
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Boryl radical-mediated halogen-atom transfer (XAT) enables the Sonogashira-like alkynylation of alkyl halides†
Javier
Corpas
 ,
Maialen
Alonso
and
Daniele
Leonori
*
,
Maialen
Alonso
and
Daniele
Leonori
*
Institute of Organic Chemistry, RWTH Aachen University, Landoltweg 1, Aachen 52056, Germany. E-mail: daniele.leonori@rwth-aachen.de
First published on 24th October 2024
Abstract
Alkynes are a crucial class of materials with application across the wide range of chemical disciplines. The alkynylation of alkyl halides presents an ideal strategy for assembling these materials. Current methods rely on the intrinsic electrophilic nature of alkyl halides to couple with nucleophilic acetylenic systems, but these methods faces limitations in terms of applicability and generality. Herein, we introduce a different approach to alkynylation of alkyl halides that proceeds via radical intermediates and uses alkynyl sulfones as coupling partners. This strategy exploits the ability of amine-ligated boryl radicals to activate alkyl iodides and bromides through halogen-atom transfer (XAT). The resulting radicals then undergo a cascade of α-addition and β-fragmentation with the sulfone reagent, leading to the construction of C(sp3)–C(sp) bonds. The generality of the methodology has been demonstrated by its successful application in the alkynylation of complex and high-value molecules.
Introduction
Alkynes play key roles in chemical synthesis, medicinal chemistry and materials science (Scheme 1A).1,2 For instance, alkynes are widely used as dipolarophiles in [3 + 2] cycloadditions, a pivotal transformation within the bioconjugation toolbox.3 Additionally, they are often employed as bioisosteres for various functionalities, such as carbonyls, 1,4-disubstituted phenyls, and cyclopropyl groups.4 Consequently, the development of methods for the modular introduction of alkyne functionalities onto organic molecules is still a highly sought-after goal in synthesis and catalysis.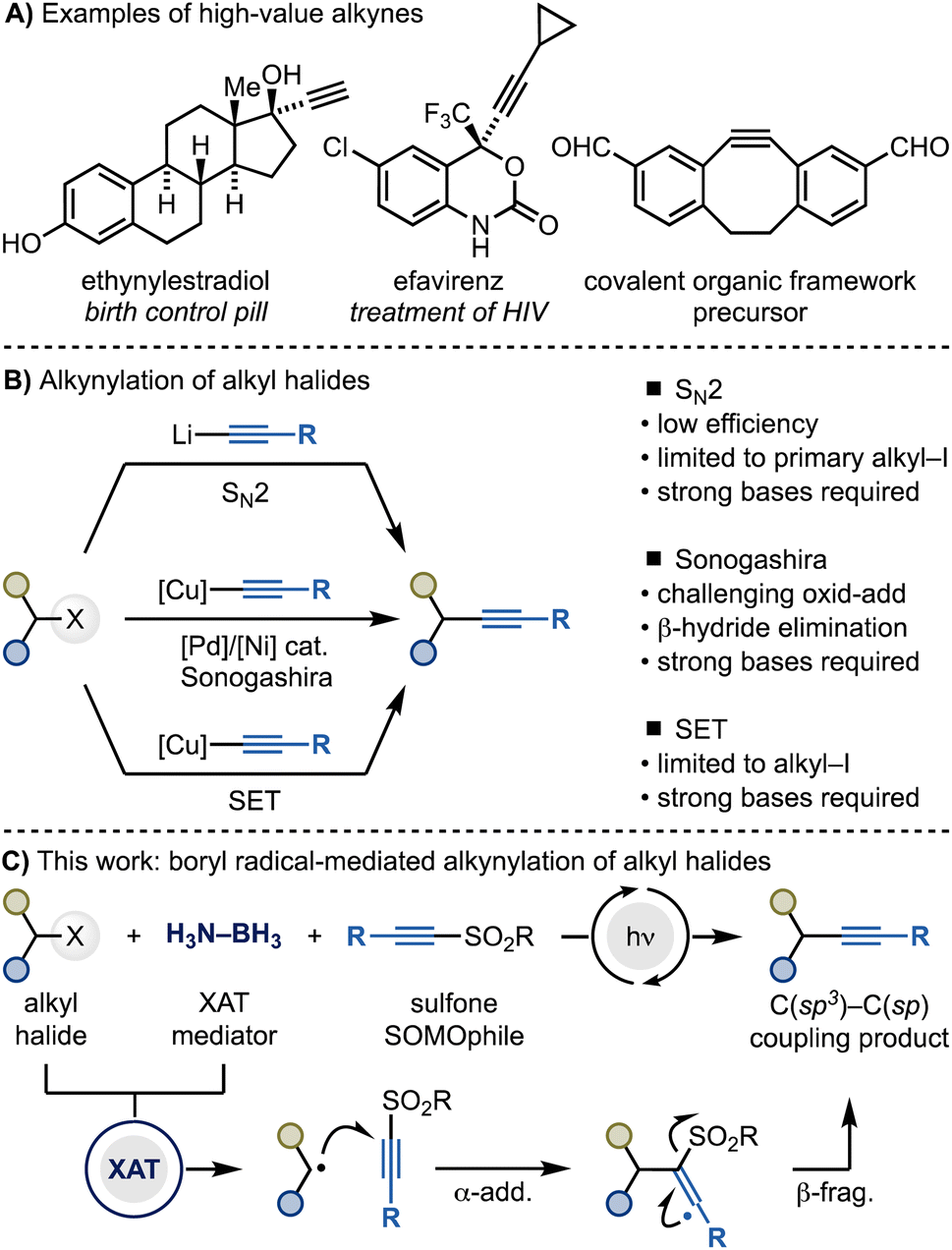 | ||
| Scheme 1 (A) Examples of high-value alkyne-containing materials. (B) General methods for the alkynylation of alkyl halides and their limitations. (C) This work demonstrates a radical approach based on XAT for the alkynylation of alkyl halides (iodides and bromides). | ||
Within this context, the alkynylation of alkyl halides would be particularly useful considering the large amounts of derivatives commercially available (Scheme 1B). However, engaging these species in C(sp3)–C(sp) bond formations is still synthetically challenging. Current methods match the intrinsic electrophilic nature of alkyl halides with nucleophilic acetylene sources for SN2 reactivity (e.g. alkynyl organolithiums), Sonogashira cross-couplings (e.g. alkynyl organocoppers) and radical manifolds (also alkynyl organocoppers). However, these strategies are often synthetically restricted to primary alkyl iodides due to challenges in either substitution reactivity (SN2 methods),5 oxidative addition and β-hydride elimination (Sonogashira cross-coupling)6 or C(sp3)–halogen bond reduction (radical methods).7,8
