
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceHalide-triggered assembly and selective bisulfate recognition in a quadruply interlocked coordination cage†
Jemma I.
Virtue
 ,
Steven
Tsoukatos
,
Martin R.
Johnston
and
Witold M.
Bloch
*
,
Steven
Tsoukatos
,
Martin R.
Johnston
and
Witold M.
Bloch
*
Institute for Nanoscale Science and Technology, College of Science and Engineering, Flinders University, Bedford Park, South Australia 5042, Australia. E-mail: witold.bloch@flinders.edu.au
First published on 24th October 2024
Abstract
Interlocked coordination cages are a class of multi-cavity architectures with applications in selective anion recognition, adaptive sensing, and catalysis. Controlling the partitioning of their cavities through ligand design and appropriate anion templates is critical to their guest binding scope, yet remains a challenge. Here, we present a thermodynamically stable [Pd2L4](BF4)4 cage assembled from a bis-monodentate ligand featuring a non-coordinating bis-pyrazole methane backbone. As a result of its idealized dimensions, NMR, ESI-MS, and X-ray analyses reveal that halides can trigger the interpenetration of this cage into a [X@Pd4L8]7+ dimer (X = Cl− or Br−) where the halide template resides only in the central pocket. The anion–cation pattern of this interlocked host facilitates exceptional binding affinity for the bisulfate anion in its two outer pockets (up to 106 M−1 in MeCN), strongly outcompeting other tetrahedral anions of similar size.
Introduction
The synthesis of interlocked systems has attracted considerable attention owing to their unique structural features, partitioned cavities, and dynamic mechanical properties.1–3 For example, interlocked molecular dimers can exhibit allosteric guest recognition,4 stimuli-responsive motion,5–7 or photocatalytic activity8–10 – unique characteristics that are not observed in their monomeric constituents.11 Despite the recent progress in the synthesis and chemistry of various interlocked structures such as catenanes,1 rotaxanes,12 Borromean rings,13 and molecular knots,14 engineering the cavities of these interlocked architectures toward selective guest recognition continues to represent a formidable challenge.15Coordination cages assembled from concave-shaped ligands and square-planar PdII cations have served as excellent precursors for interlocked molecules and other multi-cavity architectures.16,17 In the presence of a suitable anion template, two Pd2L4 monomers can interpenetrate, leading to a quadruply interlocked Pd4L8 dimer. The stability of the interlocked product arises from favourable hydrogen bonding or coulombic interactions with the anion template, which compensate for the entropic penalty of interpenetration.16,18,19
As a class of multi-cavity architectures, interpenetrated Pd4L8 dimers carry a high charge of +8 (excluding the anion template) and are thus excellent hosts for anion recognition.20 Compared to other multi-cavity cage systems that rely on synthetically demanding multi-dentate ligands,21–25 interlocked cages are more straightforward to prepare, as they are assembled from a larger number of simpler ligand components.26 In this context, it is important to note the extensive work of Clever,27 Kuroda,28 and others,19,29,30 who have demonstrated the importance of ligand length (defined herein as DN–N), sterics, counter-ion, and solvent on whether or not interpenetration occurs, the cation–anion arrangement pattern of the interlocked dimer, as well as its binding scope and selectivity towards certain guests.
For the purpose of this work, we define the three common anion–cation patterns (or motifs) for Pd4L8 interlocked cages as type I, II, and III (Fig. 1a). In the assembly of type I motifs, the monomeric Pd2L4 cage (DN–N > 15.8 Å) is a kinetic intermediate that progresses to an interpenetrated product with the same anionic guest (BF4− or NO3−) in all three cavities.38 The outer pockets of type I motifs can exhibit allosteric binding of smaller halide guests (relative to the larger, initially bound BF4− anions), leading to structural compression along the Pd⋯Pd axis and simultaneous expansion of the central cavity.4,31 Shorter bis-monodentate ligands (e.g., DN–N = 13–13.5 Å) lead to monomeric cages as the thermodynamic product when larger BF4− counter-ions are employed. In this case, a halide additive can trigger the formation of a type III interlocked motif where the same halide guest occupies the three cavities.32,33 The type II motif is an interesting and rare case, which also arises from a thermodynamically stable Pd2L4 monomeric cage. However, halides trigger the assembly of an interlocked cage where only the central pocket is occupied by the halide, leaving the two outer pockets available for exchange with larger anions. This motif was previously isolated from a Pd2L4 cage possessing bulky ligands that allowed only halides to template the dimer for steric reasons (Fig. 1a).34 Currently, this interlocked motif remains a challenge to access and derivatize,39 limiting the host–guest chemistry and further exploration of this class of multi-cavity architectures.
