
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
The carbon–iodine bond cleavage and isomerization of iodoform visualized with femtosecond X-ray liquidography†
Yongjun
Cha
 ab,
Hosung
Ki
ab,
Donghwan
Im
ab,
Yunbeom
Lee
ab,
Seonggon
Lee
ab,
Jungmin
Kim
ab,
Jae Hyuk
Lee
c,
Jeongho
Kim
d and
Hyotcherl
Ihee
*ab
ab,
Hosung
Ki
ab,
Donghwan
Im
ab,
Yunbeom
Lee
ab,
Seonggon
Lee
ab,
Jungmin
Kim
ab,
Jae Hyuk
Lee
c,
Jeongho
Kim
d and
Hyotcherl
Ihee
*ab
aDepartment of Chemistry, Korea Advanced Institute of Science and Technology (KAIST), Daejeon 34141, Republic of Korea. E-mail: hyotcherl.ihee@kaist.ac.kr
bCenter for Advanced Reaction Dynamics (CARD), Institute for Basic Science (IBS), Daejeon 34141, Republic of Korea
cPohang Accelerator Laboratory, Pohang 37673, Republic of Korea
dDepartment of Chemistry, Inha University, 100 Inha-ro, Michuhol-gu, Incheon 22212, Republic of Korea
First published on 29th October 2024
Abstract
Iodoform (CHI3) has garnered significant attention for its unique ability to induce photo-cyclopropanation of olefins by releasing an iodine radical through C–I bond cleavage. However, the detailed mechanism underlying CHI3 photodissociation is still not fully understood. Here, we elucidate the ultrafast structural dynamics of CHI3 upon photoexcitation using femtosecond time-resolved X-ray liquidography (fs-TRXL) at an X-ray free-electron laser facility. The fs-TRXL data was decomposed into the isotropic and anisotropic data. The isotropic data reveal that the formation of CHI2 and I radicals upon photolysis precedes the emergence of iso-CHI2–I. After a short induction period, two competing geminate recombination pathways of CHI2 and I radicals take place: one pathway leads to the recovery of CHI3, while the other results in the formation of iso-CHI2–I. Additionally, the anisotropic data show how the transient anisotropic distribution of both the species formed upon photoexcitation and the ground-state species depleted upon photoexcitation decays through rotational dephasing. Furthermore, the observed structural dynamics of CHI3 has distinctive differences with that of BiI3, which can be attributed to differences in their central moieties, CH and Bi. Our findings provide insights into the photoinduced reaction dynamics of CHI3, enhancing the understanding of its role in photochemical reactions.
Introduction
Haloalkanes have long captured the attention of researchers due to their diverse applications, finding utility in areas that demand halogen radicals or anions.1 Their significance stems from their innate tendency to undergo carbon–halogen bond cleavage through ultraviolet photolysis, making them effective halogen-releasing agents. Among the haloalkanes, polyhalomethanes have garnered extensive studies, primarily due to their unique ability to recombine with released halogen radicals, resulting in the formation of iso-polyhalomethanes.2–6The generation of iso-polyhalomethanes can be categorized into two primary mechanisms: (1) solvent cage-mediated geminate recombination and (2) roaming reaction. Traditionally, iso-polyhalomethanes were believed to be exclusively formed via solvent cage-mediated geminate recombination. However, recent studies on CHBr3![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 7 and BiI3,8 a polyhalomethane and a polyhalomethane-like system, respectively, have unveiled a novel isomerization process mediated by roaming reactions. In particular, the occurrence of roaming-mediated isomer formation in <200 fs time domain in the photolysis pathways of BiI3 was revealed via femtosecond time-resolved X-ray liquidography (fs-TRXL).
7 and BiI3,8 a polyhalomethane and a polyhalomethane-like system, respectively, have unveiled a novel isomerization process mediated by roaming reactions. In particular, the occurrence of roaming-mediated isomer formation in <200 fs time domain in the photolysis pathways of BiI3 was revealed via femtosecond time-resolved X-ray liquidography (fs-TRXL).