Herein, we demonstrate an alternative and general approach for C(sp3)–C(sp) bond formation of alkyl halides using alkynyl sulfones and Me3N–BH3 under photocatalytic conditions (Scheme 1C). This method exploits amine-ligated boryl radicals for the conversion of alkyl halides into the corresponding radicals, followed by their cascade α-addition–β-elimination reactions with the SOMOphilic sulfone reagent. This strategy enables the utilization of both alkyl iodides and bromides and provide a metal-free option to the synthesis of high-value alkyne materials.
Reaction design
In approaching the development of a general alkynylation reaction of alkyl halides, we identified a strategic advantage in using alkynyl SOMOphiles instead of nucleophilic acetylenes as coupling partners.9,10 Among various potential reagents, alkynyl sulfones stood out due to their ease of preparation (one step) and their stability as solid compounds.11 However, their intrinsic electrophilic nature transforms the reaction with alkyl halides into a cross-electrophile coupling,12 presenting two key challenges for any redox-based system.13 Firstly, alkynyl sulfones are more easily reduced than alkyl halides (Ered ∼ −1.5 V vs. Ered < −2.0 V vs. SCE, respectively),14 which complicates radical generation by single-electron transfer (SET). Secondly, the addition of a radical to a sulfone leads to the extrusion of a sulfinyl radical, which must be reduced to a stable sulfinate. This means that two sequential reductions are required along the reaction line, making the overall process redox imbalanced (Scheme 2A).10,13 | ||
| Scheme 2 (A) Key mechanistic differences in the use of SET vs. XAT reactivity for radical alkynylation. (B) Proposed mechanism for the XAT-based alkynylation of alkyl halides via amine-ligated boryl radicals. LB: Lewis base. | ||
To overcome the first challenge, we considered a halogen atom transfer (XAT)-based approach. Indeed, by using an appropriate abstracting radical (Y·), could achieve selective activation of alkyl halides (Scheme 2A).15 However, because the halogen-abstraction step naturally favors the use of nucleophilic radical due to polar effects, it would be crucial to suppress any Giese-type reactions between Y· and the sulfone.16
Adopting a XAT-based approach also provides a solution to the second challenge. Indeed, as XAT-mediators can be generated either by oxidation or H-atom transfer (HAT), they provide options for redox-neutral manifolds.15,17
Notably, a pioneering application of this concept was developed by Fuchs in 1998,18 using alkyl iodides and equimolar di-tin reagents under UV light in benzene. In this system, photochemical homolysis of the Sn–Sn σ-bond generated a tin radical, sustaining a radical chain through XAT on alkyl iodides, followed by radical addition to the alkynyl sulfone and regeneration of the tin-radical. However, the harsh reaction conditions and toxic reagents limited the method's applicability, especially since the more common alkyl bromides could not be activated.
