
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicencePhotocatalytic 1,3-oxyheteroarylation of aryl cyclopropanes with azine N-oxides†
Doyoung
Kim
 ab,
Hyewon
Ju
ab,
Wooseok
Lee
ab and
Sungwoo
Hong
*ab
ab,
Hyewon
Ju
ab,
Wooseok
Lee
ab and
Sungwoo
Hong
*ab
aDepartment of Chemistry, Korea Advanced Institute of Science and Technology (KAIST), Daejeon, 34141, Korea. E-mail: hongorg@kaist.ac.kr
bCenter for Catalytic Hydrocarbon Functionalizations, Institute for Basic Science (IBS), Daejeon 34141, Korea
First published on 13th November 2024
Abstract
Cyclopropanes, valuable C3 building blocks in organic synthesis, possess high strain energy and inherent stability. We present an efficient, environmentally benign 1,3-oxyheteroarylation of aryl cyclopropanes using azine N-oxides as bifunctional reagents under visible light irradiation. This metal-free method yields β-pyridyl ketones under mild conditions. Mechanistic studies reveal a photo-induced radical pathway involving single-electron oxidation of both aryl cyclopropanes and azine N-oxides, followed by stepwise ring opening. The dual oxidation mechanism accommodates diverse cyclopropane and azine N-oxide combinations based on their oxidation potentials. This green chemistry method enhances the synthetic utility of aryl cyclopropanes while introducing an efficient strategy for their difunctionalization. The methodology aligns with sustainable organic synthesis principles, offering an environmentally conscious route to valuable synthetic intermediates.
Introduction
Cyclopropane, with its high strain energy and bench stability,1 has been a versatile component in organic synthesis for decades.2 Extensive research has focused on utilizing cyclopropane as a C3 building block to develop new skeletal structures through ring-opening/1,3-difunctionalization reactions. These studies have evolved from traditional strategies, such as activating cyclopropane's C–C bond via Lewis acid or transition metal coordination, to more recent approaches.3 Recent advances in photochemistry have opened up promising avenues for creating new reactivity,4 including the induction of C–C bond cleavage.5 Notably, several studies have successfully demonstrated the ring-opening of electronically unbiased aryl cyclopropanes through single-electron oxidation under visible light.6 In this approach, electron-rich cyclopropyl arenes are oxidized by excited photocatalysts, forming aryl cyclopropane radical cations. These reactive species can undergo nucleophilic addition,7 facilitating attack on the weakened C–C bond and enabling ring opening and functionalization under environmentally friendly conditions.Despite this significant advance, important limitations remain. The approach is currently restricted to electron-rich aryl cyclopropanes, as electron-deficient variants are inadequately oxidized by the photocatalyst.6g,8a,c,d Moreover, the scope of functional groups capable of reacting with the benzylic radical intermediate has been restricted, typically resulting in products with hydrogen or oxygen substituents at the benzylic position and C–C bond formations are rarely reported.8 Notably, the incorporation of heteroaryl groups at benzylic radicals generated by such ring-opening reactions has not been reported,8d presenting an opportunity to expand the synthetic utility of aryl cyclopropanes in photocatalytic processes (Fig. 1a).
 | ||
| Fig. 1 Overview of photoredox-catalyzed 1,3-functionalization of aryl cyclopropanes. (a) Limitations of previous approaches. (b) This work. | ||
To address these challenges, we envisioned a strategy utilizing the direct oxidation of azine N-oxides by an excited-state photocatalyst to generate O-centered radicals.9 We hypothesized that these O-radicals could enable a previously inaccessible 1,3-functionalization with a broader range of aryl cyclopropanes, including electron-deficient variants. Herein, we present an efficient and environmentally benign method for the 1,3-oxyheteroarylation of various aryl cyclopropanes utilizing azine N-oxides as bifunctional reagents under visible light irradiation. Our approach leverages the ability of both aryl cyclopropanes and azine N-oxides to undergo photo-induced single-electron oxidation, generating their respective radical cations (Fig. 1b). The reaction proceeds based on the oxidation potentials of the substrates, allowing for a wide range of applications.8c,9c,d This method yields β-heteroaryl ketones, offering an efficient strategy for functionalizing these valuable building blocks. Our approach operates under mild conditions, utilizing visible light and azine N-oxides as bifunctional reagents, eliminating the need for harsh reagents or toxic metal catalysts. This method enhances the utility of aryl cyclopropanes as synthons for β-heteroaryl ketones, providing an efficient route to important building blocks and valuable synthetic intermediates.
