
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceMonitoring electrophilic intermediates in reactions of thiols in aqueous solution directly with 19F NMR†
Dmitry D.
Saraev
 and
Derek A.
Pratt
*
and
Derek A.
Pratt
*
Department of Chemistry and Biomolecular Sciences, University of Ottawa, 10 Marie Curie Pvt, Ottawa, ON K1N6N5, Canada. E-mail: dpratt@uottawa.ca
First published on 15th November 2024
Abstract
Mechanistic studies of thiol reactivity can be challenging because electrophilic reaction intermediates, such as sulfenic acids (RSOH) and sulfenyl chlorides (RSCl), are generally too reactive to be observed directly. Herein we report the design and synthesis of a sterically-encumbered fluorinated triptycene thiol which enables direct observation of reaction intermediates in aqueous buffer by 19F NMR, as demonstrated in reactions with hydrogen peroxide and hypochlorous acid. Reactions with H2O2 resulted in the formation of a persistent RSOH species, which was subsequently converted to a sulfinic acid (RSO2H) and then a sulfonic acid (RSO3H), while RSCl was found to be the intermediate in reactions with HOCl. Utilizing the same scaffold, reactions of thiol with thermally and photochemically generated singlet oxygen afforded RSO2H as the primary product. The stark difference in product profile from sterically-unencumbered thiols – which yield disulfides – implies that the reaction proceeds through a sulfenyl hydroperoxide (RSOOH) intermediate. Sulfenic acids, which were not observed in reactions of thiols with singlet oxygen, were also found to rapidly react with singlet oxygen to afford sulfinic acids, which is proposed to involve initial formation of an analogous sulfinyl hydroperoxide (RS(O)OOH). The formation and reactions of RSOOH are explored by computations. Use of the water-soluble fluorinated triptycene scaffold to probe reductive processes on RSOH (e.g., with ascorbate and/or iron) is also illustrated, wherein it was found that RSOH are surprisingly resistant to reductive heterolysis – in stark contrast with hydroperoxides – owing to their strong S–O bond.
1 Introduction
Thiol redox reactivity is central to many aspects of biology. Cysteine, the unique thiol-containing amino acid, provides proteins and peptides (i.e. glutathione) with unique catalytic,1,2 structural,3 signaling,4,5 and antioxidant6 functions. In many cases, these functions are underpinned by thiol/disulfide exchange reactions, while in others, oxidation of the thiol to a reactive intermediate is believed to be key. Of particular interest are the reactions of cysteine thiols with H2O2, the pervasive ‘reactive oxygen species’ that abounds the cell – and at elevated levels in pathological contexts. Disulfides are primarily formed in these reactions, but sulfinic acids (RSO2H) and sulfonic acids (RSO3H) can also be observed depending on conditions.7 All of these products are believed to result from the initial formation of a sulfenic acid (RSOH) from substitution of the thiolate on H2O2; the sulfenic acid then either reacts as an electrophile, undergoing substitution by another equivalent of thiolate to yield disulfide, or as a nucleophile, where it is oxidized by another equivalent of H2O2 to yield RSO2H and then RSO3H. While thiol oxidation to RSO2H is reversible in the cell, RSO3H formation is irreversible, abolishing the function of the thiol.8The reactivity of RSOH as both electrophile and nucleophile leads to their rapid self-reaction, making isolation and determination of accurate physicochemical properties and/or reaction kinetics challenging – especially under physiologically-relevant conditions (i.e. in aqueous solutions).9 Although some protein sulfenic acids are stable and can be observed by mass spectrometry,10 the protein structure can make mechanistic studies and generalizations about RSOH reactivity difficult.11,12 To provide insight to their reactivity, several persistent small-molecule RSOH have been prepared,13,14 including a sterically-encumbered cysteine sulfenic acid.15 Unfortunately, the hydrophobicity of the scaffold restricted the study of its chemical properties to largely organic solutions (i.e. 20![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 THF/H2O), which cannot be buffered for studies of the pH-dependence of the reactivity of thiols and sulfenic acids – a key consideration given that physiologically-relevant thiol chemistry is dominated by the more reactive conjugate base (thiolate), which is present to a substantial extent at pH 7.4 (thiol pKas range from 5.0 to 8.8).16
:1 THF/H2O), which cannot be buffered for studies of the pH-dependence of the reactivity of thiols and sulfenic acids – a key consideration given that physiologically-relevant thiol chemistry is dominated by the more reactive conjugate base (thiolate), which is present to a substantial extent at pH 7.4 (thiol pKas range from 5.0 to 8.8).16
We previously used a triptycene scaffold functionalized with a bridgehead fluorine atom to monitor the reaction of thiol and H2O2 in buffered methanol directly using 19F NMR.13 The triptycene moiety precludes electrophilic reactions of the sulfenic acid because a nucleophile cannot access the  orbital localized on the sulfur atom, unequivocally confirming it as the initial product. This approach not only enabled detailed kinetic measurements and unambiguous characterization of the specific-base catalysis mechanism responsible, but also the successive oxidations to sulfinic and sulfonic acids which follow.17 Unfortunately, studies of the reaction of thiols and sulfenic acids with stronger oxidants, including other biologically-relevant ‘reactive oxygen species’ such as hypochlorous acid (HOCl) and singlet oxygen (1O2), were not possible due to their competitive reactions with the (amine-based) buffers. Similarly to the reactions of thiols with H2O2, reactions of HOCl and 1O2 have been shown to yield disulfides, but the mechanisms remain unclear due, in part, to the inability to directly observe reaction intermediates. We surmised that substitution of the 9-triptycenethiol scaffold to increase water solubility would enable direct observation of any intermediates formed in the reactions of thiols with HOCl, 1O2 and/or other high energy oxidants. Moreover, it would permit analogous studies on sulfenic acids and the investigation of other reactions which have been proposed to involve RSOH intermediates. Herein we describe the preparation of such a scaffold, its use to provide insights to the reactions of thiols and sulfenic acids with H2O2, HOCl and 1O2, and describe other applications to study sulfenic acid reactivity.
orbital localized on the sulfur atom, unequivocally confirming it as the initial product. This approach not only enabled detailed kinetic measurements and unambiguous characterization of the specific-base catalysis mechanism responsible, but also the successive oxidations to sulfinic and sulfonic acids which follow.17 Unfortunately, studies of the reaction of thiols and sulfenic acids with stronger oxidants, including other biologically-relevant ‘reactive oxygen species’ such as hypochlorous acid (HOCl) and singlet oxygen (1O2), were not possible due to their competitive reactions with the (amine-based) buffers. Similarly to the reactions of thiols with H2O2, reactions of HOCl and 1O2 have been shown to yield disulfides, but the mechanisms remain unclear due, in part, to the inability to directly observe reaction intermediates. We surmised that substitution of the 9-triptycenethiol scaffold to increase water solubility would enable direct observation of any intermediates formed in the reactions of thiols with HOCl, 1O2 and/or other high energy oxidants. Moreover, it would permit analogous studies on sulfenic acids and the investigation of other reactions which have been proposed to involve RSOH intermediates. Herein we describe the preparation of such a scaffold, its use to provide insights to the reactions of thiols and sulfenic acids with H2O2, HOCl and 1O2, and describe other applications to study sulfenic acid reactivity.