Given our interest in XAT-based methodologies,15,17d,g,19 we hypothesized that the bench stable, non-toxic and inexpensive Lewis acid-base adduct Me3N–BH3, in combination with diaryl ketone photocatalysis, could address these mechanistic and practical issues.20 Specifically, we proposed a catalytic cycle featuring a diarylketone photocatalyst, leveraging HAT and SET events to generate the required amine-ligated boryl radical and enable redox neutral catalysis (Scheme 2B).
Triplet ketones, which are strong HAT mediators, have previously been shown to activate Me3N–BH3 leading to the formation of I (Scheme 2B).20a,21 This species could then homolytically activate the alkyl halide by XAT.21a The resulting radical III could then undergo α-addition, followed by β-fragmentation, with the sulfone reagent.10 This process would yield the desired product 3 and a sulfinate radical, which could close the photocatalytic cycle by SET and deprotonation with the ketyl radical (KR).
Reaction development
To validate this mechanistic hypothesis, we evaluated the reaction of alkyl iodide 1a and sulfone 2a (Scheme 3). Pleasingly, the use of Me3N–BH3 as XAT mediator in the presence of K3PO4 as the base and anthraquinone (AQ) as the photocatalyst under purple LEDs (λ = 390 nm) irradiation in EtOAc gave 3 in 86% yield (entry 1, see the ESI† for further optimization studies). Other amine-ligated boranes as well as pyr–BH3 and Ph3P–BH3 could be used in the process but they were all significantly less effective (entries 2–6). This can be mostly rationalized with their lower nucleophilic character that might retard the XAT process. Control experiments confirmed that all components as well as light irradiation are essential for reactivity (entries 9–10), while the base is mostly important to achieve high yields (entry 11). When the same reaction conditions were applied to the alkynylation of bromide 1b, the desired product 3 was obtained in poor conversion (entry 7, see the ESI†). However, conducting the reactions at 55–60 °C restored reactivity, providing 3 in 61% yield (entry 8). We believe the higher temperature might be necessary to facilitate the XAT on the stronger C(sp3)–Br bond.22 | ||
| Scheme 3 Optimization for the alkynylation of 1a with 2a. aReaction run in the dark. bReaction run without AQ. cReaction run without K3PO4. | ||
Reaction scope
With a method for radical alkynylation of alkyl halides in hand, we proceeded to explore the scope of the process. We initially examined a series of aryl-substituted alkynyl sulfones using 1a and 1b as the coupling partners (Scheme 4). This investigation demonstrated the methodology's compatibility with aromatic units substituted with both electron-donating and electron-withdrawing substituents at the para (4–9), meta (10–12) and ortho (13) positions. Notably, the reactions tolerated ester (5), CF3 (6), aryl chloride and fluoride (7 and 12) as well as free alcohol (9) and free aniline (11) functionalities. | ||
| Scheme 4 Scope of the process. Unless otherwise noted, reactions were carried out at room temperature using a fan. aReaction run at 55 °C. bReaction run using the SO2Ph instead of SO2p-Tol reagent. | ||
Next, we valuated reagents based on (hetero)aromatic systems and successfully extended the reactivity to sulfones containing 1-naphthyl (14), 9-phenanthrenyl (15) and 3-thienyl (16) groups.