 | ||
| Fig. 1 (a) The three types of Pd4L8 interlocked motifs according to the templating anion volume and ligand DN–N separation, where squares represent X-ray data from previous studies;4,29,31–37 note: in the type II motif, the outer pockets are occupied by weakly bound counter-ions. (b) This work: utilizing a bis-pyrazole methane ligand with idealized dimensions to access a type II interlocked motif with high binding selectivity for bisulfate. | ||
Mapping the ligand DN–N separation from X-ray data of reported dimeric Pd4L8 structures, type I and III motifs are observed for DN–N greater than 15.8 Å (BF4− or NO3− anion template) and less than 13.5 Å (halide template), respectively (Fig. 1a). We hypothesized that type II interlocked structures may be accessible simply by targeting an intermediate DN–N of ∼15 Å, where only halide anions induce interpenetration due to their small volume relative to the dimensional constraints of the monomeric cage. Herein, we report the coordination-driven self-assembly of a Pd2L4 cage composed of a flexible, non-coordinating bis-pyrazole methane backbone (Fig. 1b). We show that halides (Cl− and Br−) can trigger the interpenetration of this cage into an interlocked type II motif without the requirement of steric bulk at the ligand backbone. The two outer cavities of the interpenetrated dimers preferentially bind tetrahedral guests and show high selectivity toward HSO4−, an anion of substantial environmental importance.40
Results and discussion
Our interest in bis-pyrazole methane (bpm) has been driven by its straightforward derivatization41 and utility for heterometallic metal–organic framework and cage-based porous solids.42,43 Despite its appeal as a chelating backbone, we realized that when equipped with 3-ethynyl pyridine donors, its DN–N separation should fall roughly between the dimensions of type I and type III Pd4L8 structures. Indeed, preliminary DFT modelling revealed this ligand should adopt a concave geometry with a DN–N separation between 14.1–14.5 Å, depending on whether the bpm core adopts a syn or anti conformation. Therefore, bis(4-(pyridin-3-ylethynyl)-1H-pyrazol-1-yl)methane (L) was synthesized through a simple Sonogashira cross-coupling reaction (Fig. S1†). Single-crystal X-ray diffraction confirmed the concave shape of the ligand, with DN–N dimensions in close agreement with that predicted by DFT calculations (Fig. S2†).With L in hand, we hypothesized that correct stoichiometry may prevent the bpm core from chelating PdII, thus promoting a monomeric cage through self-assembly with the ligand's pyridine donors.43 Pleasingly, when combining [Pd(CH3CN)4](BF4)2 and L in a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :2 ratio in DMSO at 25 °C, 1H NMR analysis revealed a rather simple spectrum that could be assigned to a single product. The pyridyl protons of the coordinated ligand appeared downfield shifted relative to the free ligand (+0.6 ppm, proton g, Fig. 2a), whilst the pyrazole resonances remained relatively unchanged (+0.04 ppm, proton c, Fig. 2a). Importantly, the NMR data indicates that a chelate complex is not formed, which is unusual given the strong prevalence of bpm and its derivatives forming chelate complexes with PdII ions.44 Electrospray ionization mass spectrometry (ESI-MS) revealed prominent peaks corresponding to [Pd2L4 + nBF4]4−n (n = 0–2) (Fig. 2b), confirming the presence of the anticipated cage product (1). However, heating the DMSO solution of 1 at 70 °C resulted in a complex mixture, as revealed by 1H NMR spectroscopy and ESI-MS (Fig. 2a and S5†). This suggests that the cage is a kinetic intermediate, and entropically driven mixtures involving the pyrazole and pyridine donors of the ligand are energetically favorable in this solvent.
:2 ratio in DMSO at 25 °C, 1H NMR analysis revealed a rather simple spectrum that could be assigned to a single product. The pyridyl protons of the coordinated ligand appeared downfield shifted relative to the free ligand (+0.6 ppm, proton g, Fig. 2a), whilst the pyrazole resonances remained relatively unchanged (+0.04 ppm, proton c, Fig. 2a). Importantly, the NMR data indicates that a chelate complex is not formed, which is unusual given the strong prevalence of bpm and its derivatives forming chelate complexes with PdII ions.44 Electrospray ionization mass spectrometry (ESI-MS) revealed prominent peaks corresponding to [Pd2L4 + nBF4]4−n (n = 0–2) (Fig. 2b), confirming the presence of the anticipated cage product (1). However, heating the DMSO solution of 1 at 70 °C resulted in a complex mixture, as revealed by 1H NMR spectroscopy and ESI-MS (Fig. 2a and S5†). This suggests that the cage is a kinetic intermediate, and entropically driven mixtures involving the pyrazole and pyridine donors of the ligand are energetically favorable in this solvent.
 | ||
| Fig. 2 (a) 1H NMR spectra (500 MHz, DMSO-d6, 25 °C) of (i) L; (ii) cage 1, the kinetic product in DMSO; (iii) the same solution heated at 70 °C for 2 h; (b) ESI-MS spectrum of 1: [Pd2L4 + nBF4]4−n+ (n = 0–2) indicated by purple spheres (c) X-ray structure of 1. | ||
We observed a different outcome when using CD3CN as the solvent. Heating a 1:2 mixture of [Pd(CH3CN)4](BF4)2 and L at 70 °C for 2 h resulted in monomeric cage 1 as the only product (Fig. 3a). More complex mixtures could be obtained when an excess of PdII was used (Fig. S4†), confirming the importance of stoichiometry in the assembly of 1. Allowing the CD3CN solution of 1 to stand at 25 °C for 72 h resulted in the formation of large block-shaped crystals. Single-crystal X-ray analysis confirmed the [Pd2L4](BF4)4 structure, wherein the bpm core remains non-coordinated despite its syn-conformation in the solid state (Fig. 2c). The DN–N separation of 1 measures 15.2 Å, which reflects the flexibility of the bpm ligand afforded by the rotational freedom around the methylene hinge. Additionally, the crystal packing of 1 sheds light on its crystallization, where numerous close π-stacking interactions (closest contact = 3.30 Å) occur between the pyrazole and pyridine rings of neighbouring cage molecules (Fig. S66†).