2 Results & discussion
2.1. Synthesis of thiol 1 and its oxidation products (6, 7, 8)
To improve the water-solubility of the fluorinated 9-triptycenethiol used in our previous work, we incorporated pendant tetraethylene glycol (TEG) units on the triptycene backbone as in 1. The synthesis of 1 was carried out in 7 steps from 9-bromoanthracene with an overall yield of 9% (Fig. 1A). Lithium-bromide exchange on 9-bromo-10-fluorotriptycene (2)17 and substitution of the resulting triptycenyl lithium on methyl disulfide afforded methyl sulfide 3 in 92% yield. Selective bromination of 3 was challenging; the Lewis base-catalyzed approach developed by Nishii and Miura18 was most successful, leading to a mixture of dibrominated products 4a (meta, meta to the bridgehead C–S bond) and 4b (meta, para). Interestingly, despite its striking similarity, the fluorinated triptycene 3 did not assist the reaction on its own and the TpSMe used by Nishii and Miura18 had to be added for the reaction to proceed to completion. Indeed, the presence of the fluorine atom also precluded access to the tribrominated triptycene, which was readily obtained under these reaction conditions in its absence. Both isomers of 4 were then subjected to Ullmann-type coupling conditions in the presence of Cu(OAc)2 using TEG as both nucleophile and solvent, sluggishly affording a corresponding isomeric mixture of 5 in 78% yield. The methyl sulfide was then cleaved using freshly prepared lithium thioethoxide in absolute DMPU at 120 °C to obtain the target thiol 1 in 51% yield. Reverse phase HPLC could resolve 1a from 1b, enabling its unequivocal characterization, but most of our experiments were carried out on the mixture as it afforded a second data set in each independent experiment (vide infra). | ||
| Fig. 1 (A) Preparation of thiols 1, and (B) corresponding sulfenic acids 6, sulfinic acids 7 and sulfonic acids 8. | ||
The sulfenic acids (6) and sulfinic acids (7) derived from 1 were prepared in 46% and 22% yield, respectively, from the direct oxidation of thiols 1 by H2O2 (2 eq.) in H2O/THF (1:1) containing K2HPO4 and separated by flash column chromatography. The sulfonic acids (8) were prepared similarly by the overnight reaction of 1 with 5 eq. of H2O2 (Fig. 1B).
2.2. Reactions of 1 with H2O2
To obtain reaction kinetics for the oxidation of thiols 1 by H2O2 in water, 1 was treated with an excess of H2O2 in phosphate-buffered D2O/THF (3:1, v/v) at pD 7.8 (pH 7.4),19 and their consumption was monitored by 19F NMR along with the corresponding appearance of product sulfenic, sulfinic and sulfonic acids (quantified relative to added 2,2,2-trifluoroethanol) as shown in Fig. 2. Since the two regioisomers of 1 and their corresponding sulfur oxyacids have distinct 19F NMR shifts, two data sets were collected per experiment (Fig. 2B). These data sets were fit to a simple kinetic model for successive bimolecular reactions of 1, 6 and 7 with H2O2 (Fig. 2C), from which observed rate constants (kobs) of 0.10, 0.013 and 0.0014 M−1 s−1 were derived for the reactions of thiol, sulfenic and sulfinic acid, respectively. Not only is this the first example of these reactions being followed directly in aqueous solution but, as far as we are aware, these are the first rate constants for RSOH and sulfinic acid oxidation by H2O2 measured in aqueous solution.‡
 | ||
| Fig. 2 (A) Reaction scheme for oxidation of thiols 1 by H2O2, including pKa values determined for each of 1, 6 and 7 and absolute rate constants obtained for the reactions of the conjugate bases of each of 1, 6, and 7 with H2O2 in 50 mM phosphate-buffered pD 7.8 D2O/THF (3:1). The rate constants are derived from the mean of the individual rate constants determined for each of the a and b isomers (see ESI†). (B) Representative time-resolved 19F NMR spectra obtained upon treatment of 1 (5 mM) with 25 mM H2O2 in 50 mM phosphate-buffered pD 7.8 D2O/THF (3:1). (C) Representative reaction profiles of 1a (2.8 mM) and 1b (2.2 mM) with 5 eq. H2O2 in pD 7.8 D2O/THF (3:1), fits of the data to a simple kinetic model for successive bimolecular reactions of 1, 6 and 7 with H2O2 and the corresponding observed rate constants (in M−1 s−1) derived from the fits. | ||
Previously measured kobs for the reactions of the water-insoluble triptycene thiol, sulfenic acid and sulfinic acid with H2O2 in methanol at  (ca. pH = 7.66 in H2O)20 were 2.5 × 10−3, 9.5 × 10−4 and 7.3 × 10−4 M−1 s−1, respectively. Given that these reactions are specific-base catalyzed (Fig. 2A), we suspected that the origin of the large solvent effects on the reactions of the thiol (40×) and sulfenic acid (13×) is the more favourable pre-equilibrium in aqueous solution relative to methanol. To derive absolute rate constants, the pKas of thiol 1 (8.3 ± 0.1) and sulfenic acid 6 (10.4 ± 0.1) were measured in D2O/THF (3 : 1) (the values for 1a and 6a were within the margin of error on the measurements made with the mixtures and corresponding values in H2O afforded 8.0 ± 0.1 and 10.1 ± 0.1, respectively). Expectedly, deprotonation of thiol and sulfenic acid were found to be much more favourable in water than in methanol (pKa = 11.6 and 12.8, respectively).17 With these pKa values at hand, we derived absolute (pH-independent) rate constants for the reactions of thiolate (k = 0.43 M−1 s−1, compared to 0.13 M−1 s−1 in methanol) and sulfenate (k = 5.2 M−1 s−1, compared to 0.75 M−1 s−1 in methanol) with H2O2. As expected from the alpha effect21 and our previous data,17 sulfenate anions were found to be more reactive nucleophiles than thiolate anions. Sulfinate was found to be much less reactive as a nucleophile (k = 1.4 × 10−3 M−1 s−1 compared to 5.8 × 10−4 M−1 s−1 in methanol) consistent with the greater relative stability of the anion (pKa ∼217vs. 10 for the corresponding RSOH). Overall, the rate constant determined for 1 is toward the low end of the range of those which have previously been reported (derived from consumption of thiol(ate)s),§ which span from 0.16 M−1 s−1 (for N-acetylcysteine) to 4.5 M−1 s−1 (for penicillamine) at pH 7.4,12 which is presumably due to the electron-withdrawing effect of the 10-fluorotriptycenyl scaffold.
(ca. pH = 7.66 in H2O)20 were 2.5 × 10−3, 9.5 × 10−4 and 7.3 × 10−4 M−1 s−1, respectively. Given that these reactions are specific-base catalyzed (Fig. 2A), we suspected that the origin of the large solvent effects on the reactions of the thiol (40×) and sulfenic acid (13×) is the more favourable pre-equilibrium in aqueous solution relative to methanol. To derive absolute rate constants, the pKas of thiol 1 (8.3 ± 0.1) and sulfenic acid 6 (10.4 ± 0.1) were measured in D2O/THF (3 : 1) (the values for 1a and 6a were within the margin of error on the measurements made with the mixtures and corresponding values in H2O afforded 8.0 ± 0.1 and 10.1 ± 0.1, respectively). Expectedly, deprotonation of thiol and sulfenic acid were found to be much more favourable in water than in methanol (pKa = 11.6 and 12.8, respectively).17 With these pKa values at hand, we derived absolute (pH-independent) rate constants for the reactions of thiolate (k = 0.43 M−1 s−1, compared to 0.13 M−1 s−1 in methanol) and sulfenate (k = 5.2 M−1 s−1, compared to 0.75 M−1 s−1 in methanol) with H2O2. As expected from the alpha effect21 and our previous data,17 sulfenate anions were found to be more reactive nucleophiles than thiolate anions. Sulfinate was found to be much less reactive as a nucleophile (k = 1.4 × 10−3 M−1 s−1 compared to 5.8 × 10−4 M−1 s−1 in methanol) consistent with the greater relative stability of the anion (pKa ∼217vs. 10 for the corresponding RSOH). Overall, the rate constant determined for 1 is toward the low end of the range of those which have previously been reported (derived from consumption of thiol(ate)s),§ which span from 0.16 M−1 s−1 (for N-acetylcysteine) to 4.5 M−1 s−1 (for penicillamine) at pH 7.4,12 which is presumably due to the electron-withdrawing effect of the 10-fluorotriptycenyl scaffold.