The scope of alkyl halides was then explored using 2a as the sulfone coupling partner. Pleasingly, we successfully engaged a range of saturated heterocyclic fragments commonly found in medicinal chemistry libraries.23 These included 3-tetrahydropyran (17), 3-N-Boc-pryrrolidine (18) as well as 2-N-Boc-azetidine (19), 2-oxetane (20) and 2-indenol (23) derivatives. Complex alkyl halides can be conveniently accessed through halo-lactonization processes, which upon alkynylation yielded tricyclic systems 21 and 22 in high diastereoselectivity (dr exo![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :endo > 20:1).
:endo > 20:1).
We then screened primary alkyl halides and demonstrated the chemistry's compatibility with alkyl chloride (24), free alcohol (25), olefin (26) as well as acetal (27–28) and amino acid (29) functionalities. We were then keen on determining if the method could be used for the functionalization of hindered halides. This was demonstrated by engaging a series of derivatives of neo-pentyl nature (31, 33). Moreover, we showcased the reactivity on a series of tertiary halides featuring adamantyl (34), bicyclo[1.1.0]pentyl (35) and cubyl (36) units. The formation of 35 and 36 is particularly noteworthy, given the growing importance of these scaffolds as bioisosteric replacement units in modern medicinal chemistry campaigns.24
The radical nature of the transformation was also harnessed as part of radical 5-exo-trig cyclization–alkynylation sequences using alkyl halides with tethered alkene functionalities (37). These processes took place in good yield and high diastereoselectivity (see the ESI†).
As a final element of substrate scope, we attempted the functionalization of complex alkyl halides. These species were obtained from the corresponding alcohol in one step by Appel reaction. Pleasingly, we successfully applied this method to the alkynylation of carbohydrate derivatives based on α-D-galactopyranose (40), D-glucose (41) and α-D-glucofuranose (42) moieties. The chemistry was also applied to a derivative of Corey's lactone (39), the alkaloid nortropine (38) and the neurosteroid androsterone (43). Finally, we prepared a sulfonylated ethynylestradiol reagent that was successfully coupled with 1a to give 44. These examples highlight the ability of the method to engage alkyl-substituted acetylenic sulfones as reactive coupling partners.
Notably, this boryl radical-mediated XAT approach can also be used with other sulfone reagents for the transfer of additional functional groups. A preliminary demonstration of this potential is demonstrated by the reaction of 1a with 2p–r under identical reaction conditions to give products of olefination (45), cyanation (46) and chlorination (47).
Conclusions
The alkynylation of alkyl halides remains a challenging transformation. The results presented here demonstrate that by converting alkyl halides into the corresponding radicals, efficient C(sp3)–C(sp) bond formation can be achieved using alkynyl sulfones as the coupling partners. This method leverages amine-ligated boryl radicals for halide activation by halogen-atom transfer (XAT), enabling effective functionalization of primary, secondary and tertiary sites. The broad applicability of this approach has been showcased with the successful alkynylation of alkyl bromides, as well as the functionalization of complex and high-value molecules. We anticipate that this strategy will find utility in the preparation of alkyne-containing materials and further stimulate the development of novel methodologies employing boryl radical-mediated XAT.Data availability
The data that support the findings of this study are available in the ESI† of this article.Author contributions
J. C.: conceptualization, investigation, writing – original draft. M. A.: investigation. D. L.: conceptualization, supervision, funding, writing – review & editing.Conflicts of interest
There are no conflicts to declare.Acknowledgements
D. L. acknowledges the European Research Council for a grant (101086901). J. C. acknowledges the Marie Curie actions for a Fellowship (project 101104383-DES-B-CAT). M. A. acknowledges Sanofi for additional support.Notes and references
- F. Diederich, P. J. Stang and R. R. Tykwinski, Acetylene Chemistry: Chemistry, Biology and Material Science, Wiley-VCH, Weinheim, 2005 Search PubMed.
- J. Lehmann, M. H. Wright and S. A. Sieber, Chem.–Eur. J., 2016, 22, 4666–4678 CrossRef CAS.
- E. Mons, I. D. C. Jansen, J. Loboda, B. R. van Doodewaerd, J. Hermans, M. Verdoes, C. A. A. van Boeckel, P. A. van Veelen, B. Turk, D. Turk and H. Ovaa, J. Am. Chem. Soc., 2019, 141, 3507–3514 CrossRef CAS.
- T. T. Talele, J. Med. Chem., 2020, 63, 5625–5663 CrossRef CAS.