Results and discussion
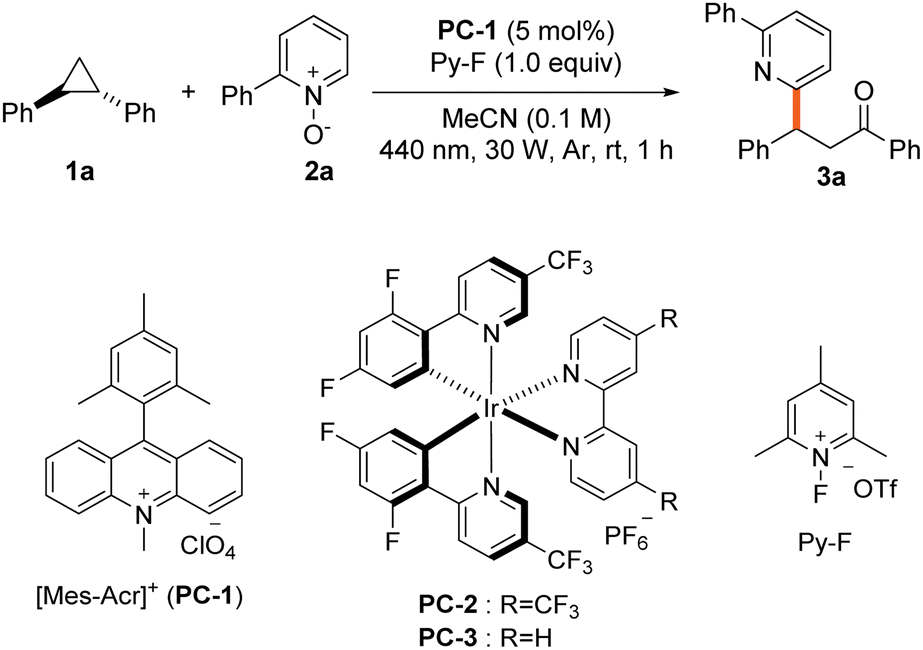
We evaluated pyridine N-oxide's efficacy as a nucleophile for aryl cyclopropane ring opening using 1,2-diphenylcyclopropane (1a) and 2-phenylpyridine N-oxide (2a) as model substrates under visible light irradiation with a photocatalyst (Table 1). Optimal conditions were identified: 9-mesityl-10-methylacridinium perchlorate (PC-1) as photocatalyst and 1-fluoro-2,4,6-trimethylpyridinium triflate (Py-F) as oxidant in acetonitrile under blue light, yielding 79% of the 1,3-oxyheteroarylated product (entry 1). Bond formation preferentially occurred at the C6 position of the pyridine core over the C4 position. Alternative oxidants like tBuOOH and (PhS)2 reduced yields (entry 2). Additionally, during our systematic oxidant screening studies, we consistently observed the formation of alcohol as a side product (up to 15% yield). When using Py-F, less than 5% of this byproduct is generated in trace amounts (Table S1†). Iridium catalyst PC-2 gave comparable yields, while PC-3 showed poor reactivity (entry 3). Moderately polar solvents such as acetone performed well, but polar aprotic solvent (DMSO), nonpolar aprotic solvent (pentane) inhibited the reaction (entry 4). The reaction tolerated water and air, achieving 66% and 56% yields respectively (entry 5). Using O2 from ambient air as the oxidant led to modest product formation, while the reaction was completely suppressed under pure oxygen atmosphere, suggesting that excessive oxygen may deactivate the photocatalytic cycle through quenching processes (entry 6, Table S5†). Control experiment indicate that the both the photocatalyst and light are essential for the reaction (entry 7).|
|
||
|---|---|---|
| Entry | Variations | Yieldb |
| a Reaction conditions: 1a (0.05 mmol), 2a (1.5 equiv.), PC (5 mol%), oxidant (1.0 equiv.) in solvent (0.5 mL) under irradiation with 440 nm LEDs (30 W) at rt for 1 h under argon atmosphere. b Yields were determined by 1H NMR spectroscopy using 1,3,5-trimethoxybenzene as an internal standard. c Isolated yield. d 2 mol% of PC used. | ||
| 1 | None | 79% (78%)c |
| 2 | t BuOOH, (PhS)2 instead of Py-F | 55%, 27% |
| 3 | PC-2, PC-3 instead of PC-1 | 75%,d 7%d |
| 4 | Acetone, DMSO, n-pentane instead of MeCN | 78%, trace, 16% |