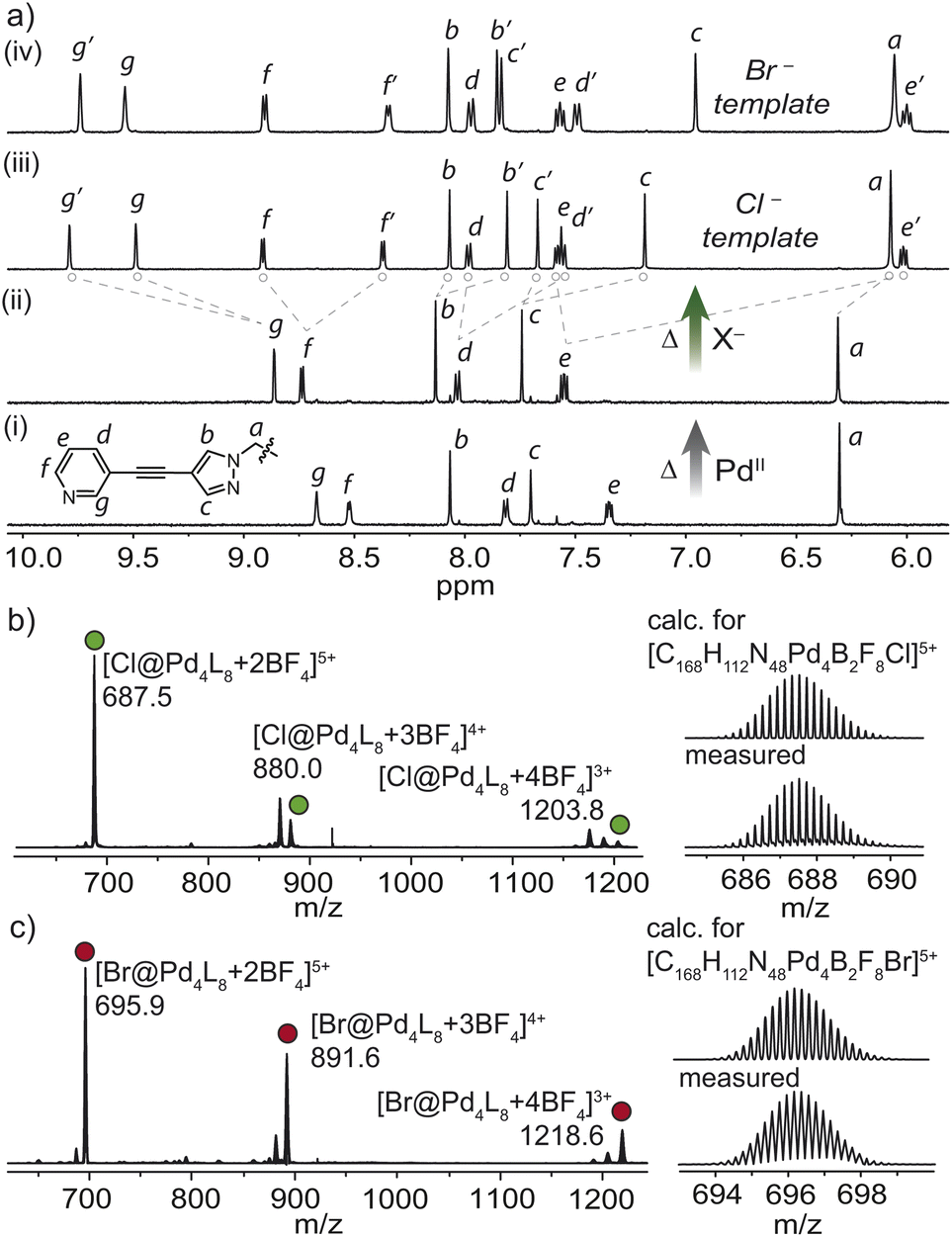 | ||
| Fig. 3 (a) 1H NMR spectra (600 MHz, CD3CN, 25 °C) of (i) L; (ii) cage 1, the thermodynamic product in CD3CN; (iii) Cl@Pd4L8 – Cl@2 and (iv) Br@Pd4L8 – Br@2; (b) ESI-MS spectrum of Cl@2 (green spheres); (c) ESI-MS spectrum of Br@2 (red spheres). | ||
The isolation of 1 is worth highlighting, given the tendency of bpm derivatives to chelate PdII.43,44 Previous work by Crowley and co-workers showed that the assembly of di-pyridyl ligands bearing pendant chelating groups results in chelate complexes with PdII, rather than Pd2L4 cages.45 In the case of 1, the Pd2L4 cage may be entropically favored due to the preservation of the rotational freedom of the non-coordinated bpm core.
Given the intermediate DN–N distance of 1, we next investigated its propensity to interpenetrate in the presence of halides. Indeed, heating 1 in the presence of 0.5 equivalents of Cl− resulted in the clean formation of a new interpenetrated assembly denoted as Cl@2. Characteristic two-fold splitting of the ligand resonances in the 1H NMR spectrum along with major peaks in the ESI-MS spectrum, assigned to [Cl@Pd4L8 + nBF4]8−n (n = 3–5) confirmed the interlocked product (Fig. 3a and b). Using 1.1 equivalents of Cl− in the synthesis of Cl@2 resulted in a mixture of the same interpenetrated product and free ligand (Fig. S8†). 1H–1H NOESY of Cl@2 revealed several cross-peaks that confirmed the interpenetrated structure, such as a contact between pyridyl proton e′ and pyrazole proton c (Fig. S12†).