2.3. Reactions of 1 with HOCl
At physiological pH, thiols react rapidly with HOCl (k > 108 M−1 s−1),22 with sulfenyl chlorides being proposed as the initial products, but which are not observed due to their rapid reaction with thiols to yield disulfides.23 We observed instant formation of a single product (13a) upon addition of limiting NaOCl to a solution of 1a in pD 7.8 D2O/THF (Fig. 3A). The 19F NMR chemical shift (−205.9 ppm) did not correspond to the sulfenic acid 6a (−206.6 ppm), implying formation of the sulfenyl chloride. However, when thiol 1a was treated with N-chlorosuccinimide in chloroform, a product (14a) with a distinct 19F NMR chemical shift (−206.4 ppm) was obtained (Fig. 3B).24 High-resolution mass spectrometry indicated that 14a was the sulfenyl chloride. While 14a was stable over extended periods in pD 7.8 D2O/THF buffer, addition of 1a readily yielded 13a, which we have characterized as the disulfide of 1a (Fig. 3C). Since substitution of thiolate 1a on sulfenyl chloride 14a to form the disulfide is unlikely (the nucleophile cannot access the orbital localized on the sulfur atom), the thiolate presumably reacts with the sulfenyl chloride by electron transfer, resulting in the formation of thiyl radicals which dimerize to give the disulfide (Fig. 3B). An analogous mechanism has been proposed for the observation of disulfides from the reaction of sulfenyl chlorides and amines.25 The intervention of thiyl radicals in the reaction of non-hindered thiols with HOCl has previously been demonstrated by Davies,26 who characterized DMPO adducts of thiyl radicals by EPR, suggesting that this pathway is not unique to the reaction of 9-triptycenethiols.
orbital localized on the sulfur atom), the thiolate presumably reacts with the sulfenyl chloride by electron transfer, resulting in the formation of thiyl radicals which dimerize to give the disulfide (Fig. 3B). An analogous mechanism has been proposed for the observation of disulfides from the reaction of sulfenyl chlorides and amines.25 The intervention of thiyl radicals in the reaction of non-hindered thiols with HOCl has previously been demonstrated by Davies,26 who characterized DMPO adducts of thiyl radicals by EPR, suggesting that this pathway is not unique to the reaction of 9-triptycenethiols.
 | ||
| Fig. 3 (A) Representative 19F NMR spectra obtained before and after treatment of 1a (17 mM) with 0.35 eq. of NaOCl in 50 mM phosphate-buffered pD 7.8 D2O/THF (3:1). (B) Preparation of sulfenyl chloride 14a from 1a and representative 19F NMR in CDCl3. (C) Representative 19F NMR spectra obtained before and after treatment of 14a (17 mM) with 0.65 eq. of 1a in 50 mM phosphate-buffered pD 7.8 D2O/THF (3:1). (D) Proposed mechanism of disulfide formation upon thiol oxidation by hypochlorous acid. | ||
2.4. Reactions of 1 with 1O2
Similarly to their reactions with H2O2 and HOCl, thiols have been shown to yield disulfides upon reaction with 1O2.27 To provide insight to the mechanism of this reaction, we investigated the reaction of 1 with 1O2 derived from decomposition of the endoperoxide of sodium naphthalene-1,4-dipropionate (NDPO2) at 37 °C (t1/2 = 23 min).28,29 The reactions were again monitored by 19F NMR at 37 °C in pD 7.8 phosphate buffered D2O/THF (3:1, v/v), but now in the presence of 4 eq. of NDPO2in lieu of either H2O2 or HOCl. After 33 minutes, the conversion of thiol was ∼45%, giving primarily sulfinates 7 (∼25%). Importantly, disulfide 13a was not observed. Other products included two presumed pairs of unknown isomers (∼10%) downfield of 8 at δ = −204.55 ppm and −205.25 ppm (Fig. 4A). Control experiments suggest that the formation of these unknowns can be attributed to a reaction occurring in THF in the presence of 1O2 (see ESI Fig. S6†). The missing ∼10% in the mass balance suggests that some signal suppression from the formation of persistent paramagnetic species is taking place (see ESI Fig. S3†).30,31
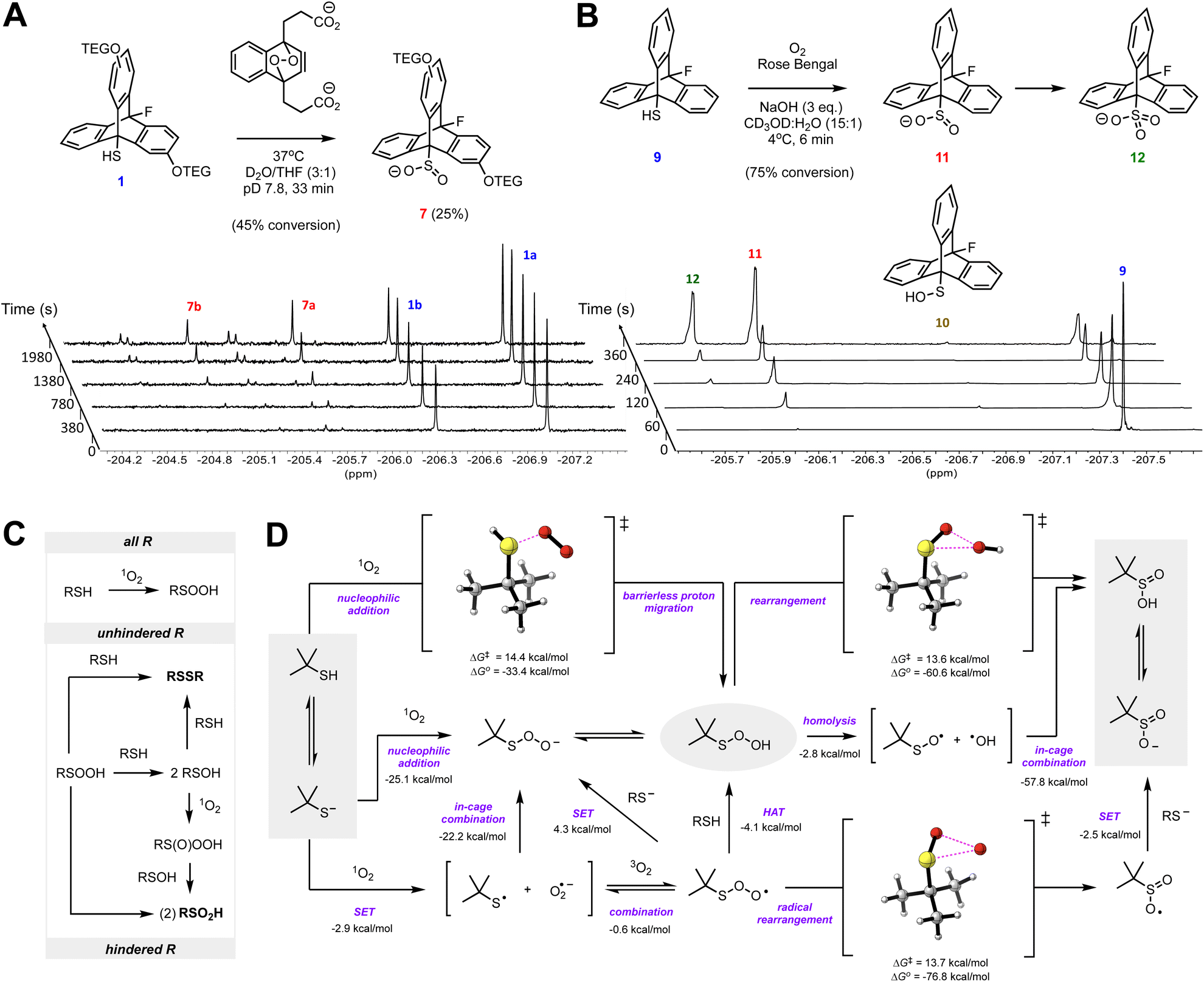 | ||
| Fig. 4 (A) Representative time-resolved 19F NMR spectra obtained upon treatment of 1 (5 mM) with 4 eq. of NDPO2 in 50 mM phosphate-buffered pD 7.8 D2O/THF (3:1). (B) Representative time-resolved 19F NMR spectra obtained of the reaction of 9 (20 mM) with photochemically generated 1O2 in CD3OD/H2O (15:1) containing 60 mM NaOH. (C) Scheme accounting for the different product distributions arising from reaction of hindered and unhindered thiols with 1O2. (D) Mechanistic possibilities and CBS-QB3 calculated free energies (298 K) of the reaction of a model (t-butyl) thiol with 1O2 (SET = single electron transfer; HAT = hydrogen atom transfer). Data calculated using a polarizable continuum model using the integral equation formalism (IEFPCM) parameterized for water are given; associated gas phase values are given in the ESI.† | ||
Consistent with the foregoing experiments with 1, when thiol 9 was subjected to Rose Bengal-photosensitized oxygenation in 15:1 CD3OD:H2O containing 3 eq. NaOH, the corresponding sulfinate 11 and sulfonate 12 were formed (Fig. 4B) (see Fig. S4 of the ESI† for corresponding 1H NMR spectra). Traces of disulfide 15 could be detected under these conditions (as a precipitate), suggesting that thiyl radical formation is possible under the reaction conditions, albeit as a minor reaction pathway.¶ Likewise, while RSOH 6 was not detected in D2O/THF, a miniscule amount of 10 could be detected in CD3OD/D2O (Fig. 4B).