| 5 | H2O (20 μL of), under open air | 66%, 56% |
| 6 | No Py-F under Ar, air, O2 | 9%, 21%, n.d. |
| 7 | No PC, light | Trace |
When performed on a larger scale (5.2 mmol), the reaction afforded the desired product in comparable yield. The substrate scope was investigated under optimized conditions. Various azine N-oxides afforded 1,3-oxyheteroarylated products (Table 2). Simple pyridine N-oxide demonstrated good reactivity (3b). 2-Substituted pyridine N-oxides performed well with both electron-withdrawing (halogens and CF3) and electron-donating (methoxy) groups (3c–3e). N-Oxides with aryl substituents at the pyridine C2 position showed high efficiency, regardless of electronic properties (3f and 3g). Notably, N-oxides with alkyl substituents at the pyridine C2 position exhibited particularly high reactivity (3h). Bipyridine, commonly used as a ligand, also provided satisfactory results (3i), while moderate conversion was observed with a thiophene-containing substrate (3j). Substituted pyridine N-oxides with aryl or alkyl groups at the C4 position performed well (3k and 3l). Efficient conversions were also achieved with electron-withdrawing groups, demonstrating broad functional group tolerance (3m and 3n). For 3-substituted pyridine N-oxides bearing amide (3p) or phenyl (3q) groups, radical addition preferentially occurs at the C6 position due to significant steric hindrance. In contrast, with 3-fluorine substituted pyridine N-oxide (3o), electronic effects dominate the regioselectivity, favoring addition at the more electrophilic C2 position. The reaction was successful with various azine N-oxides including pyrimidine, pyridazine, quinoline, and quinoxaline (3r–3u). For pyrimidine N-oxide, bond formation favored the C4 position (3r). N-Oxides containing three rings, such as phenanthridine (a DNA intercalating agent), also demonstrated good efficiency (3v and 3w). Late-stage functionalization was performed on several biorelevant azine N-oxides. Derivatives of pyriproxyfen and vismodegib showed good reactivity (3x and 3z), with excellent conversion obtained from bisacodyl-derived N-oxide (3y). Quinoxyfen and roflumilast derivatives also performed well (3aa and 3ab).