CHI3, along with CH2I2, is a substance used in the photo-cyclopropanation of olefins and is representative of polyhalomethanes in research.9–11 As a triiodo-molecular system, CHI3 resembles BiI3, showing valence isoelectronicity, and serves as an appropriate comparative molecular system for observing isomerization mechanisms. Previous studies have revealed the existence of iso-CHI2–I during the photolysis process of CHI3 in cyclohexane.12–15 However, capturing the isomerization process of CHI3via time-resolved X-ray liquidography (TRXL)16–21 in third-generation synchrotrons posed challenges due to limited temporal resolution, typically limited to approximately 100 picoseconds.22 By this time point, both isomer and radical intermediates were already coexisting, and the initial processes leading to the formation of each intermediate could not be identified. Fortunately, the development of X-ray free-electron laser (XFEL) facilities enabled us to utilize femtosecond time resolution X-ray sources with more intense X-ray pulses. This breakthrough allowed us to directly observe the iodine dissociation process and the formation of the isomer in the sub-picosecond temporal regime (Fig. 1).
 | ||
| Fig. 1 Schematic diagrams for the photoinduced reaction dynamics of CHI3 in cyclohexane and BiI3 in acetonitrile. (a) The photoinduced reaction dynamics of CHI3 in cyclohexane. Two intermediates present after 100 ps had been revealed by a previously conducted TRXL experiment at a synchrotron facility. Three major questions that can be answered by conducting an fs-TRXL experiment at an XFEL facility are: (1) whether roaming reaction occurs, (2) whether geminate recombination occurs, and (3) whether there are any other isomers. (b) The photoinduced reaction dynamics of BiI3 in acetonitrile. BiI3 shows distinct three pathways: (1) roaming-mediated early isomer formation, (2) geminate recombination of radicals, and (3) late isomer formation via relaxation of early isomer. In both (a) and (b), all chemical species are illustrated with colored spheres representing each element: white (H), grey (C), purple (I), and green (Bi). | ||
In this study, we present the results of our investigation into the photoinduced reaction dynamics of CHI3 in cyclohexane using femtosecond time-resolved X-ray liquidography (fs-TRXL). Specifically, we focus on the ultrafast structural dynamics during C–I bond cleavage and the formation of iso-CHI2–I through geminate recombination of radical species. We also analyzed the anisotropic signal component generated by the photoselective excitation caused by linearly polarized pump pulses, resolving its decay kinetics and structural origin. Furthermore, we draw comparisons with BiI3, which exhibits roaming-mediated formation of an early isomer. Despite the similarities in structure and number of valence electrons, the photolysis of CHI3, unlike that of BiI3, did not result in the roaming-mediated formation of an early isomer. This comparison highlights significant differences in the photoinduced reaction dynamics of CHI3 and BiI3, which are likely attributed to differences in the properties of the central moieties, carbon hydride and bismuth.
Results and discussion
Experimental data, ΔS(q, t) of fs-TRXL
We obtained the difference scattering patterns of photoexcited CHI3 in cyclohexane for various time delays. The obtained difference scattering patterns are decomposed into isotropic (ΔS0(q, t)) and anisotropic (ΔS2(q, t)) components via an established method.23,24 The isotropic signal components are further processed via the projection to extract the perpendicular component (PEPC)25 method to remove the kinetic contributions of solvent heating and experimental artifacts arising from the fluctuation of experimental conditions, such as the fluctuation in the thickness of sample and X-ray intensity. The resulting PEPC-treated signal is denoted as ΔS⊥0(q, t), where the symbol ⊥ indicates PEPC-treated data (see ESI† for details). qΔS⊥0(q, t), ΔS⊥0(q, t) multiplied by q to emphasize the signal in high-q region, is depicted in Fig. 2a as a contour plot. For the time delays less than 500 fs, a sudden rise and peak shift are observed, indicating the ultrafast structural dynamics during C–I bond cleavage. These changes are well-resolved with the temporal resolution of the experiment, which is represented by the instrument response function (IRF) and determined to be approximately 180 fs (180 ± 7 fs). After t = 500 fs, slow and smooth changes in signals are observed, suggesting that the initially formed intermediates undergo further reactions. | ||