- The direct SN2 between deprotonated alkynes and secondary alkyl halides is a very low-efficient process, see: (a) H. Lind and A. J. Deutschman, J. Org. Chem., 1967, 32, 326–329 CrossRef; (b) A. B. Holmes and G. E. Jones, Tetrahedron Lett., 1980, 21, 3111–3112 CrossRef CAS; (c) J. M. Chong and S. Wong, Tetrahedron Lett., 1986, 27, 5445–5448 CrossRef CAS.
- For selected examples, see: (a) M. Eckhardt and G. C. Fu, J. Am. Chem. Soc., 2003, 125, 13642–13643 CrossRef CAS PubMed; (b) G. Altenhoff, S. Würtz and F. Glorius, Tetrahedron Lett., 2006, 47, 2925–2928 CrossRef CAS; (c) O. Vechorkin, D. Barmaz, V. Proust and X. Hu, J. Am. Chem. Soc., 2009, 131, 12078–12079 CrossRef CAS PubMed; (d) J. Yi, X. Lu, Y.-Y. Sun, B. Xiao and L. Liu, Angew. Chem., Int. Ed., 2013, 52, 12409–12413 CrossRef CAS PubMed.
- For selected examples, see: (a) A. Hazra, M. T. Lee, J. F. Chiu and G. Lalic, Angew. Chem., Int. Ed., 2018, 57, 5492–5496 CrossRef CAS PubMed; (b) X.-Y. Dong, Y.-F. Zhang, C.-L. Ma, Q.-S. Gu, F.-L. Wang, Z.-L. Li, S.-P. Jiang and X.-Y. Liu, Nat. Chem., 2019, 11, 1158–1166 CrossRef CAS PubMed; (c) Y.-X. Cao, X.-Y. Dong, J. Yang, S.-P. Jiang, S. Zhou, Z.-L. Li, G.-Q. Chen and X.-Y. Liu, Adv. Synth. Catal., 2020, 362, 2280–2284 CrossRef CAS; (d) H.-D. Xia, Z.-L. Li, Q.-S. Gu, X.-Y. Dong, J.-H. Fang, X. Y. Du, L.-L. Wang and X.-Y. Liu, Angew. Chem., Int. Ed., 2020, 59, 16926–16932 CrossRef CAS PubMed; (e) F.-L. Wang, C.-J. Yang, J.-R. Liu, N.-Y. Yang, X.-Y. Dong, R.-Q. Jiang, X.-Y. Chang, Z.-L. Li, G.-X. Xu, D.-L. Yuan, Y.-S. Zhang, Q.-S. Gu, X. Hong and X.-Y. Liu, Nat. Chem., 2022, 14, 949–957 CrossRef CAS PubMed; (f) X. Zeng, C. Wang, W. Yan, J. Rong, Y. Song, Z. Xiao, A. Cai, S. H. Liang and W. Liu, ACS Catal., 2023, 13, 2761–2770 CrossRef CAS PubMed; (g) S. R. Narra, M. Z. Bacho, M. Hattori and N. Shibata, Adv. Sci., 2024, 11, 2306554 CrossRef CAS PubMed . For examples using other metals, see:; (h) L. Huang, A. M. Olivares and D. J. Weix, Angew. Chem., 2017, 129, 12063–12067 CrossRef; (i) J. M. Smith, T. Qin, R. R. Merchant, J. T. Edwards, L. R. Malins, Z. Liu, G. Che, Z. Shen, S. A. Shaw, M. D. Eastgate and P. S. Baran, Angew. Chem., Int. Ed., 2017, 56, 11906–11910 CrossRef CAS; (j) F. Zhurkin, W. Parisot and G. Lefèvre, Adv. Synth. Catal., 2024, 366, 1782–1787 CrossRef CAS.
- For selected reviews, see: (a) X. Mo, R. Guo and G. Zhang, Chin. J. Chem., 2023, 41, 481–489 CrossRef CAS; (b) X. Zhao and W. Liu, Synthesis, 2024, 56, 1244–1258 CrossRef CAS.
- (a) J. P. Brand and J. Waser, Chem. Soc. Rev., 2012, 41, 4165–4179 RSC; (b) F. Le Vaillant and J. Waser, Chem. Sci., 2019, 10, 8909–8923 RSC; (c) E. Le Du and J. Waser, Chem. Commun., 2023, 59, 1589–1604 RSC.