| a Reaction conditions: 1a (0.05 mmol), 2 (1.5 equiv.), [Mes-Acr]+ (5 mol%), Py-F (1.0 equiv.) in MeCN (0.5 mL) under irradiation with 440 nm LEDs (30 W) at room temperature for 1 h under argon atmosphere. Regioisomeric ratios were measured by 1H NMR spectroscopy. Isolated yield. a 3 h for reaction time. b 18 h for reaction time. |
|---|

|
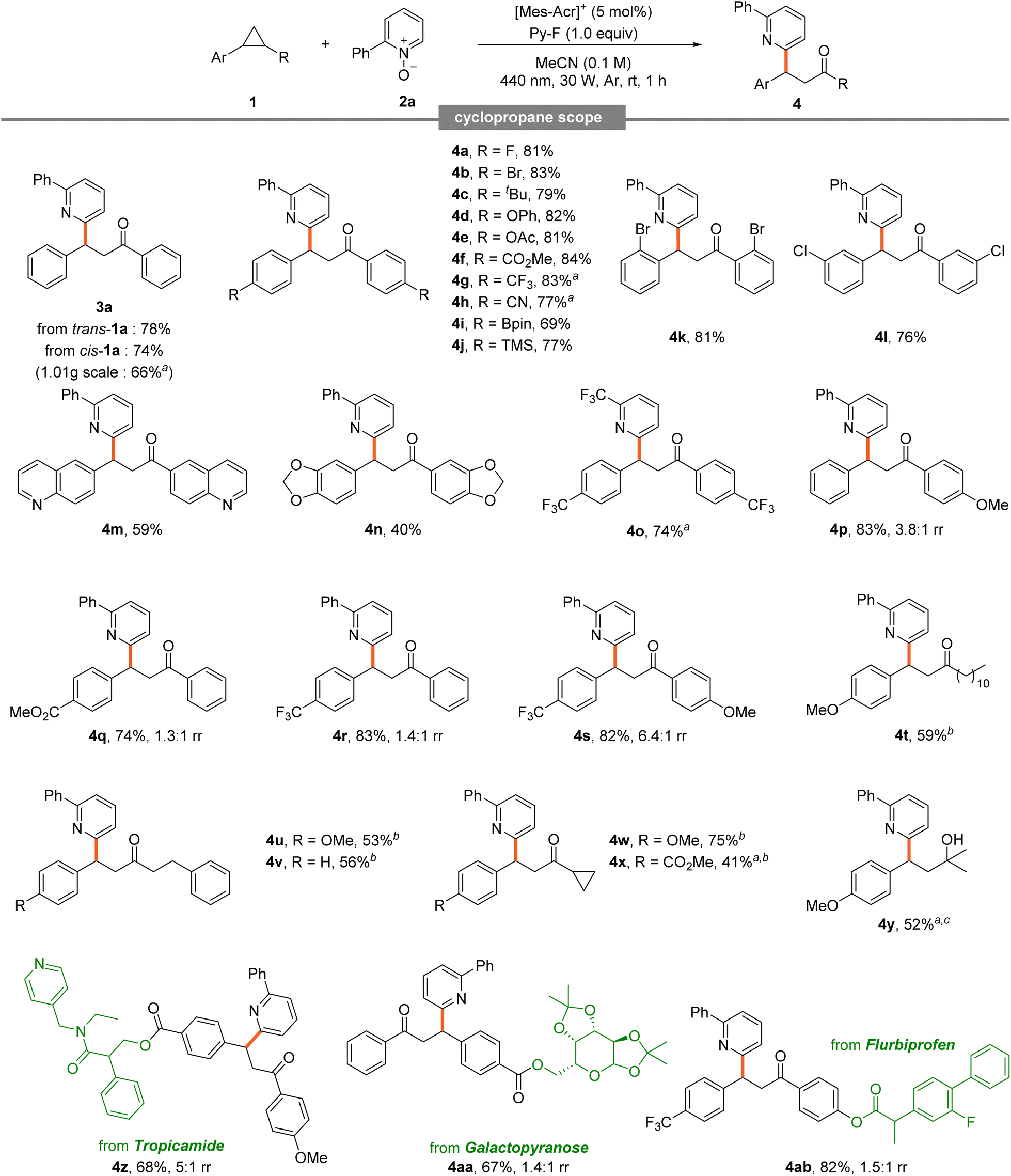
A broad substrate scope was obtained from various cyclopropanes (Table 3). Diphenyl cyclopropane 1a exhibited excellent reactivity in both cis and trans forms, even at large scale (3a). Symmetric diaryl cyclopropanes generally showed good efficiency. Notably high conversions were observed for those with halogens at the para position (4a and 4b). Substrates bearing bulky t-butyl groups and electron-rich phenoxy and acetyl groups exhibited excellent reactivity (4c–4e). Notably, good conversions were also achieved with electron-withdrawing groups such as ester, trifluoromethyl, and cyano suggesting limited electronic influence at the para position (4f–4h). This clearly demonstrates that the reaction proceeds successfully even in electron-deficient cyclopropanes, where oxidation was previously ineffective in existing studies.
| a Reaction conditions: 1 (0.05 mmol), 2a (1.5 equiv.), [Mes-Acr]+ (5 mol%), Py-F (1.0 equiv.) in MeCN (0.5 mL) under irradiation with 440 nm LEDs (30 W) at room temperature for 1 h under argon atmosphere. Regioisomeric ratios were measured by 1H NMR spectroscopy. Isolated yield. a 3 h for reaction time. b PC-2 (2 mol%), acetone (0.5 mL) were used. cPC-3 (2 mol%), (4-OMePhS)2 (50 mol%), DCM (0.5 mL) were used. |
|---|
|
Symmetric diaryl cyclopropanes with ortho and meta halogen substitutions underwent efficient reactions (4k and 4l). Cyclopropanes containing Bpin and aryl silane, common coupling partners, performed well (4i and 4j). Those with quinoline and piperonyl moieties, frequently observed in biomolecules, also reacted well, demonstrating tolerance to diverse chemical moieties (4m and 4n). Efficient conversion was achieved with CF3-substituted pyridine N-oxide and an electron-deficient, trifluoromethyl-substituted diaryl cyclopropane (4o). Notably, substrates like this trifluoromethyl-substituted diaryl cyclopropane typically possess higher oxidation potentials, making them resistant to oxidation.