The drastically different product distribution arising from the reaction of 1 (and 9) with 1O2 compared to the precedent32 suggests that nucleophilic substitution on an oxidized sulfur intermediate is responsible for disulfide formation from reactions of thiols and 1O2, in general. While at first glance it would seem that RSOH is that intermediate, and although we found that RSOH 10 was readily photooxidized to RSO2H 11 (see ESI Fig. S7†), it is difficult to provide a compelling mechanistic rationale for the direct formation of RSOH from RSH and 1O2. Instead, we posit that a sulfenyl hydroperoxide (RSOOH) is the immediate product, and when the reaction with a thiolate cannot take place at sulfur to give a disulfide (i.e. for 1 and 9), the reaction takes place at oxygen to give two equivalents of RSOH. Alternatively, RSOOH can rearrange to RSO2H. If formed, RSOH can react with 1O2 to afford a sulfinyl hydroperoxide (RS(O)OOH) which can oxidize another equivalent of RSOH to yield two equivalents of RSO2H (Fig. 4C).
Formation of RSOO− from thiolate (RS−) and 1O2 is predicted by CBS-QB3 (ref. 33) to be highly exergonic (ΔG° = −25.1 kcal mol−1 for t-BuS− as a model thiolate). No TS could be computed for the nucleophilic addition pathway, presumably due to the use of a single determinant wavefunction approach. A TS could, however, be computed for the addition of RSH to 1O2, which was associated with a barrier of ΔG‡ = 14.7 kcal mol−1 (Fig. 4D). This may be taken as an upper bound for the thiolate reaction since the barrier to addition of the better nucleophile is sure to be lower. Another possibility involves electron transfer from RS− to 1O2 to form a radical pair that can combine or, if cage escape occurs, oxygenation of the thiyl radical to yield a thiylperoxyl radical (RSOO˙) that could be subsequently reduced via either H-atom transfer or electron transfer. H-atom transfer from a thiol to RSOO˙ is predicted to be exergonic (ΔG° = −4.1 kcal mol−1) and proceed via an accessible barrier (ΔG‡ = 18.8 kcal mol−1). However, our calculations indicate that the highly exergonic (ΔG° = −76.8 kcal mol−1) rearrangement of RSOO˙ to a sulfonyl radical  is much more likely. Earlier calculations had suggested this would require traversing a prohibitively high barrier (ΔG‡ = 45 kcal mol−1),34 but we have found this barrier to be significantly lower (ΔG‡ = 13.7 kcal mol−1) using the higher accuracy CBS-QB3 methodology. Regardless, the lack of disulfide formed in our experiments argues that thiyl radicals are not key intermediates.
is much more likely. Earlier calculations had suggested this would require traversing a prohibitively high barrier (ΔG‡ = 45 kcal mol−1),34 but we have found this barrier to be significantly lower (ΔG‡ = 13.7 kcal mol−1) using the higher accuracy CBS-QB3 methodology. Regardless, the lack of disulfide formed in our experiments argues that thiyl radicals are not key intermediates.
The sulfinate observed when disulfide formation is suppressed could also arise from concerted rearrangement of RSOOH – a highly exergonic reaction (ΔG° = −60.6 kcal mol−1) with an easily accessible barrier (ΔG‡ = 13.6 kcal mol−1). Again, this barrier is likely an upper bound given that the H-bond between the migrating OH group and an explicit solvent molecule will become stronger in the TS (this cannot be captured in the polarizable solvent continuum employed in the calculation). Alternatively, a step-wise rearrangement can be envisioned. The calculated O–O BDE of RSOOH was found to be a meagre 10.2 kcal mol−1, driven by the stability of the sulfinyl radical, suggesting O–O bond homolysis followed by in-cage recombination to form a sulfinic acid is possible. Earlier spin-trapping studies during thiol photooxygenation suggested the formation of ˙OH, presumably due to cage-escape following RSOOH thermolysis (Fig. 4E).30 The lack of significant signal suppression in our NMR experiments, suggests that homolysis does not occur to a significant extent and that nucleophilic chemistry predominates. Regardless, the predicted rapid rearrangement chemistry presumably prevents the direct observation of RSOOH on the timescale of the NMR experiments.
The implication of RSOOH in disulfide formation from thiols exposed to 1O2 compels consideration that RSOOH could also be intermediates in disulfide formation in the radical-mediated oxidation (autoxidation) of thiols, given the rapid reaction of thiyl radicals with 3O2 (∼108 M−1 s−1).35 Indeed, sulfur oxyacids are major products under some conditions,36,37 and may derive from RSOOH as a common intermediate to disulfide formation. However, as indicated above, given that the (unimolecular) rearrangement of RSOO˙ to  is predicted to proceed with a smaller barrier than the (biomolecular) H-atom transfer to RSOO˙ from RSH (13.7 kcal mol−1vs. 18.8 kcal mol−1, respectively), the formation of RSOOH under radical conditions is unlikely. Instead, the partitioning of RS˙ and RSOO˙ between thiyl dimerization and thiylperoxyl rearrangement pathways is likely key in determining whether disulfides or sulfur oxyacids are the major products.
is predicted to proceed with a smaller barrier than the (biomolecular) H-atom transfer to RSOO˙ from RSH (13.7 kcal mol−1vs. 18.8 kcal mol−1, respectively), the formation of RSOOH under radical conditions is unlikely. Instead, the partitioning of RS˙ and RSOO˙ between thiyl dimerization and thiylperoxyl rearrangement pathways is likely key in determining whether disulfides or sulfur oxyacids are the major products.
2.5. Other applications of the new scaffold
Since comparably very little remains known of RSOH reactivity (relative to RSH, RSO2H and RSO3H), we surmised that 6 may prove generally useful as a mechanistic probe. It has been shown previously that ascorbate acts as the cofactor for 1-Cys peroxiredoxins (Prx), and it was proposed to reduce the RSOH of 1-Cys Prx back to its active thiol form after oxidation.38 However, other protein sulfenic acids are resistant to reduction by ascorbate.39 Previously employed indirect bifunctional assays estimated rate constants ranging from 2.2 to 4 × 102 M−1 s−1, depending on the protein RSOH. To provide additional insight to this reactivity, we incubated 6 with 3 eq. of ascorbate|| in pD 7.8 D2O/THF (3:1) and monitored the reaction by 19F NMR (Fig. 5A). No reaction was observed over 72 hours, suggesting an amino acid sidechain in the Prx active site is required for activation of the sulfenic acid to reduction (the conjugate acid of an essential histidine residue is a possibility), or that a more sophisticated redox cascade is at play.
 | ||
| Fig. 5 (A) 1-Cys peroxiredoxins (Prx) reduce hydrogen peroxide and hydroperoxides by nucleophilic substitution by the cysteine thiol(ate). The resultant sulfenic acids are converted back to the starting thiol by ascorbate to complete the catalytic cycle. The lack of reactivity of RSOH 6 to ascorbate implies that active site residues are essential to enzyme turnover. (B) Lipid peroxidation is auto-initiated in the presence of Fe2+ due to reductive cleavage of the O–O bonds in product hydroperoxides, which yields initiating radicals. No reaction is observed for RSOH 10, implying that the process is less favourable for RSOH than it is for the corresponding ROOH. | ||
Labile iron(II) is often invoked as key to the auto-initiation of cellular (phospho)lipid peroxidation and associated ferroptosis due to its ability to reduce lipid hydroperoxides to form initiating lipid alkoxyl radicals.40,41 Since thiyl radicals are significantly more thermodynamically stable than alkoxyl radicals (i.e. t-BuSH and t-BuOH BDEs are 86 and 106 kcal mol−1, respectively),42 we anticipated that RSOH would readily undergo the same reaction (Fig. 5B). To our great surprise, 10 did not react with Fe(II) (or Fe(III) in the presence of ascorbic acid) in methanol. At first glance, we surmised that this may have to do with some impairment of the reaction by the triptycene backbone. However, CBS-QB3 calculations predict that the S–O bond in a model (t-butyl) RSOH is 27 kcal mol−1 stronger than the O–O bond in the corresponding hydroperoxide – despite the 20 kcal mol−1 difference in the stabilities of the product thiyl and alkoxyl radicals. The significant stabilization of RSOH relative to ROOH can be rationalized by lesser lone pair/lone pair repulsion in RSOH due to the longer S–O bond and more diffuse S lone pairs and also greater lone-pair/ hyperconjugation in RSOH compared to ROOH. Based on these calculated thermodynamics and the lack of reactivity of 6 and ascorbate with or without added iron, it is clear that reductive heterolysis of RSOH is a highly unfavourable process in instances where there is no acid available to activate the RSOH.
hyperconjugation in RSOH compared to ROOH. Based on these calculated thermodynamics and the lack of reactivity of 6 and ascorbate with or without added iron, it is clear that reductive heterolysis of RSOH is a highly unfavourable process in instances where there is no acid available to activate the RSOH.
