle, G. Veronesi, B. Gallet, H. Stryhanyuk, P. Benettoni, M. Schmidt, R. Tucoulou, M. Passarelli, S. Bohic, P. Clode and N. Musat, Trends Cell Biol., 2020, 30, 173–188 CrossRef CAS PubMed.
- S. G. Lehmann, D. Toybou, A.-E. Pradas del Real, D. Arndt, A. Tagmount, M. Viau, M. Safi, A. Pacureanu, P. Cloetens, S. Bohic, M. Salomé, H. Castillo-Michel, B. Omaña-Sanz, A. Hofmann, C. Vulpe, J.-P. Simonato, C. Celle, L. Charlet and B. Gilbert, Proc. Natl. Acad. Sci. U. S. A., 2019, 116, 14893–14898 CrossRef CAS PubMed.
- S. Ünsal, R. Girod, C. Appel, D. Karpov, M. Mermoux, F. Maillard, V. A. Saveleva, V. Tileli, T. J. Schmidt and J. Herranz, J. Am. Chem. Soc., 2023, 145, 7845–7858 CrossRef PubMed.
- J. J. Conesa, A. C. Carrasco, V. Rodríguez-Fanjul, Y. Yang, J. L. Carrascosa, P. Cloetens, E. Pereiro and A. M. Pizarro, Angew. Chem., Int. Ed., 2020, 59, 1270–1278 CrossRef CAS PubMed.
- A. Procopio, E. Malucelli, A. Pacureanu, C. Cappadone, G. Farruggia, A. Sargenti, S. Castiglioni, D. Altamura, A. Sorrentino, C. Giannini, E. Pereiro, P. Cloetens, J. A. M. Maier and S. Iotti, ACS Cent. Sci., 2019, 5, 1449–1460 CrossRef CAS PubMed.
- S. W. T. Price, K. Geraki, K. Ignatyev, P. T. Witte, A. M. Beale and J. F. W. Mosselmans, Angew. Chem., Int. Ed., 2015, 54, 9886–9889 CrossRef CAS PubMed.
- S. Kalirai, U. Boesenberg, G. Falkenberg, F. Meirer and B. M. Weckhuysen, ChemCatChem, 2015, 7, 3674–3682 CrossRef CAS PubMed.
- K. W. Bossers, R. Valadian, S. Zanoni, R. Smeets, N. Friederics, J. Garrevoet, F. Meirer and B. M. Weckhuysen, J. Am. Chem. Soc., 2020, 142, 3691–3695 CrossRef CAS PubMed.
- S. Aksu, I. W. C. E. Arends and F. Hollmann, Adv. Synth. Catal., 2009, 351, 1211–1216 CrossRef CAS.
- S. Kara, D. Spickermann, J. H. Schrittwieser, A. Weckbecker, C. Leggewie, I. W. C. E. Arends and F. Hollmann, ACS Catal., 2013, 3, 2436–2439 CrossRef CAS.
- R. R. Melo, R. C. Alnoch, A. F. L. Vilela, E. M. Souza, N. Krieger, R. Ruller, H. H. Sato and C. Mateo, Molecules, 2017, 22, 1088 CrossRef PubMed.
- J. M. Bolivar, C. Mateo, V. Grazu, A. V. Carrascosa, B. C. Pessela and J. M. Guisan, Process Biochem., 2010, 45, 1692–1698 CrossRef CAS.
- C. Mateo, J. M. Bolivar, C. A. Godoy, J. Rocha-Martin, B. C. Pessela, J. A. Curiel, R. Muñoz, J. M. Guisan and G. Fernández-Lorente, Biomacromolecules, 2010, 11, 3112–3117 CrossRef CAS PubMed.
- C. Mateo, V. Grazu, J. M. Palomo, F. Lopez-Gallego, R. Fernandez-Lafuente and J. M. Guisan, Nat. Protoc., 2007, 2, 1022–1033 CrossRef CAS PubMed.
- A. I. Benitez-Mateos, M. L. Contente, S. Velasco-Lozano, F. Paradisi and F. Lopez-Gallego, ACS Sustain. Chem. Eng., 2018, 6, 13151–13159 CrossRef CAS.
- M. L. Contente and F. Paradisi, Nat. Catal., 2018, 1, 452–459 CrossRef CAS.
- C. Ceccarelli, Z.-X. Liang, M. Strickler, G. Prehna, B. M. Goldstein, J. P. Klinman and B. J. Bahnson, Biochemistry, 2004, 43, 5266–5277 CrossRef CAS PubMed.
- Purchased from Sigma-Aldrich, Ref: SAE0050.
- S. Velasco-Lozano, J. Santiago-Arcos, J. A. Mayoral and F. López-Gallego, ChemCatChem, 2020, 12, 3030–3041 CrossRef CAS.
- J. B. Costa, M. J. Lima, M. J. Sampaio, M. C. Neves, J. L. Faria, S. Morales-Torres, A. P. M. Tavares and C. G. Silva, Chem. Eng. J., 2019, 355, 974–985 CrossRef CAS.
- A. I. Benitez-Mateos, C. Huber, B. Nidetzky, J. M. Bolivar and F. López-Gallego, ACS Appl. Mater. Interfaces, 2020, 12, 56027–56038 CrossRef CAS PubMed.
- J. Santiago-Arcos, S. Velasco-Lozano, E. Diamanti, A. I. Benitez-Mateos, D. Grajales-Hernández, F. Paradisi and F. López-Gallego, ACS Sustainable Chem. Eng., 2024, 12, 9474–9489 CrossRef CAS PubMed.
- M. Skiba, G. Guedes, D. Karpov, N. Feliu, A. L. Cortajarena, W. J. Parak and C. Sanchez-Cano, Int. J. Mol. Sci., 2024, 25, 528 CrossRef CAS PubMed.
- K. F. Kalz, R. Kraehnert, M. Dvoyashkin, R. Dittmeyer, R. Gläser, U. Krewer, K. Reuter and J. D. Grunwaldt, ChemCatChem, 2017, 9, 17–29 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Materials and methods, additional figures and tables. See DOI: https://doi.org/10.1039/d4sc05136j |
| This journal is © The Royal Society of Chemistry 2024 |