The successful transformation of such a challenging substrate underscores the robustness of this method. Unsymmetric diaryl cyclopropanes showed good reactivity, with excellent conversions observed for phenyl rings substituted with methoxy, ester, or trifluoromethyl groups (4p–4s). When electron-donating and electron-withdrawing groups were on different rings, good efficiency and improved regioselectivity were observed, indicating that the electronic difference between the two rings can influence the regioselectivity of the reaction.6d The substrate scope extended to unsymmetric aryl alkyl cyclopropanes. Alkyl substituents containing undecyl, homobenzyl, and cyclopropyl showed good conversion (4t–4x). For molecules with two cyclopropyl rings, ring-opening occurred selectively at the aryl-substituted ring. Dimethyl-substituted cyclopropane, which cannot form a ketone, yielded alcohol as the product (4y). Late-stage functionalization of biorelevant cyclopropanes derived from tropicamide, galactopyranose, and flurbiprofen showed good conversions (4z–4ab).
To elucidate the reaction mechanism, a series of experiments were conducted. Control experiments ruled out a two-electron pathway involving nucleophilic addition by the N-oxide (Table 1, entry 7). The reaction was inhibited by the radical trapping agent TEMPO, with HRMS detection of a TEMPO adduct confirming a radical pathway (Fig. 2a). Light on-off experiments revealed no additional product formation in the absence of light, suggesting that a radical chain mechanism is unlikely to be operative in this system (Fig. S5†). To investigate the potential oxidation of the in situ generated alcohol intermediate to a ketone, we explored various reaction conditions (Fig. 2b). Our experiments revealed that with only the photocatalyst present, ketone conversion was minimal. Similarly, in the presence of pyridine N-oxide 2a alone, the conversion remained minimal and correlated with the amount of photocatalyst used. Notably, without the photocatalyst, no conversion occurred. Interestingly, when Py-F was introduced to the reaction, we observed a 27% yield of 3a, accompanied by a significant reduction in the alcohol intermediate. Under our standard conditions, which included Py-F, 3a was produced with a 78% yield, and the alcohol 5a was almost entirely consumed. These observations suggest that the alcohol compound may function as an intermediate in the reaction pathway. The necessity of both the photocatalyst cycle and N-oxide for effective conversion indicates that N-oxide plays a crucial role in the oxidation of alcohol to the ketone product. Stern–Volmer quenching experiments identified which substrate undergoes radical initiation upon light exposure. Cyclic voltammetry was then employed to determine the oxidation potentials of the substrates (Fig. 2c and S16–S19†). Both 1a and 2a were found to quench the photocatalyst. Subsequent quenching measurements involving electron-deficient cyclopropane and N-oxide revealed ineffective photocatalyst quenching. However, successful synthesis of products from combinations such as 1a with 2d and 1h with 2a suggests two concurrent pathways: (1) ring opening through an SN2 reaction of the pyridine N-oxide with the cyclopropane radical cation formed by cyclopropane oxidation, and (2) ring opening through an SH2 reaction involving the O-centered radical generated by pyridine N-oxide oxidation.
 | ||
| Fig. 2 Control experiments and mechanistic studies. (a) Radical trapping experiments with TEMPO. (b) Oxidation of the alcohol intermediate to ketone. (c) Stern–Volmer quenching experiments. | ||
Based on the experimental results and previous literature,6,8 we propose two distinct mechanisms (Fig. 3). Initially, photoexcitation of the photocatalyst facilitates single-electron oxidation of either the cyclopropane or the pyridine N-oxide. Nucleophilic attack by the pyridine N-oxide 2a on the oxidized cyclopropane intermediate or the O-centered radical of N-oxide to cyclopropane results in cleavage of the C–C bond, forming intermediate Int 1. The common pathway proceeds with the benzylic radical in Int 1 adding to the C-6 position of the pyridine ring, leading to cyclization and formation of Int 2.9a,10 Subsequent N–O bond cleavage and rearomatization produce Int 3, which undergoes intermolecular HAT or 1,2-HAT to form Int 4.11 Finally, oxidation of the benzylic radical by the pyridine radical cation yields a benzylic cation, which forms the ketone product 3a through further deprotonation. In an alternative mechanism, the formation of Int 3 proceeds as before (Fig. S20 in the ESI†). However, instead of undergoing 1,2-HAT, Int 3 is reduced by the PC radical, resulting in the formation of the alcohol intermediate. Concurrently, from the second PC cycle, the O-centered radical N-oxide is regenerated. This radical acts as an HAT catalyst, facilitating the conversion of alcohol to Int 4. Finally, Int 4 is oxidized to yield the ketone product 3a.