- D. Ge, X. Wangb and X.-Q. Chu, Org. Chem. Front., 2021, 8, 5145–5164 RSC.
- (a) C. C. Chen and J. Waser, Org. Lett., 2015, 17(3), 736–739 CrossRef CAS; (b) J. Meesin, P. Katrun, C. Pareseecharoen, M. Pohmakotr, V. Reutrakul, D. Soorukram and C. Kuhakarn, J. Org. Chem., 2016, 81, 2744–2752 CrossRef CAS PubMed; (c) M. A. Guerrero-Robles, M. A. Vilchis-Reyes, E. M. Ramos-Rivera and C. Alvarado, ChemistrySelect, 2019, 4, 13698–13708 CrossRef CAS.
- (a) A. R. Tripathy, A. Kumar, R. Rahmathulla, A. K. Jha and V. R. Yatham, Org. Lett., 2022, 24, 5186–5519 CrossRef CAS PubMed; (b) J. Yin, X. Zhang, L. Zhao, M. Luo, L. Guo, C. Yang and W. Xia, Org. Chem. Front., 2023, 10, 4679–4686 RSC.
- For selected examples, see: (a) J. Yang, J. Zhang, L. Qi, C. Hu and Y. Chen, Chem. Commun., 2015, 51, 5275–5278 RSC; (b) C. Gao, J. Li, J. Yu, H. Yanga and H. Fu, Chem. Commun., 2016, 52, 7292–7294 RSC; (c) J. Schwarz and B. König, ChemPhotoChem, 2017, 1, 237–242 CrossRef CAS; (d) M. Ociepa, J. Turkowska and D. Gryko, ACS Catal., 2018, 8, 11362–11367 CrossRef CAS; (e) L. Wang, Y. Xia, K. Bergander and A. Studer, Org. Lett., 2018, 20, 5817–5820 CrossRef CAS PubMed; (f) N. Iqbal, N. Iqbal, S. S. Han and E. J. Cho, Org. Biomol. Chem., 2019, 17, 1758–1762 RSC; (g) M. Li, T. Liu, J. Li, H. He, H. Dai and J. Xie, J. Org. Chem., 2021, 86, 12386–12393 CrossRef CAS PubMed; (h) L. Capaldo and D. Ravelli, Org. Lett., 2021, 23, 2243–2247 CrossRef CAS PubMed; (i) Y. Yang, C. G. Daniliuc and A. Studer, Angew. Chem., Int. Ed., 2021, 60, 2145–2148 CrossRef CAS; (j) N. L. Frye, C. G. Daniliuc and A. Studer, Angew. Chem., Int. Ed., 2022, 61, e202115593 CrossRef CAS; (k) L. Xia, M. Jin, Y. Jiao and S. Yu, Org. Lett., 2022, 24, 364–368 CrossRef CAS PubMed; (l) M. Swain, T. B. Bunnell, J. Kim and O. Kwon, J. Am. Chem. Soc., 2022, 144, 14828–14837 CrossRef CAS PubMed; (m) X. Ma, L. Wang, X. Meng, W. Li, Q. Wang, Y. Gu and L. Qiu, Org. Biomol. Chem., 2023, 21, 6693–6696 RSC; (n) H. Wu, S. Chen, D. Xiao, F. Li, K. Zhou, X. Yin, C. Liu, X. He and Y. Shang, Org. Lett., 2023, 25, 1166–1171 CrossRef CAS PubMed.
- (a) H. G. Roth, N. A. Romero and D. A. Nicewicz, Synlett, 2016, 27, 714–723 CAS; (b) A. Luque, J. Groß, T. J. B. Zähringer, C. Kerzig and T. Opatz, Chem.–Eur. J., 2022, 28, e202104329 CrossRef CAS PubMed; (c) X. Sun and K. Zheng, Nat. Commun., 2023, 14, 6825 CrossRef CAS PubMed.
- F. Juliá, T. Constantin and D. Leonori, Chem. Rev., 2022, 122, 2292–2352 CrossRef PubMed.
- (a) B. Giese and S. Lachhein, Angew. Chem., Int. Ed., 1982, 21, 768–775 CrossRef; (b) U. Wille, Chem. Rev., 2013, 113, 813–853 CrossRef CAS PubMed.