F− and I− proved to be poor templates for cage dimerization, resulting in mixtures dominated by free ligand and monomeric cage 1. On the other hand, heating 1 with 0.5 equivalents of Br− yielded Br@2 almost quantitatively, as shown by 1H NMR spectroscopy and ESI-MS (Fig. 3a and c). Whereas only a single broad 19F resonance corresponding to solvated BF4− was observed for Cl@2, the 19F NMR spectrum for the Br− templated dimer showed two distinctive resonances at −151 and −146 ppm (Fig. 4c). These were assigned to the free and encapsulated BF4− anion, respectively, and 19F exchange spectroscopy (EXSY) confirmed their exchange at 25 °C (Fig. 4d). The tighter binding of the BF4− anions in the outer pockets of Br@2 can be explained by the larger Br− template, which resides in the central pocket, pushing the interpenetrating cages closer together, diminishing the available volume of the outer cavities. DOSY analysis suggested that these differences are subtle, with both templated dimers diffusing at ∼4.6 × 10−10 m2 s−1 (Fig. S10 and S15†).
 | ||
| Fig. 4 X-ray structures of (a) Cl@2 and (b) Br@2 with Pd⋯Pd separation distances shown. Note: DN–N = 14.8 Å, and the anions residing in the cavities are shown as space-filling models; (c) 19F NMR spectra (565 MHz, CD3CN, 25 °C) of (i) Br@2 and (ii) Cl@2; (d) 19F–19F EXSY spectrum (376.5 MHz, CD3CN, 25 °C) of Br@2. | ||
Single crystals of both interlocked structures were isolated by slow vapor diffusion of chloroform into solutions of Cl@2 and Br@2 in MeCN, allowing us to unequivocally confirm their structures and compare their dimensions through X-ray analysis. Both the Cl− and Br− templated dimers crystallize in an orthorhombic space group (Pbcn for Cl@2 and Ccc2 for Br@2) with half of the double cage in the respective asymmetric unit. In contrast to the X-ray structure of 1, the bis-pyrazole methane moieties in X@2 are present in the anti-conformation, with two identical rotational conformers comprising each Pd2L4 unit. This highlights the rotational freedom of the bpm core, which is supported by the number of 1H NMR resonances of the monomeric and dimeric cage species. The interlocked structures host one halide in the smaller central cavity and a BF4− in each of the two larger outer pockets. Although the intermolecular H⋯F contacts between the host and guest do not differ significantly between the two structures (Fig. S73†), the distance between the outermost PdII and the next inner PdII is shorter for Br@2 by 0.07 Å. VOIDOO calculations provided a clearer picture of the effect of the larger Br− template. These revealed that the outer cavities of Cl@2 possess a volume of 128.5 Å3, whilst the same cavities in the Br− templated dimer reduce to 108.5 Å3. A higher packing coefficient of 51% for the BF4− anion in Br@2 (compared to 43% for Cl@2) is consistent with its tighter binding. On the other hand, the volume of the central cavity measures 12.7 Å3 and 15.8 Å for Cl@2 and Br@2 respectively, indicative of extremely tight binding of the respective halides. This was further supported by the high stability of X@2 in the presence of AgBF4 (Fig. S37 and S53†).
Since the bpm ligand lacks bulky groups that would prevent interpenetration in the presence of BF4−, we propose that the intermediate DN–N separation of the ligand facilitates the assembly of a Pd2L4 monomeric cage that later can be triggered by halides to form X@2 – a rare example of a type II interlocked motif. It is worth noting that Pd2L4 cages that accommodate BF4− anions typically require a DN–N distance >8 Å.17,46,47 Thus, we hypothesize based on the dimensions of X@2, that a BF4− anion in the central cavity would result in outer cavities too small to accommodate BF4− anions favorably, or the ligand would incur an energetic penalty in bond-strain to expand its DN–N. These scenarios may also disrupt the favorable C–H π interactions (proton e and adjacent pyrazole ring, DC–H⋯N = 2.72 Å, 174°), that stabilize the interlocked complex. Interestingly, NO3− (V = 40.7 Å3) could not template the interlocked dimer in the presence of the BF4− counterions (V = 54.8 Å3) of cage 1 (Fig. 5a). In contrast, the self-assembly of L with Pd(NO3)2 led directly to a mixture of the dimeric cage and free ligand, and the monomeric cage could not be detected in this experiment (Fig. 5b). ESI-MS corroborated this result with prominent peaks assignable to 3NO3@2 (Fig. 5c). The clear transition from a type II to a typical type I interlocked motif in the presence of smaller NO3− counter ions further underscores the importance of the DN–N separation in restricting the volume of the central and outer cavities.