 | ||
| Fig. 3 Proposed reaction mechanism. | ||
Conclusions
In summary, we have successfully demonstrated the visible light-mediated ring opening of aryl cyclopropanes, yielding 1,3-oxyheteroarylated products. This method offers several significant advantages, including the functionalization of aryl cyclopropanes under mild conditions and broad tolerance for diverse functional groups. The sustainability of this approach is enhanced by the use of readily accessible azine N-oxides, which function as efficient bifunctional reagents. Mechanistic studies reveal that both aryl cyclopropane and azine N-oxides can quench the photocatalyst through distinct pathways, confirming the involvement of radical processes in this reaction. This efficient approach not only expands the synthetic utility of aryl cyclopropanes but also aligns with the principles of green chemistry, offering a sustainable pathway for the synthesis of complex molecular scaffolds. The method's efficiency, mild conditions, and use of visible light as an environmentally benign energy source represent a significant advancement in sustainable organic synthesis.Data availability
The data supporting this article have been included as part of the ESI.†Author contributions
D. K., H. J., W. L. performed the experiments, analyzed the data, and wrote the manuscript. S. H. directed the project and wrote the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This research was supported financially by Institute for Basic Science (IBS-R010-A2).Notes and references
- (a) A. de Meijere, Angew. Chem., Int. Ed., 1979, 18, 809–826 CrossRef; (b) K. B. Wiberg, Angew. Chem., Int. Ed., 1986, 25, 312–322 CrossRef.
- (a) C. Ebner and E. M. Carreira, Chem. Rev., 2017, 117, 11651–11679 CrossRef CAS PubMed; (b) H. N. C. Wong, M. Y. Hon, C. W. Tse, Y. C. Yip, J. Tanko and T. Hudlicky, Chem. Rev., 1989, 89, 165–198 CrossRef CAS; (c) H.-U. Reissig and R. Zimmer, Chem. Rev., 2003, 103, 1151–1196 CrossRef CAS PubMed.
- (a) T. F. Schneider, J. Kaschel and D. B. Werz, Angew. Chem., Int. Ed., 2014, 53, 5504–5523 CrossRef CAS PubMed; (b) M. A. Cavitt, L. H. Phun and S. France, Chem. Soc. Rev., 2014, 43, 804–818 RSC; (c) H. K. Grover, M. R. Emmett and M. A. Kerr, Org. Biomol. Chem., 2015, 13, 655–671 RSC; (d) E. Budynina, K. Ivanov, I. Sorokin and M. Melnikov, Synthesis, 2017, 49, 3035–3068 CrossRef CAS; (e) F. Doraghi, S. Karimian, O. H. Qareaghaj, M. J. Karimi, B. Larijani and M. Mahdavi, J. Organomet. Chem., 2024, 1005, 122963 CrossRef CAS; (f) K. Ghosh and S. Das, Org. Biomol. Chem., 2021, 19, 965–982 RSC; (g) G. Fumagalli, S. Stanton and J. F. Bower, Chem. Rev., 2017, 117, 9404–9432 CrossRef CAS PubMed; (h) A. S. Adhikari and N. Majumdar, Eur. J. Org Chem., 2024, 27, e202301225 CrossRef CAS; (i) E. Budynina, K. Ivanov, I. Sorokin and M. Melnikov, Synthesis, 2017, 49, 3035–3068 CrossRef CAS; (j) A. Ratzenböck, M. Kobras, A. Rustler and O. Reiser, Chem.–Eur. J., 2024, 30, e202401332 CrossRef PubMed.