3 Conclusions
A newly synthesized 10-fluorotriptycene thiol enables visualization of transient reactive intermediates (e.g., RSOH and RSCl) and can be used to directly observe transformations of thiols and sulfenic acids in aqueous buffer by 19F NMR. Reactions of thiols and sulfenic acids with H2O2 were found to proceed with kinetics that were slightly faster than in methanol, largely due to more favourable ionization to the reactive conjugate bases. Reaction of thiol with HOCl did not yield a sulfenic acid, but presumably a sulfenyl chloride. The sulfenyl chloride could be independently prepared by reaction of the thiol with N-chlorosuccinimide and was found to rapidly yield disulfide when treated with thiol(ate) in buffer – attributed to dimerization of thiyl radicals formed upon electron transfer from thiolate to sulfenyl chloride. Product distributions of the reaction of thiols with 1O2 enable us to propose sulfenyl and sulfinyl hydroperoxides as intermediates en route to disulfides, sulfinic and sulfonic acids. Through product analyses and CBS-QB3 calculated reaction energetics, the myriad of one- and two-electron processes that these short-lived intermediates undergo are herein summarized. A lack of reactivity of RSOH 6 and 10 to one-electron reduction strongly supports that reductive heterolysis of RSOH does not occur to a significant extent without enzyme (acid) catalysis and highlights the energetic contribution of lone pair repulsion in hydroperoxides to their relative instability. We anticipate that this scaffold will be a useful tool compound for mechanistic investigations of thiol and sulfenic acid reactivity.4 Experimental section
4.1. General
Reagents and solvents were obtained from commercial suppliers and used as received, unless indicated otherwise. Hydrogen peroxide concentration was determined by manganometric titration. Sodium hypochlorite concentration was determined by iodometry. 1H, 13C and 19F NMR spectra were recorded on a Bruker AVANCE spectrometer at the specified frequency. 19F NMR spectra in D2O were referenced using 2,2,2-trifluoroethanol (TFE) (δ −77.0 ppm) as an internal standard, 19F NMR spectra in CDCl3, CD3OD, and C6D6 were referenced to trifluorotoluene (δ −63.72 ppm). Product ratios were obtained through integration of their 19F NMR signals. For quantification, 19F NMR spectra were phased and baseline corrected. For NMR characterization of isomeric mixtures, the signals corresponding to each isomer were assigned based on those observed for HPLC-purified isomer 1a, and the corresponding signals observed when it was oxidized with H2O2. Preparative HPLC was carried on a Waters 2996 HPLC system, using a preparative Waters XBridge Prep C18 5 μm (19 × 150 mm) column. Column chromatography was carried out with 40–63 μm, 230–400 mesh silica gel purchased from SiliCycle. All calculations were carried out using the Gaussian 09 quantum chemistry package using the CBS-QB3 complete basis set approach.334.2. Synthesis
4.2.2.1 2,7-Dibromo-10-fluoro-9,10-[1′,2′]benzenoanthracen-9(10H)-yl(methyl)sulfane (4a). 33% yield. 1H NMR (600 MHz, CDCl3) δ 7.74–7.71 (m, 2H), δ 7.61–7.57 (m, 2H), δ 7.47–7.43 (m, 2H), δ 7.31–7.24 (m, 2H), δ 7.19–7.13 (m, 2H), δ 2.56 (s, 3H) 13C NMR (151 MHz, CDCl3) δ 145.9, 143.6, 143.2, 142.9, 142.8, 142.7, 140.7, 140.5, 128.9, 126.2, 126.1, 122.8, 122.4, 120.5, 119.0, 97.8, 59.9, 14.1. HRMS (EI) m/z calculated for [M] C21H13Br2FS 473.9087, found 473.9089.
4.2.2.2 2,6-Dibromo-10-fluoro-9,10-[1′,2′]benzenoanthracen-9(10H)-yl(methyl)sulfane (4b). 28% yield. 1H NMR (600 MHz, CDCl3) δ 7.74–7.71 (m, 2H), δ 7.61–7.57 (m, 2H), δ 7.47–7.43 (m, 2H), δ 7.31–7.24 (m, 2H), δ 7.19–7.13 (m, 2H), δ 2.54 (s, 3H) 13C NMR (151 MHz, CDCl3) δ 145.7, 143.4, 143.1, 142.6, 140.7, 140.5, 128.9, 126.2, 126.1, 124.4, 122.8, 122.4, 120.5, 119.9, 118.9, 97.0, 59.9, 14.0. HRMS (EI) m/z calculated for [M] C21H13Br2FS 473.9087, found 473.9089.
:5b = 57:43) as a viscous yellow oil (1.27 g, 1.81 mmol, 78% total yield).
4.2.3.1 2,7-Di(tetraethylene glycol)-10-fluoro-9,10-[1′,2′]benzenoanthracen-9(10H)-yl(methyl)sulfane (5a). 1H NMR (600 MHz, CDCl3) δ 7.58–7.52 (m, 2H), 7.46–7.40 (m, 2H), 7.43 (d, J = 8.2 Hz, 2H), 7.23–7.16 (m, 2H), 7.14–7.06 (m, 2H), 6.64–6.57 (m, 2H), 4.08 (t, J = 4.8 Hz, 4H), 3.80 (t, J = 4.8 Hz, 4H), 3.73–3.61 (m, 20H), 3.57 (t, J = 4.5 Hz, 4H), 2.56 (s, 3H). 13C NMR (151 MHz, CDCl3) δ 156.9, 143.3, 141.5, 137.0, 136.1, 125.7, 125.5, 122.5, 122.2, 119.3, 118.2, 111.3, 110.8, 110.2, 106.2, 97.0, 72.5, 70.8, 70.6, 70.3, 69.7, 67.8, 61.7, 60.6, 60.0, 14.2. HRMS (ESI+) m/z calculated for [M + Na]+ C36H45FNaO10S 725.2772, found 725.2767.
4.2.3.2 2,6-Di(tetraethylene glycol)-10-fluoro-9,10-[1′,2′]benzenoanthracen-9(10H)-yl(methyl)sulfane (5b). 1H NMR (600 MHz, CDCl3) δ 7.58–7.52 (m, 2H), 7.46–7.40 (m, 2H), 7.43 (d, J = 8.2 Hz, 2H), 7.23–7.16 (m, 2H), 7.14–7.06 (m, 2H), 6.64–6.57 (m, 2H), 4.08 (t, J = 4.8 Hz, 4H), 3.80 (t, J = 4.8 Hz, 4H), 3.73–3.61 (m, 20H), 3.57 (t, J = 4.5 Hz, 4H), 2.54 (s, 3H). 13C NMR (151 MHz, CDCl3) δ 157.0, 156.9, 143.3, 137.0, 125.7, 125.5, 123.6, 122.5, 122.2, 119.5, 119.3, 118.3, 111.3, 110.8, 110.0, 97.0, 72.5, 70.8, 70.6, 70.3, 69.7, 67.8, 61.7, 60.6, 60.0, 14.2. HRMS (ESI+) m/z calculated for [M + Na]+ C36H45FNaO10S 725.2772, found 725.2767.
:1b = 57:43) as a viscous pale-yellow oil (174 mg, 51% total yield). The 1a isomer could be separated by reverse phase HPLC using 3:1 MeOH:H2O as the mobile phase.