- (a) P. Zhang, C. C. Le and D. W. C. MacMillan, J. Am. Chem. Soc., 2016, 138, 8084–8087 CrossRef CAS PubMed; (b) C. Chatgilialoglu, C. Ferreri, Y. Landais and V. I. Timokhin, Chem. Rev., 2018, 118, 6516–6572 CrossRef CAS PubMed; (c) M. N. Lavagnino, T. Liang and D. W. C. MacMillan, Proc. Natl. Acad. Sci. U. S. A., 2020, 117, 21058–21064 CrossRef CAS PubMed; (d) T. Constantin, M. Zanini, A. Regni, N. S. Sheikh, F. Juliá and D. Leonori, Science, 2020, 367, 1021–1026 CrossRef CAS PubMed; (e) A. Cai, W. Yan, C. Wang and W. Liu, Angew. Chem. Int. Ed., 2021, 60, 27070–27077 CrossRef CAS PubMed; (f) A. Cai, W. Yan and W. Liu, J. Am. Chem. Soc., 2021, 143, 9952–9960 CrossRef CAS PubMed; (g) A. Ruffoni, R. C. Mykura, M. Bietti and D. Leonori, Nat. Synth., 2022, 1, 682–695 CrossRef.
- J. Xiang and P. L. Fuchs, Tetrahedron Lett., 1998, 39, 8597–8600 CrossRef CAS.
- For selected examples, see: (a) B. Górski, A.-L. Barthelemy, J. J. Douglas, F. Juliá and D. Leonori, Nat. Catal., 2021, 4, 623–630 CrossRef; (b) H. Zhao, A. J. McMillan, T. Constantin, R. C. Mykura, F. Juliá and D. Leonori, J. Am. Chem. Soc., 2021, 143, 14806–14813 CrossRef CAS PubMed; (c) Z. Zhang, B. Górski and D. Leonori, J. Am. Chem. Soc., 2022, 144, 1986–1992 CrossRef CAS PubMed; (d) T. Constantin, B. Górski, M. J. Tilby, S. Chelli, F. Juliá, J. Llaveria, K. J. Gillen, H. Zipse, S. Lakhdar and D. Leonori, Science, 2022, 377, 1323–1328 CrossRef CAS PubMed; (e) L. Caiger, H. Zhao, T. Constantin, J. J. Douglas and D. Leonori, ACS Catal., 2023, 13, 4985–4991 CrossRef CAS.
- (a) Z. Zhang, M. J. Tilby and D. Leonori, Nat. Synth., 2024, 3, 1221–1230 CrossRef; (b) Z. Zhang, L. Poletti and D. Leonori, J. Am. Chem. Soc., 2024, 146, 22424–22430 CrossRef CAS PubMed.
- (a) J. A. Baban and B. P. Roberts, J. Chem. Soc., Perkin Trans. 2, 1984, 1717–1722 RSC; (b) J. A. Baban, V. P. J. Marti and B. P. Roberts, J. Chem. Soc., Perkin Trans. 2, 1985, 1723–1733 RSC.
- (a) T. Wan, L. Capaldo, D. Ravelli, W. Vitullo, F. J. de Zwart, B. de Bruin and T. Noël, J. Am. Chem. Soc., 2023, 145, 991–999 CrossRef CAS; (b) L. Capaldo, T. Wan, R. Mulder, J. Djossoua and T. Nöel, Chem. Sci., 2024, 15, 14844–14850 RSC.
- E. Vitaku, D. T. Smith and J. T. Njardarson, J. Med. Chem., 2014, 57, 10257–10274 CrossRef CAS PubMed.
- (a) E. G. Tse, S. D. Houston, C. M. Williams, G. P. Savage, L. M. Rendina, I. Hallyburton, M. Anderson, R. Sharma, G. S. Walker, R. S. Obach and M. H. Todd, J. Med. Chem., 2020, 63, 11585–11601 CrossRef CAS PubMed; (b) S. Cuadros, J. Paut, E. Anselmi, G. Dagousset, E. Magnier and L. Dell'Amico, Angew. Chem., Int. Ed., 2024, 63, e202317333 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Procedures, optimization, characterization of compounds and spectroscopic data. See DOI: https://doi.org/10.1039/d4sc06516f |
| This journal is © The Royal Society of Chemistry 2024 |