- (a) R. Liu, S. P. M. Chia, Y. Y. Goh, H. W. Cheo, B. Fan, R. Li, R. Zhou and J. Wu, Eur. J. Org Chem., 2020, 1459–1465 CrossRef CAS; (b) F. Rammal, D. Gao, S. Boujnah, A. Gaumont, A. A. Hussein and S. Lakhdar, Org. Lett., 2020, 22, 7671–7675 CrossRef CAS PubMed; (c) F. Yuan, D.-M. Yan, P.-P. Gao, D.-Q. Shi, W.-J. Xiao and J.-R. Chen, ChemCatChem, 2021, 13, 543–547 CrossRef CAS; (d) A. Tlili and S. Lakhdar, Angew. Chem., Int. Ed., 2021, 60, 19526–19549 CrossRef CAS PubMed; (e) M. Nakagawa, K. Nagao, Z. Ikeda, M. Reynolds, I. Ibáñez, J. Wang, N. Tokunaga, Y. Sasaki and H. Ohmiya, ChemCatChem, 2021, 13, 3930–3933 CrossRef CAS; (f) U. Karmakar, H. S. Hwang, Y. Lee and E. J. Cho, Org. Lett., 2022, 24, 6137–6141 CrossRef CAS PubMed; (g) C. Hyeon Ka, D. Seul Lee and E. J. Cho, ChemPhotoChem, 2022, 6, e202200136 CrossRef CAS; (h) S. Pan, M. T. Passia, X. Wang, P. Wu, D. Ma and C. Bolm, Adv. Synth. Catal., 2023, 365, 31–36 CrossRef CAS.
- (a) C. Wang, X. Ren, H. Xie and Z. Lu, Chem.–Eur. J., 2015, 21, 9676–9680 CrossRef CAS PubMed; (b) Ł. Woźniak, G. Magagnano and P. Melchiorre, Angew. Chem., Int. Ed., 2018, 57, 1068–1072 CrossRef PubMed; (c) D. Staveness, T. M. Sodano, K. Li, E. A. Burnham, K. D. Jackson and C. R. J. Stephenson, Chem, 2019, 5, 215–226 CrossRef CAS PubMed; (d) M.-M. Wang, T. V. T. Nguyen and J. Waser, Chem. Soc. Rev., 2022, 51, 7344–7357 RSC; (e) Y. Tong, N. Hao, L. Zhang, J. Wei, Z. Zhang, Q. Fu, D. Yi, Y. Mou, J. Wang, X. Pan, L. Yang, S. Wei, L. Zhong and J. Lu, Org. Chem. Front., 2022, 9, 2129–2134 RSC; (f) M. Vellakkaran, T. Kim and S. Hong, Angew. Chem., Int. Ed., 2022, 61, e202113658 CrossRef CAS PubMed; (g) M. Ji, Z. Wu and C. Zhu, Chem. Commun., 2019, 55, 2368–2371 RSC; (h) T. Feng, C. Liu, Z. Wu, X. Wu and C. Zhu, Chem. Sci., 2022, 13, 2669–2673 RSC; (i) Q.-Q. Zhao, X.-S. Zhou, S.-H. Xu, Y.-L. Wu, W.-J. Xiao and J.-R. Chen, Org. Lett., 2020, 22, 2470–2475 CrossRef CAS PubMed; (j) S. Budde, F. Goerdeler, J. Floß, P. Kreitmeier, E. F. Hicks, O. Moscovitz, P. H. Seeberger, H. M. L. Davies and O. Reiser, Org. Chem. Front., 2020, 7, 1789–1795 RSC; (k) M. Kumar, S. Verma, V. Mishra, O. Reiser and A. K. Verma, J. Org. Chem., 2022, 87, 6263–6272 CrossRef CAS PubMed.
- (a) L. Ge, D.-X. Wang, R. Xing, D. Ma, P. J. Walsh and C. Feng, Nat. Commun., 2019, 10, 4367 CrossRef PubMed; (b) D. Petzold, P. Singh, F. Almqvist and B. König, Angew. Chem., Int. Ed., 2019, 58, 8577–8580 CrossRef CAS PubMed; (c) H. Liu, Y. Li, D.-X. Wang, M.-M. Sun and C. Feng, Org. Lett., 2020, 22, 8681–8686 CrossRef CAS PubMed; (d) L. Ge, C. Zhang, C. Pan, D.-X. Wang, D.-Y. Liu, Z.-Q. Li, P. Shen, L. Tian and C. Feng, Nat. Commun., 2022, 13, 5938 CrossRef CAS PubMed; (e) X. Qiao, Y. Lin, D. Huang, H. Ji, C. Chen, W. Ma and J. Zhao, J. Org. Chem., 2022, 87, 13627–13642 CrossRef CAS PubMed; (f) T. V. T. Nguyen, M. D. Wodrich and J. Waser, Chem. Sci., 2022, 13, 12831–12839 RSC; (g) C. Pan, Y. Xu, B. Zhang, L. Ge, C. Zhang and C. Feng, Cell Rep. Phys. Sci., 2023, 4, 101233 CrossRef CAS.