 | ||
| Fig. 5
1H NMR spectra (600 MHz, CD3CN, 25 °C) of (a) 1 + 0.5 equivalents of NO3−, heated at 70° for 2 h; (b) a 2:1 mixture of L and Pd(NO3)2, heated at 70° for 3 h. Red colour denotes free ligand; (c) ESI-MS spectrum of 3NO3@2 (blue spheres). | ||
Next, we examined the anion binding capabilities of the halide-templated dimeric cages with a range of monoanionic guests in CD3CN. As tetrabutylammonium salts of ClO4−, ReO4− and NO3− were added in increasing equivalents to Cl@2, the inner pointing protons of the outer pocket (g, c, f′ and e′) underwent shifting with fast exchange kinetics relative to the 1H NMR time scale (Fig. S20–S26†). A Job plot analysis indicated that ClO4−, ReO4− bind with the expected 1:2 host–guest stoichiometry (Fig. S21–S27†). This was further corroborated by a single-crystal structure of (ClO4)2Cl@2, which confirmed that after treating Cl@2 with 2.1 equivalents of tetrabutylammonium perchlorate, the BF4− anions are exchanged for ClO4− anions in the two outer cavities of the interlocked host (Fig S75†). Fitting the NMR data to a 1:2 binding model based on shifts in proton g gave K1 and K2 values of 3.4 ± 0.8 × 104 M−1; K2 = 1.5 ± 0.03 × 103 M−1 for ClO4− and K1 = 1.2 ± 0.4 ×104 M−1; K2 = 3.8 ± 0.2 × 103 M−1 for ReO4−.48 This indicates non-cooperative binding since these values are close to the statistically expected K2 = K1/4. For the NO3− guest, inner (g, f′, e′ and c) and outer (f and c′) pointing protons concurrently underwent downfield shifting suggesting non-specific binding.34 Larger PF6− and OTf− anions were observed to bind only to the outside of the cage (Table 1, Fig. S32 and S33†), whilst charge-dense anions I− and Br− decomposed Cl@2 at low equivalents (Fig. S35 and S36†).
| Guest | X = Cl− | X = Br− | ||
|---|---|---|---|---|
| K 1 (M−1) | K 2 (M−1) | K 1 (M−1) | K 2 (M−1) | |
| a Binding was indicative by shifts in proton g. b Cage decomposition. c Non-specific binding. d Determined using proton c. e Determined by ITC in MeCN. f Outside binding. IE = intermediate exchange. For % uncertainty, see Tables S1 and S2. | ||||
| Br−b | — | — | — | — |
| I−b | — | — | — | — |
| NO3−c | 5.8 × 103 | 2.1 × 103 | IE | IE |
| ClO4−d | 2.9 × 104 | 1.2 × 103 | 8.0 × 103 | 1.7 × 103 |
| HSO4−e | 3.3 × 106 | 3.1 × 104 | 5.3 × 105 | 1.5 × 103 |
| ReO4− | 1.2 × 104 | 3.8 × 103 | IE | IE |
| H2PO4− | — | — | — | — |
| PF6−f | — | — | — | — |
| OTF−f | — | — | — | — |
To compare the effect of the halide template in the inner cavity, the anion-binding behavior of Br@2 was investigated (Table 1). 1H NMR analysis revealed host–guest binding for ClO4− and ReO4−, although intermediate exchange kinetics hindered the fitting of the NMR data, particularly for proton g (Fig. S38 and S41†). Closer inspection of the data revealed that pyrazole proton c (which also points towards the outer cavity) is affected more by the binding of ReO4−, compared to ClO4−; an effect further amplified in the case of the Br− template. For the ReO4− anion, we also observed a notable shift in proton e′, suggesting that the larger ReO4− guest does not reside directly between the PdII centers, but rather adjacent to the pyrazole units of the ligand (Fig S57†). This difference in binding may relate to the relative size of these two anions (59.1 Å3vs. 55.4 Å3) with the ReO4− guests being too large to align with the Pd4 axis. Pyrazole proton c could be monitored to compare the binding of the ClO4− guest for Br@2 and Cl@2. Fitting the isotherm data yielded K1 = 8.0 ± 0.2 × 103 M−1 and K2 = 1.7 ± 0.1 × 103 M−1 – an order of magnitude lower than that obtained by monitoring the same proton for Cl@2: K1 = 2.9 ± 0.4 × 104 M−1 and K2 = 1.2 ± 0.02 × 103 M−1. The weaker binding of Br@2 compared to Cl@2 can be explained by the smaller outer cavities that result in stronger competition between the tightly bound BF4− and the ClO4− guests.
Next, we examined whether the selective binding of X@2 toward tetrahedral anions can be extended to bisulfate (HSO4−) – an anion of environmental importance and considerable concern due to its contamination of agricultural fields.40 It is worth noting that selective receptors for bisulfate are scarce,49–51 reflecting challenges in their design and synthesis. Given that HSO4− is a tetrahedral anion with a volume of 58.9 Å3 (falling between that of ClO4− and ReO4−) we hypothesized that it should be readily encapsulated by the interlocked host. Indeed, upon the addition of 2 equivalents of HSO4− to Cl@2, a significant shift of protons g and f′ (+0.48 and + 0.3 ppm respectively) which point into the outer pockets, was observed to occur slowly relative to the 1H NMR timescale (Fig. S29 and S47†).‡ In contrast to the ReO4− guest, the pyrazole proton c showed minimal shifting, suggesting that the bisulfate guest is in line with the Pd4 axis, positioned directly between the PdII of the outer cavities. Evidence for a (HSO4)2X@2 host–guest complex was obtained from ESI-MS analysis (Fig. S64 and S67†). Since initial estimates indicated that the binding affinity of HSO4− was an order of magnitude greater than that of ClO4−, (thus approaching the accuracy limit of 1H NMR spectroscopy), we turned to Isothermal Titration Calorimetry (ITC).