4.2.4.1 2,7-Di(tetraethylene glycol)-10-fluoro-9,10-[1′,2′]benzenoanthracen-9(10H)-ylmercaptan (1a). δ 156.9 (s), 144.6 (d, J = 6.0 Hz), 143.8 (d, J = 20 Hz), 136.2 (d, J = 20 Hz), 126.1 (s), 125.5 (s), 121.4 (d, J = 2 Hz), 119.1 (d, J = 6.0 Hz), 118.0 (d, J = 6 Hz), 110.4 (s), 110.2 (s), 97.5 (d, J = 205 Hz), 72.5 (s), 70.8 (s), 70.6 (s), 70.3 (s), 69.7 (s), 67.8 (s), 61.7 (s), 57.5 (d, J = 2 Hz). 19F NMR (471 MHz, D2O/THF) δ −207.1. HRMS (ESI+) m/z calculated for [M + Na]+ C36H45FNaO10S 711.2632, found 711.2615.
4.2.4.2 2,6-Di(tetraethylene glycol)-10-fluoro-9,10-[1′,2′]benzenoanthracen-9(10H)-ylmercaptan (1b). 1H NMR (600 MHz, CDCl3) δ 7.63 (m, 1H), 7.57–7.49 (m, 2H), 7.43 (m, 1H), 7.31 (m, 1H), 7.18 (d, J = 2.4 Hz, 1H), 7.17–7.04 (m, 2H), 6.62 (dd, J = 8.4, 2.4 Hz, 1H), 6.58 (dd, J = 8.4, 2.4 Hz, 1H), 4.09 (t, J = 4.8 Hz, 4H), 3.80 (t, J = 4.8 Hz, 4H), 3.73–3.61 (m, 20H), 3.57 (t, J = 4.5 Hz, 4H), 2.42 (s, 1H). 13C NMR (151 MHz, CDCl3) δ 157.1 (s), 157.0 (s), 145.0 (d, J = 20 Hz), 143.5 (d, J = 20 Hz), 143.2 (d, J = 6 Hz), 135.4 (d, J = 20 Hz), 125.8 (s), 125.7 (s), 122.5 (d, J = 2 Hz), 121.1 (d, J = 2 Hz), 119.3 (d, J = 2 Hz), 118.2 (d, J = 2 Hz), 110.6 (s), 110.5 (s), 110.3 (s), 110.0 (d, J = 2 Hz), 97.5 (d, J = 205 Hz), 72.5 (s), 70.8 (s), 70.6 (s), 70.3 (s), 70.3 (s), 69.7 (s), 67.8 (s), 61.7 (s), 57.5 (d, J = 2 Hz). 19F NMR (471 MHz, D2O/THF) δ −206.3. HRMS (ESI+) m/z calculated for [M + Na]+ C36H45FNaO10S 711.2632, found 711.2615.
:1b 57:43) were dissolved in THF (1 mL) and added to a solution of dibasic potassium phosphate (30.4 mg, 0.194 mmol) in HPLC-grade H2O (900 μL). The mixture was thoroughly mixed to ensure homogeneity before 100 μL of H2O2 as a 1.94 M solution in D2O/H2O was slowly added while stirring. Progress of the reaction was monitored by 19F NMR. Upon complete consumption of the starting material the reaction was concentrated in vacuo until almost no water remained. The residue was purified by column chromatography (gradient elution 4–10% MeOH/CHCl3) to obtain the isomeric sulfenic acids 6 (28.2 mg, 46% total yield, 6a:6b = 58:42) and sulfinates 7 (19.1 mg, 22% total yield, 7a:7b = 57:43). Isomerically pure 6a was obtained using the same protocol starting from thiol 1a. To obtain isomerically pure 7a, sulfenic acid 6a (5.8 mg, 0.00823 mmol) was dissolved in a 10 mM solution of NaOH in CD3OD (0.75 mL) in an NMR tube. 25 μL of H2O2 as a 300 mM solution in CD3OD was then added in 5 μL portions until quantitative conversion of 6a to 7a was observed by 19F NMR.
4.2.5.1 2,7-Di(tetraethylene glycol)-10-fluoro-9,10-[1′,2′]benzenoanthracen-9(10H)-ylsulfenic acid (6a). 1H NMR (600 MHz, CD3OD) δ 7.53–7.48 (m, 2H), 7.41–7.38 (d, J = 8.2 Hz, 2H), 7.23 (t, J = 2.1 Hz, 2H), 7.14–7.05 (m, 2H), 6.67 (dd, J = 8.2, 2.1 Hz, 2H), 4.08 (m, 4H), 3.77 (m, 4H), 3.67–3.63 (m, 4H), 3.63–3.57 (m, 12H), 3.56–3.52 (m, 4H), 3.50–3.46 (m, 4H). 13C NMR (151 MHz, CD3OD) δ 158.4 (s), 146.2 (d, J = 20 Hz), 144.5 (d, J = 6 Hz), 142.7 (d, J = 6 Hz), 138.3 (d, J = 20 Hz), 128.8 (s), 126.6 (d, J = 6 Hz), 123.3 (d, J = 2.5 Hz), 119.8 (d, J = 6 Hz), 118.8 (d, J = 6 Hz), 112.2 (d, J = 2.5 Hz), 111.4 (s), 111.4 (s), 99.0 (d, J = 200 Hz), 73.6 (s), 71.7 (s), 71.6 (s), 71.5 (s), 71.3 (s), 71.3 (s), 70.8 (s), 68.9 (s), 64.5 (d, J = 2.5 Hz), 62.2 (s). 19F NMR (471 MHz, D2O/THF) δ −206.65. HRMS (ESI+) m/z calculated for [M + Na]+ C36H45FNaO11S 727.2564, found 727.2592.
4.2.5.2 2,6-Di(tetraethylene glycol)-10-fluoro-9,10-[1′,2′]benzenoanthracen-9(10H)-ylsulfenic acid (6b). 1H NMR (600 MHz, CD3OD) δ 7.53–7.47 (m, 2H), 7.50–7.43 (m, 2H), 7.31–7.27 (m, 1H), 7.17–7.05 (m, 3H), 6.68 (dd, J = 8.2, 2.4 Hz, 1H), 6.62 (dd, J = 8.2, 2.4 Hz, 1H) 4.16 (m, 4H), 3.85 (m, 4H), 3.74–3.57 (m, 20H), 3.54 (t, J = 4.5 Hz, 4H). 13C NMR (151 MHz, CD3OD) δ 158.6 (s), 158.3 (s), 147.5 (d, J = 20 Hz), 145.7 (d, J = 20 Hz), 145.0 (d, J = 6 Hz), 143.2 (d, J = 6 Hz), 137.8 (d, J = 6 Hz), 135.3 (d, J = 20 Hz), 126.8 (s), 126.5 (s), 125.4 (s), 124.5 (d, J = 2.5 Hz), 123.1 (d, J = 2.5 Hz), 120.0 (d, J = 6 Hz), 119.0 (d, J = 6 Hz), 111.9 (d, J = 2.5 Hz), 111.7 (s), 111.3 (s), 99.0 (d, J = 200 Hz), 74.5 (s), 72.6 (s), 72.4 (s), 72.2 (s), 71.7 (s), 69.8 (s), 64.1 (d, J = 2.5 Hz), 63.1 (s). 19F NMR (471 MHz, D2O/THF) δ −205.95. HRMS (ESI+) m/z calculated for [M + Na]+ C36H45FNaO11S 727.2564, found 727.2592.
4.2.5.3 2,7-Di(tetraethylene glycol)-10-fluoro-9,10-[1′,2′]benzenoanthracen-9(10H)-ylsulfinate (7a). 1H NMR (500 MHz, CD3OD) δ 8.22 (br, 2H), 7.96 (br, 1H), 7.46 (m, 1H), 7.37 (d, J = 8.2 Hz, 2H), 7.11–7.04 (m, 2H), 6.67 (dd, J = 8.2 Hz, 2.1 Hz, 2H), 4.15 (m, 4H), 3.81 (m, 4H), 3.71–3.56 (m, 20H), 3.54–3.49 (m, 4H). 13C NMR (126 MHz, CD3OD) δ 157.3 (s), 157.2 (s), 149.1 (d, J = 20 Hz), 147.7 (d, J = 6 Hz), 143.8 (d, J = 6 Hz), 142.6 (d, J = 6 Hz), 140.2 (d, J = 20 Hz), 128.8 (s), 126.0 (d, J = 20 Hz), 125.4 (d, J = 2.5 Hz), 119.7 (d, J = 6 Hz), 118.6 (d, J = 6 Hz), 113.9 (d, J = 2.5 Hz), 111.1 (s), 99.1 (d, J = 200 Hz), 72.3 (d, J = 2 Hz), 72.1 (s), 70.3 (s), 70.2 (s), 70.1 (s), 70.0 (s), 69.3 (s), 67.5 (s), 60.6 (s). 19F NMR (471 MHz, D2O/THF) δ −206.1. HRMS (ESI−) m/z calculated for [M–H]− C36H44FO12S 719.2538, found 719.2526.