- (a) J. P. Dinnocenzo, T. R. Simpson, H. Zuilhof, W. P. Todd and T. Heinrich, J. Am. Chem. Soc., 1997, 119, 987–993 CrossRef CAS; (b) J. P. Dinnocenzo, H. Zuilhof, D. R. Lieberman, T. R. Simpson and M. W. McKechney, J. Am. Chem. Soc., 1997, 119, 994–1004 CrossRef CAS.
- (a) Z. Zuo, C. G. Daniliuc and A. Studer, Angew. Chem., Int. Ed., 2021, 60, 25252–25257 CrossRef CAS PubMed; (b) Z. Zuo and A. Studer, Org. Lett., 2022, 24, 949–954 CrossRef CAS PubMed; (c) Y. Xu, H.-X. Gao, C. Pan, Y. Shi, C. Zhang, G. Huang and C. Feng, Angew. Chem., Int. Ed., 2023, 62, e202310671 CrossRef CAS PubMed; (d) D. J. Li, X. L. Liu, Y. Zhao and F. Pan, Org. Lett., 2024, 26, 8063–8068 CrossRef CAS PubMed , this work was published while our paper was in preparation..
- (a) W. Zhou, T. Miura and M. Murakami, Angew. Chem., Int. Ed., 2018, 57, 5139–5142 CrossRef CAS PubMed; (b) J.-H. Xu, W.-B. Wu and J. Wu, Org. Lett., 2019, 21, 5321–5325 CrossRef CAS PubMed; (c) B. Wang, C. Ascenzi Pettenuzzo, J. Singh, G. E. Mccabe, L. Clark, R. Young, J. Pu and Y. Deng, ACS Catal., 2022, 12, 10441–10448 CrossRef CAS; (d) M. Schlegel, S. Qian and D. A. Nicewicz, ACS Catal., 2022, 12, 10499–10505 CrossRef CAS PubMed; (e) C. Ascenzi-Pettenuzzo, J. Nganda and Y. Deng, ChemCatChem, 2023, 15, e202300953 CrossRef CAS; (f) L. Laze, B. Quevedo-Flores, I. Bosque and J. C. Gonzalez-Gomez, Org. Lett., 2023, 25, 8541–8546 CrossRef CAS PubMed; (g) B. Wang, J. Singh and Y. Deng, Org. Lett., 2023, 25, 9219–9224 CrossRef CAS PubMed; (h) C. Ascenzi Pettenuzzo, L. Liu, J. Singh, G. Cuffel and Y. Deng, ChemRxiv, 2024 DOI:10.26434/chemrxiv-2024-b32c5.
- (a) Y. Moon, W. Lee and S. Hong, J. Am. Chem. Soc., 2020, 142, 12420–12429 CrossRef CAS PubMed; (b) H. Im, W. Choi and S. Hong, Angew. Chem., Int. Ed., 2020, 59, 17511–17516 CrossRef CAS PubMed; (c) W. Lee, Y. Koo, H. Jung, S. Chang and S. Hong, Nat. Chem., 2023, 15, 1091–1099 CrossRef CAS PubMed.
- (a) C. Che, Q. Huang, H. Zheng and G. Zhu, Chem. Sci., 2016, 7, 4134–4139 RSC; (b) J. Zhang, D. Liu, S. Liu, Y. Ge, Y. Lan and Y. Chen, iScience, 2020, 23, 100755 CrossRef CAS PubMed; (c) L.-J. Zhong, H.-Y. Wang, X.-H. Ouyang, J.-H. Li and D.-L. An, Chem. Commun., 2020, 56, 8671–8674 RSC; (d) Y. Chen, D. Liu and J. Zhang, Synlett, 2021, 32, 356–361 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental procedure, characterization of new compounds (1H and 13C NMR spectra). See DOI: https://doi.org/10.1039/d4sc06723a |
| This journal is © The Royal Society of Chemistry 2024 |