Titrating Cl@2 with the bisulfate anion produced an isotherm that was fitted to a multiple-sites model. Values for the binding sites (n1 = 1; n2 = 1) confirmed the expected 1:2 host–guest stoichiometry (Fig. 6a and b). Data fitting yielded anion-exchange equilibrium constants of K1 = 3.3 × 106 M−1 and K2 = 3.1 × 104 M−1 which indeed is an order of magnitude higher than ClO4− and ReO4−. The thermodynamic data revealed that anion exchange at both sites of Cl@2 is enthalpically and entropically driven (Fig. 6c). The binding of HSO4− was also observed for Br@2, although unsurprisingly, the binding constants were an order of magnitude lower compared to Cl@2 (Table 1). For Br@2, the exchange of the second BF4− anion for HSO4− appears to be somewhat entropically disfavoured, suggesting the host may lose degrees of freedom (presumably manifested by the flexible bis-pyrazole ligand) when accommodating the larger HSO4− anion. Given that K2 is ∼100 and ∼350 times smaller than K1 for Cl@2 and Br@2 respectively, the host–guest binding occurs with negative cooperativity. Interestingly, both interlocked cages showed no binding affinity toward dihydrogen phosphate (H2PO4−), which is remarkable given it is also a tetrahedral anion albeit with a marginally higher volume than HSO4− (63.1 Å3vs. 58.9 Å3, Fig. S33 and S50†). This suggests that the anion volume plays an important role in determining the cooperativity of this semi-flexible interlocked host. As observed in the X-ray structure of (ClO4)2Cl@2, the exchange of BF4− for the larger ClO4− guest results in a 0.16 Å compression along the Pd4 axis (Fig. S75†), which may reduce the available volume in the second outer cavity after the first binding event. This effect is likely to be amplified for anions approaching a volume of ∼60 Å3 which can explain the negative cooperativity in the case of HSO4−.
 | ||
| Fig. 6 (a) Heat flow produced upon titrating a MeCN solution of Cl@2 with tetrabutylammonium bisulfate; (b) binding isotherm according to the cumulative heat of injection and equivalents of the guest; (c) thermodynamic parameters of guest exchange (BF4− for HSO4−) (d) 1H NMR spectra (600 MHz, CD3CN, 25 °C) of a competition experiment: (i) Cl@2; (ii) Cl@2 + 2 equiv. of ClO4−; (iii) Cl@2 + 2 equiv. of ClO4− and HSO4−; (iv) Cl@2 + 2 equiv. of HSO4−. | ||
Finally, we performed competition experiments to assess the selectivity of X@2 for HSO4− over ClO4− and ReO4−. Upon titrating two equivalents of ClO4− and HSO4− to Cl@2, diagnostic downfield shifts of proton g indicated that the (HSO4)2Cl@2 complex dominates the mixture (Fig. 6d and S58†). In this experiment, the presence of ClO4− resulted only in the shifting of proton f, which points outside the cage cavity (Fig. 6d). In nearly all cases, the competing ClO4− or ReO4− complexes could not be detected by 1H NMR spectroscopy (Fig. S58–S61†). This is fully consistent with the higher binding strength of HSO4− and further confirms that the interlocked cage is a highly selective receptor for HSO4−. The stability of the HSO4− host–guest complex may relate to the guest's optimal size as well as the possibility of forming O–H⋯π interactions between the acidic proton of the guest and the electron-rich pyrazole rings of the host.52,53
Conclusions
In summary, we have reported the anion-templated assembly of a novel interlocked coordination cage with two outer pockets poised to bind HSO4− anions selectively. First, we showed that a thermodynamically stable monomeric cage composed of a non-coordinating bpm backbone could be isolated through appropriate solvent choice and stoichiometric control. The intermediate dimensions of this Pd2L4 cage (as defined by the ligand's DN–N of ∼15 Å) facilitated the formation of a rare type II interlocked dimer in the presence of Cl− or Br− templates, and BF4− counter ions. The self-assembly experiments with NO3− counterions further highlighted the importance of counter ion volume in accessing type I or type II motifs. Whilst this is only one more piece of the puzzle to understand the anion–cation patterns of catenation, it may open up opportunities to design and access a greater range of multi-cavity cage structures with guest-accessible cavities.Comparing the host–guest chemistry between the Br− and Cl− templated dimer revealed large differences in anion binding strength (up to an order of magnitude in favor of Cl@2), despite the small difference in the volume of the templating anion. These findings may be relevant for tuning cavity dimensions and thus enhancing guest binding selectivity in this class of interlocked dimers. Finally, ITC and 1H NMR experiments revealed strong and selective binding of HSO4− by X@2, outcompeting similarly sized anions in binary mixtures. These results may inspire future studies on interlocked hosts for biphasic anion separation systems51 or selective ion channels.54,55
Data availability
The data supporting this article have been included as part of the ESI.†Author contributions
J. I. V. conducted the NMR analysis and host–guest studies. S. T. conducted ligand synthesis and contributed to data analysis. M. R. J. aided in 2D NMR measurements and performed EXSY analysis. W. M. B. conceived the idea, solved and refined the X-ray structures, performed ITC analysis, and wrote the manuscript. All authors commented and provided input into the final version of the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
W. M. B gratefully acknowledges Flinders University EMCR start-up grant and the Australian Research Council (DE190100327) for financially supporting this project. Aspects of this research were undertaken on the MX1 (ref. 56) and MX2 (ref. 57) beamlines at the Australian Synchrotron, Victoria, Australia. J. I. V. acknowledges support from an ATSE Elevate PhD Scholarship.Notes and references
- G. Gil-Ramírez, D. A. Leigh and A. J. Stephens, Angew. Chem., Int. Ed., 2015, 54, 6110–6150 CrossRef.
- R. Zhu, J. Ding, L. Jin and H. Pang, Coord. Chem. Rev., 2019, 389, 119–140 CrossRef CAS.
- S. Mena-Hernando and E. M. Pérez, Chem. Soc. Rev., 2019, 48, 5016–5032 RSC.