4.2.5.4 2,6-Di(tetraethylene glycol)-10-fluoro-9,10-[1′,2′]benzenoanthracen-9(10H)-ylsulfinate (7b). 1H NMR (500 MHz, CD3OD) δ 8.50 (br, 1H), 7.97 (br, 1H), 7.47 (m, 1H), 7.36 (m, 1H), 7.14–7.04 (m, 3H), 6.71–6.62 (m, 2H), 4.15 (m, 4H), 3.81 (m, 4H), 3.71–3.56 (m, 20H), 3.54–3.49 (m, 4H). 13C NMR (126 MHz, CD3OD) δ 157.5 (s), 157.4 (s), 145.1 (d, J = 20 Hz), 144.6 (d, J = 20 Hz), 142.0 (d, J = 6 Hz), 141.6 (d, J = 6 Hz), 137.1 (d, J = 20 Hz), 126.1 (s), 125.1 (s), 125.0 (d, J = 2.5 Hz), 119.4 (d, J = 6 Hz), 118.0 (d, J = 6 Hz), 117.0 (d, J = 6 Hz), 116.8 (d, J = 6 Hz), 114.3 (d, J = 2.5 Hz), 113.1 (d, J = 2.5 Hz), 110.5 (s), 110.3 (s), 99.9 (d, J = 200 Hz), 72.4 (d, J = 2.5 Hz), 72.1 (s), 70.3 (s), 70.2 (s), 70.1 (s), 70.0 (s), 69.3 (s), 67.5 (s), 60.6 (s). 19F NMR (471 MHz, D2O/THF) δ −205.4. HRMS (ESI−) m/z calculated for [M–H]− C36H44FO12S 719.2538, found 719.2526.
:47 isomeric mixture of 1a:1b) or purified 1a were dissolved in THF (1 mL) and added to a solution of dibasic potassium phosphate (15 mg, 0.086 mmol) in HPLC-grade H2O (900 μL). 114 μL of H2O2 as a 1.94 M solution in D2O/H2O was added all at once. After stirring overnight, the reaction was concentrated in vacuo until almost no water remained. The residue was purified by column chromatography (10% MeOH/CHCl3) to obtain the isomeric sulfonates 8 (30 mg, quantitative yield, 8a:8b = 53:47).
4.2.6.1 2,7-Di(tetraethylene glycol)-10-fluoro-9,10-[1′,2′]benzenoanthracen-9(10H)-ylsulfonate (8a). 1H NMR (500 MHz, CD3OD) δ 8.23 (m, 1H), 7.91 (t, J = 2.1 Hz, 2H), 7.51–7.45 (m, 1H), 7.41–7.35 (m, 2H), 7.14–7.04 (m, 2H), 6.71–6.63 (m, 2H), 4.15 (m, 4H), 3.81 (m, 4H), 3.71–3.56 (m, 20H), 3.54–3.49 (m, 4H). 13C NMR (126 MHz, CD3OD) δ 156.5 (s), 145.1 (d, J = 20 Hz), 141.5 (d, J = 6 Hz), 140.0 (d, J = 6 Hz), 137.5 (d, J = 20 Hz), 125.1 (s), 124.9 (d, J = 2 Hz), 124.8 (s), 117.9 (d, J = 6 Hz), 116.9 (d, J = 6 Hz), 113.0 (d, J = 2 Hz), 110.4 (s), 97.5 (d, J = 200 Hz), 73.6 (d, J = 2 Hz), 73.4 (s), 71.6 (s), 71.5 (s), 71.4 (s), 71.3 (s), 71.2 (s), 68.9 (s), 62.2 (s), 62.0 (s). 19F NMR (471 MHz, D2O/THF) δ −205.9. HRMS (ESI−) m/z calculated for [M–H]− C36H44FO13S 735.2503, found 735.2491.
4.2.6.2 2,6-Di(tetraethylene glycol)-10-fluoro-9,10-[1′,2′]benzenoanthracen-9(10H)-ylsulfonate (8b). 1H NMR (500 MHz, CD3OD) δ 8.23 (m, 1H), 8.12 (dd, J = 8.4, 1.4 Hz, 1H), 7.89 (t, J = 2.1 Hz, 1H), 7.51–7.45 (m, 1H), 7.41–7.35 (m, 1H), 7.14–7.04 (m, 3H), 6.71–6.63 (m, 2H), 4.15 (m, 4H), 3.81 (m, 4H), 3.71–3.56 (m, 20H), 3.54–3.49 (m, 4H). 13C NMR (126 MHz, CD3OD) δ 156.7 (s), 156.6 (s), 146.4 (d, J = 20 Hz), 144.5 (d, J = 20 Hz), 142.0 (d, J = 6 Hz), 140.0 (d, J = 6 Hz), 137.0 (d, J = 20 Hz), 132.5 (d, J = 6 Hz), 126.2 (d, J = 2 Hz), 125.0 (s), 124.9 (d, J = 2 Hz), 124.7 (d, J = 2 Hz), 118.0 (d, J = 6 Hz), 117.0 (d, J = 6 Hz), 112.7 (d, J = 2 Hz), 110.4 (s), 110.0 (s), 97.5 (d, J = 200 Hz), 73.6 (d, J = 2 Hz), 73.4 (s), 71.6 (s), 71.5 (s), 71.4 (s), 71.3 (s), 71.2 (s), 68.9 (s), 62.2 (s), 62.0 (s). 19F NMR (471 MHz, D2O/THF) δ −205.25. HRMS (ESI−) m/z calculated for [M–H]− C36H44FO13S 735.2503, found 735.2487.
4.3 19F NMR-monitored reactions
:1) was added dropwise, while mixing, revealing stoichiometric conversion of 14a to 13a.
Data availability
Additional experimental procedures and reaction monitoring data, NMR spectra and computational data are available in the ESI.†Author contributions
D. D. S. designed and executed the experiments and carried out quantum chemical calculations. D. A. P. conceived the project and contributed to experimental design. D. D. S. and D. A. P. wrote the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by grants from the Natural Sciences and Engineering Council of Canada and the Canada Foundation for Innovation to D. A. P. Computational investigations were enabled by Compute Ontario and the Digital Research Alliance of Canada. D. D. S. thanks the Government of Ontario for support in the form of an Ontario Graduate Scholarship.Notes and references
- B. Morgan, D. Ezeriņa, T. N. E. Amoako, J. Riemer, M. Seedorf and T. P. Dick, Nat. Chem. Biol., 2013, 9, 119–125 CrossRef CAS PubMed.
- G. J. McBean, M. Aslan, H. R. Griffiths and R. C. Torrão, Redox Biol., 2015, 5, 186–194 CrossRef CAS PubMed.
- J.-M. Lv, S.-Q. Lü, Z.-P. Liu, J. Zhang, B.-X. Gao, Z.-Y. Yao, Y.-X. Wu, L. A. Potempa, S.-R. Ji, M. Long and Y. Wu, Sci. Rep., 2018, 8, 1494 CrossRef PubMed.
- S. García-Santamarina, S. Boronat and E. Hidalgo, Biochemistry, 2014, 53, 2560–2580 CrossRef PubMed.
- S. G. Rhee, Science, 2006, 312, 1882–1883 CrossRef PubMed.
- Z. Wu, U. Barayeu, D. Schilling, T. P. Dick and D. A. Pratt, Curr. Opin. Chem. Biol., 2023, 76, 102353 CrossRef CAS PubMed.
- Y.-C. Chang, C.-N. Huang, C.-H. Lin, H.-C. Chang and C.-C. Wu, Proteomics, 2010, 10, 2961–2971 CrossRef CAS PubMed.
- S. Akter, L. Fu, Y. Jung, M. Lo Conte, J. R. Lawson, W. T. Lowther, R. Sun, K. Liu, J. Yang and K. S. Carroll, Nat. Chem. Biol., 2018, 14, 995–1004 CrossRef CAS PubMed.