- S. Freye, J. Hey, A. Torras-Galán, D. Stalke, R. Herbst-Irmer, M. John and G. H. Clever, Angew. Chem., Int. Ed., 2012, 51, 2191–2194 CrossRef CAS PubMed.
- S. La Cognata, A. Miljkovic, R. Mobili, G. Bergamaschi and V. Amendola, Chempluschem, 2020, 85, 1145–1155 CrossRef CAS PubMed.
- A. Bessaguet, Q. Blancart-Remaury, P. Poinot, I. Opalinski and S. Papot, Angew. Chem., Int. Ed., 2023, 62, e202216787 CrossRef CAS PubMed.
- H.-Y. Zhou, Y. Han and C.-F. Chen, Mater. Chem. Front., 2020, 4, 12–28 RSC.
- Y. Li, H. Jiang, W. Zhang, X. Zhao, M. Sun, Y. Cui and Y. Liu, J. Am. Chem. Soc., 2024, 146, 3147–3159 CrossRef CAS PubMed.
- S. Pullen, S. Löffler, A. Platzek, J. J. Holstein and G. H. Clever, Dalton Trans., 2020, 49, 9404–9410 RSC.
- A. Garci, J. A. Weber, R. M. Young, M. Kazem-Rostami, M. Ovalle, Y. Beldjoudi, A. Atilgan, Y. J. Bae, W. Liu, L. O. Jones, C. L. Stern, G. C. Schatz, O. K. Farha, M. R. Wasielewski and J. Fraser Stoddart, Nat. Catal., 2022, 5, 524–533 CrossRef CAS.
- F. J. Rizzuto, W. J. Ramsay and J. R. Nitschke, J. Am. Chem. Soc., 2018, 140, 11502–11509 CrossRef CAS PubMed.
- A. Saura-Sanmartin and C. A. Schalley, Chem, 2023, 9, 823–846 CAS.
- Y. Lu, H.-N. Zhang and G.-X. Jin, Acc. Chem. Res., 2018, 51, 2148–2158 CrossRef CAS PubMed.
- S. D. P. Fielden, D. A. Leigh and S. L. Woltering, Angew. Chem., Int. Ed., 2017, 56, 11166–11194 CrossRef CAS PubMed.
- M. Denis and S. M. Goldup, Nat. Rev. Chem., 2017, 1, 61 CrossRef CAS.
- M. Han, D. M. Engelhard and G. H. Clever, Chem. Soc. Rev., 2014, 43, 1848–1860 RSC.
- J. E. M. Lewis, Chem. Commun., 2022, 58, 13873–13886 RSC.
- M. Frank, M. D. Johnstone and G. H. Clever, Chem.–Eur. J., 2016, 22, 14104–14125 CrossRef CAS PubMed.
- W. M. Bloch, J. J. Holstein, B. Dittrich, W. Hiller and G. H. Clever, Angew. Chem., Int. Ed., 2018, 57, 5534–5538 CrossRef CAS.
- F. J. Rizzuto, L. K. S. von Krbek and J. R. Nitschke, Nat. Rev. Chem., 2019, 3, 204–222 CrossRef.
- D. Preston, J. E. M. Lewis and J. D. Crowley, J. Am. Chem. Soc., 2017, 139, 2379–2386 CrossRef CAS PubMed.
- R. A. S. Vasdev, D. Preston and J. D. Crowley, Chem.–Asian J., 2017, 12, 2513–2523 CrossRef CAS PubMed.
- K. Li, Z. Li, J. Yuan, M. Chen, H. Zhao, Z. Jiang, J. Wang, Z. Jiang, Y. Li, Y.-T. Chan, P. Wang and D. Liu, Chem. Sci., 2024, 15, 8913–8921 RSC.
- S. S. Mishra, S. Krishnaswamy and D. K. Chand, J. Am. Chem. Soc., 2024, 146, 4473–4488 CrossRef CAS PubMed.
- K. Yazaki, M. Akita, S. Prusty, D. K. Chand, T. Kikuchi, H. Sato and M. Yoshizawa, Nat. Commun., 2017, 8, 15914 CrossRef CAS PubMed.
- Y. Yamauchi, M. Yoshizawa and M. Fujita, J. Am. Chem. Soc., 2008, 130, 5832–5833 CrossRef CAS PubMed.
- G. H. Clever and P. Punt, Acc. Chem. Res., 2017, 50, 2233–2243 CrossRef CAS PubMed.
- R. Sekiya, M. Fukuda and R. Kuroda, J. Am. Chem. Soc., 2012, 134, 10987–10997 CrossRef CAS PubMed.
- Y.-H. Li, J.-J. Jiang, Y.-Z. Fan, Z.-W. Wei, C.-X. Chen, H.-J. Yu, S.-P. Zheng, D. Fenske, C.-Y. Su and M. Barboiu, Chem. Commun., 2016, 52, 8745–8748 RSC.
- S. M. Jansze, M. D. Wise, A. V Vologzhanina, R. Scopelliti and K. Severin, Chem. Sci., 2017, 8, 1901–1908 RSC.
- S. Löffler, J. Lübben, L. Krause, D. Stalke, B. Dittrich and G. H. Clever, J. Am. Chem. Soc., 2015, 137, 1060–1063 CrossRef PubMed.
- R. Zhu, J. Lübben, B. Dittrich and G. H. Clever, Angew. Chem., Int. Ed., 2015, 54, 2796–2800 CrossRef CAS PubMed.
- R. Zhu, I. Regeni, J. J. Holstein, B. Dittrich, M. Simon, S. Prévost, M. Gradzielski and G. H. Clever, Angew. Chem., Int. Ed., 2018, 57, 13652–13656 CrossRef CAS PubMed.
- S. Freye, R. Michel, D. Stalke, M. Pawliczek, H. Frauendorf and G. H. Clever, J. Am. Chem. Soc., 2013, 135, 8476–8479 CrossRef CAS PubMed.
- M. Frank, L. Krause, R. Herbst-Irmer, D. Stalke and G. H. Clever, Dalton Trans., 2014, 43, 4587–4592 RSC.
- D. Luo, B. Pan, J. Zhang, C. Ma, Y. Su and Q. Gan, Chin. Chem. Lett., 2021, 32, 1397–1399 CrossRef CAS.
- M. Frank, J. M. Dieterich, S. Freye, R. A. Mata and G. H. Clever, Dalton Trans., 2013, 42, 15906–15910 RSC.
- M. Fukuda, R. Sekiya and R. Kuroda, Angew. Chem., Int. Ed., 2008, 47, 706–710 CrossRef CAS.
- R. Zhu, W. M. Bloch, J. J. Holstein, S. Mandal, L. V Schäfer and G. H. Clever, Chem.–Eur. J., 2018, 24, 12976–12982 CrossRef CAS PubMed.
- S. Paul, T. Majumdar and A. Mallick, Dalton Trans., 2021, 50, 1531–1549 RSC.
- A. S. Potapov and A. I. Khlebnikov, Polyhedron, 2006, 25, 2683–2690 CrossRef CAS.
- W. M. Bloch, A. Burgun, C. J. Coghlan, R. Lee, M. L. Coote, C. J. Doonan and C. J. Sumby, Nat. Chem., 2014, 6, 906–912 CrossRef CAS PubMed.
- M. T. Yong, O. M. Linder-Patton and W. M. Bloch, Inorg. Chem., 2022, 61, 12863–12869 CrossRef CAS.
- G. Durá, M. C. Carrión, F. A. Jalón, B. R. Manzano and A. M. Rodríguez, Eur. J. Inorg. Chem., 2015, 2015, 5874–5885 CrossRef.
- J. E. M. Lewis, A. B. S. Elliott, C. J. McAdam, K. C. Gordon and J. D. Crowley, Chem. Sci., 2014, 5, 1833–1843 RSC.
- W. M. Bloch, S. Horiuchi, J. J. Holstein, C. Drechsler, A. Wuttke, W. Hiller, R. A. Mata and G. H. Clever, Chem. Sci., 2023, 14, 1524–1531 RSC.
- A. P. Birvé, H. D. Patel, J. R. Price, W. M. Bloch and T. Fallon, Angew. Chem., Int. Ed., 2022, 61, e202115468 CrossRef PubMed.
- P. Thordarson, Chem. Soc. Rev., 2011, 40, 1305–1323 RSC.
- H. T. Ngo, X. Liu and K. A. Jolliffe, Chem. Soc. Rev., 2012, 41, 4928–4965 RSC.
- N. Busschaert, C. Caltagirone, W. Van Rossom and P. A. Gale, Chem. Rev., 2015, 115, 8038–8155 CrossRef CAS PubMed.
- C.-L. Deng, J. P. Bard, J. A. Lohrman, J. E. Barker, L. N. Zakharov, D. W. Johnson and M. M. Haley, Angew. Chem., Int. Ed., 2019, 58, 3934–3938 CrossRef CAS PubMed.
- M. Sandro and J. J. Rebek, Chem.–Eur. J., 1998, 4, 1016–1022 CrossRef.
- M. Kazim, L. Guan, A. Chopra, R. Sun, M. A. Siegler and T. Lectka, J. Org. Chem., 2020, 85, 9801–9807 CrossRef CAS PubMed.
- B. P. Benke, P. Aich, Y. Kim, K. L. Kim, M. R. Rohman, S. Hong, I.-C. Hwang, E. H. Lee, J. H. Roh and K. Kim, J. Am. Chem. Soc., 2017, 139, 7432–7435 CrossRef CAS PubMed.
- E. G. Percástegui, Chem. Commun., 2022, 58, 5055–5071 RSC.
- N. P. Cowieson, D. Aragao, M. Clift, D. J. Ericsson, C. Gee, S. J. Harrop, N. Mudie, S. Panjikar, J. R. Price, A. Riboldi-Tunnicliffe, R. Williamson and T. Caradoc-Davies, J. Synchrotron Radiat., 2015, 22, 187–190 CrossRef CAS PubMed.
- D. Aragao, J. Aishima, H. Cherukuvada, R. Clarken, M. Clift, N. P. Cowieson, D. J. Ericsson, C. L. Gee, S. Macedo, N. Mudie, S. Panjikar, J. R. Price, A. Riboldi-Tunnicliffe, R. Rostan, R. Williamson and T. T. Caradoc-Davies, J. Synchrotron Radiat., 2018, 25, 885–891 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. CCDC 2372761–2372764 and 2386148. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d4sc04913f |
| ‡ The 1H NMR spectra indicated that protonation of the dimeric cages does not occur when bisulfate is added; the cage remained stable at higher equivalents (judged by no free ligand being released) and the pyrazole protons (b and c) remained relatively sharp after the addition of >2 equivalents of HSO4− to Cl@2 and Br@2 (Fig. S29 and S47†). Moreover, titrating HSO4− in a CD3CN solution of L did not result in protonation of the ligand (Fig. S56†). |
| This journal is © The Royal Society of Chemistry 2024 |