- J. M. M. Pople and J. M. Chalker, Curr. Opin. Chem. Biol., 2021, 60, 55–65 CrossRef CAS PubMed.
- N. J. Kettenhofen and M. J. Wood, Chem. Res. Toxicol., 2010, 23, 1633–1646 Search PubMed.
- V. Gupta and K. S. Carroll, Biochim. Biophys. Acta, Gen. Subj., 2014, 1840, 847–875 CrossRef CAS PubMed.
- C. C. Winterbourn and D. Metodiewa, Free Radical Biol. Med., 1999, 27, 322–328 CrossRef CAS PubMed.
- N. Nakamura, J. Am. Chem. Soc., 1983, 105, 7172–7173 CrossRef CAS.
- T. Yoshimura, E. Tsukurimichi, S. Yamazaki, S. Soga, C. Shimasaki and K. Hasegawa, J. Chem. Soc. Chem. Commun., 1992, 1337–1338 RSC.
- T. Sano, R. Masuda, S. Sase and K. Goto, Chem. Commun., 2021, 57, 2479–2482 RSC.
- C. C. Winterbourn and M. B. Hampton, Free Radical Biol. Med., 2008, 45, 549–561 CrossRef CAS PubMed.
- J.-P. R. Chauvin and D. A. Pratt, Angew. Chem., Int. Ed., 2017, 56, 6255–6259 CrossRef CAS PubMed.
- Y. Nishii, M. Ikeda, Y. Hayashi, S. Kawauchi and M. Miura, J. Am. Chem. Soc., 2020, 142, 1621–1629 CrossRef CAS PubMed.
- A. Krȩżel and W. Bal, J. Inorg. Biochem., 2004, 98, 161–166 CrossRef PubMed.
- I. Canals, F. Z. Oumada, M. Rosés and E. Bosch, J. Chromatogr. A, 2001, 911, 191–202 CrossRef CAS PubMed.
- G. Klopman, K. Tsuda, J. B. Louis and R. E. Davis, Tetrahedron, 1970, 26, 4549–4554 CrossRef CAS.
- C. Storkey, M. J. Davies and D. I. Pattison, Free Radical Biol. Med., 2014, 73, 60–66 CrossRef CAS PubMed.
- X. L. Armesto, M. Canle L, M. I. Fernández, M. V García and J. A. Santaballa, Tetrahedron, 2000, 56, 1103–1109 CrossRef CAS.
- P. Nagy and M. T. Ashby, J. Am. Chem. Soc., 2007, 129, 14082–14091 CrossRef CAS PubMed.
- N. T. Ioffe, M. I. Kalinkin, I. N. Rozhkov and I. L. Knunyants, Bull. Acad. Sci. USSR, Div. Chem. Sci., 1969, 18, 1929–1930 CrossRef.
- M. J. Davies and C. L. Hawkins, Free Radical Res., 2000, 33, 719–729 CrossRef CAS PubMed.
- T. P. A. Devasagayam, A. R. Sundquist, P. Di Mascio, S. Kaiser and H. Sies, J. Photochem. Photobiol., B, 1991, 9, 105–116 CrossRef CAS PubMed.
- A. W. M. Nieuwint, J. M. Aubry, F. Arwert, H. Kortbeek, S. Herzberg and H. Joenje, Free Radical Res. Commun., 1985, 1, 1–9 CrossRef CAS PubMed.
- P. Di Mascio and H. Sies, J. Am. Chem. Soc., 1989, 111, 2909–2914 CrossRef CAS.
- G. R. Buettner, FEBS Lett., 1984, 177, 295–299 CrossRef CAS PubMed.
- A. J. McGrath, G. E. Garrett, L. Valgimigli and D. A. Pratt, J. Am. Chem. Soc., 2010, 132, 16759–16761 CrossRef CAS PubMed.
- T. P. A. Devasagayam, A. R. Sundquist, P. Di Mascio, S. Kaiser and H. Sies, J. Photochem. Photobiol., B, 1991, 9, 105–116 CrossRef CAS PubMed.
- J. A. Montgomery Jr, M. J. Frisch, J. W. Ochterski and G. A. Petersson, J. Chem. Phys., 2000, 112, 6532–6542 CrossRef.
- C. Chatgilialoglu and M. Guerra, in Sulfur-Centered Reactive Intermediates in Chemistry and Biology, ed. C. Chatgilialoglu and K.-D. Asmus, Springer US, Boston, MA, 1990, pp. 31–36 Search PubMed.
- J. Mönig, K.-D. Asmus, L. G. Forni and R. L. Willson, Int. J. Radiat. Biol. Relat. Stud. Phys., Chem. Med., 1987, 52, 589–602 CrossRef PubMed.
- A. V Kachur, C. J. Koch and J. E. Biaglow, Free Radical Res., 1998, 28, 259–269 CrossRef PubMed.
- H. Berger, Recl. Trav. Chim. Pays-Bas, 1963, 82, 773–789 CrossRef CAS.
- G. Monteiro, B. B. Horta, D. C. Pimenta, O. Augusto and L. E. S. Netto, Proc. Natl. Acad. Sci. U. S. A., 2007, 104, 4886–4891 CrossRef CAS PubMed.
- L. Turell, H. Botti, S. Carballal, G. Ferrer-Sueta, J. M. Souza, R. Durán, B. A. Freeman, R. Radi and B. Alvarez, Biochemistry, 2008, 47, 358–367 CrossRef CAS PubMed.
- M. Conrad and D. A. Pratt, Nat. Chem. Biol., 2019, 15, 1137–1147 CrossRef CAS PubMed.
- D. D. Saraev, Z. Wu, H.-Y. H. Kim, N. A. Porter and D. A. Pratt, ACS Chem. Biol., 2023, 18, 2073–2081 CrossRef CAS PubMed.
- W. M. Haynes, CRC Handbook of Chemistry and Physics, Taylor & Francis, 93rd edn, 2012 Search PubMed.
- J. N. Cvengroš Stefan, B. Anne and S. Hans-Günther, Synlett, 2008, 2008, 1993–1998 CrossRef.
- S. Hoops, S. Sahle, R. Gauges, C. Lee, J. Pahle, N. Simus, M. Singhal, L. Xu, P. Mendes and U. Kummer, Bioinformatics, 2006, 22, 3067–3074 CrossRef CAS PubMed.
- J. M. Aubry, J. Am. Chem. Soc., 1985, 107, 5844–5849 CrossRef CAS.
- P. S. Surdhar and D. A. Armstrong, J. Phys. Chem., 1987, 91, 6532–6537 CrossRef CAS.
- W. H. Koppenol, D. M. Stanbury and P. L. Bounds, Free Radical Biol. Med., 2010, 49, 317–322 CrossRef CAS PubMed.
- T. P. A. Devasagayam, P. Di Mascio, S. Kaiser and H. Sies, Biochim. Biophys. Acta, Gene Struct. Expression, 1991, 1088, 409–412 CrossRef CAS PubMed.
- V. Anschau, G. Ferrer-Sueta, R. L. Aleixo-Silva, R. Bannitz Fernandes, C. A. Tairum, C. C. C. Tonoli, M. T. Murakami, M. A. de Oliveira and L. E. S. Netto, Free Radical Biol. Med., 2020, 156, 207–216 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available: Experimental procedures, characterization data, NMR spectra, additional kinetic data, computational data. See DOI: https://doi.org/10.1039/d4sc04871g |
| ‡ Kinetic studies on reactions of protein sulfenic acids have been conducted, however, the approach did not involve direct visualization of the RSOH intermediate. See ref. 39. |
| § It was assumed that the sulfinic acid is fully deprotonated at this pD, based on previously measured pKa values.17 |
| ¶ Reduction potentials in water (E0(RS˙/RS−) = +0.84 V vs. NHE46 and E0(1O2/O2˙−) = +0.81 vs. NHE47) suggest that the reaction of 1O2 and RS− is essentially thermoneutral. This may indicate an even stronger oxidant than 1O2 is formed in the reaction. Earlier studies support this notion, as it has been shown that thiols significantly exacerbate the damage to DNA caused by 1O2 and that hydroxyl radicals may be formed in the reaction.48 |
| || Three equivalents of ascorbate were selected for this reaction based on the reported rate constants.49 If reduction were to occur, it would be possible to monitor by 19F NMR under these conditions. |
| This journal is © The Royal Society of Chemistry 2024 |