| Fig. 2 Isotropic fs-TRXL signals of CHI3 with theoretical fit results, and the corresponding concentration profiles and kinetic model. (a) Experimental data qΔS⊥0(q, t) plotted in a contour map with time (ps) and q (Å−1) as x- and y-axis, respectively. (b) Corresponding theoretical fit results derived from the structural analysis and the exponential kinetics model. (c) Residual obtained by subtracting theoretical fit results from the corresponding experimental data. All panels share a common color scale representing the amplitude of the signal in arbitrary unit on the very right side. (d) The concentration profiles of the intermediate species. (e) Kinetic model of CHI3 photodissociation process. A more comprehensive version is in Fig. S7.† The structural refinement was applied for the early time period (t ≤ 500 fs), and the LCF was used for the later time period (t > 500 fs). The results from these two approaches are stitched together and shown in (b). In (d), the plots are presented in a linear time scale up to 1.8 ps, then are presented in a logarithmic time scale extending to 100 ps. The kinetic model fit results (solid lines) well describe the linear combination fit results (dots with one-standard-deviation error bars). The decrease in both total and radical concentrations is clearly shown in (d). | ||
Overall reaction pathways and kinetics of photoexcited CHI3
First, we examined whether the data can be well described by only the two intermediates ( and iso-CHI2–I) that were reported to be populated at time delays longer than 100 ps via a TRXL study at a synchrotron.22 For this purpose, we performed linear combination fitting (LCF) analysis, where the data at each time delay is fitted with a linear combination of basis components. Specifically, species-associated difference scattering curves (SADSs) corresponding to the two intermediates were PEPC-treated in the same manner as the experimental data. The PEPC-treated SADSs were then used to fit the experimental data via linear combination. The solute structures used to calculate the SADSs, namely those for the species CHI3, iso-CHI2–I, and CHI2, were adapted from experimentally refined structures reported in the previous study.22 If the data is well described by the linear combination of the known solute components, it confirms that no other intermediates beyond the known ones contribute to the reaction pathway. Conversely, if the fit is unsatisfactory, it indicates the presence of additional short-lived intermediates significantly contributing to the signal, beyond the known intermediates. The results of the LCF analysis are presented in Fig. S1.† It is observed that the data after 500 fs time delay is well described by a combination of signals corresponding to the two intermediates. This confirms that, at least after the 500 fs time delay, there are no additional short-lived intermediates.
and iso-CHI2–I) that were reported to be populated at time delays longer than 100 ps via a TRXL study at a synchrotron.22 For this purpose, we performed linear combination fitting (LCF) analysis, where the data at each time delay is fitted with a linear combination of basis components. Specifically, species-associated difference scattering curves (SADSs) corresponding to the two intermediates were PEPC-treated in the same manner as the experimental data. The PEPC-treated SADSs were then used to fit the experimental data via linear combination. The solute structures used to calculate the SADSs, namely those for the species CHI3, iso-CHI2–I, and CHI2, were adapted from experimentally refined structures reported in the previous study.22 If the data is well described by the linear combination of the known solute components, it confirms that no other intermediates beyond the known ones contribute to the reaction pathway. Conversely, if the fit is unsatisfactory, it indicates the presence of additional short-lived intermediates significantly contributing to the signal, beyond the known intermediates. The results of the LCF analysis are presented in Fig. S1.† It is observed that the data after 500 fs time delay is well described by a combination of signals corresponding to the two intermediates. This confirms that, at least after the 500 fs time delay, there are no additional short-lived intermediates.
Through the LCF analysis, we extracted how the contributions of  and iso-CHI2–I vary over time, revealing the concentration changes of each species. This information is depicted in Fig. 2d (empty circles). The following insights can be gleaned upon examination of the data. At early time points (<1.5 ps), radicals
and iso-CHI2–I vary over time, revealing the concentration changes of each species. This information is depicted in Fig. 2d (empty circles). The following insights can be gleaned upon examination of the data. At early time points (<1.5 ps), radicals  are formed first. During this period, the contribution of radicals is dominant, while the contribution of the isomer is negligible. Subsequently, between 2 ps and 40 ps, a decrease in the concentration of
are formed first. During this period, the contribution of radicals is dominant, while the contribution of the isomer is negligible. Subsequently, between 2 ps and 40 ps, a decrease in the concentration of  is observed, accompanied by an increase in the concentration of iso-CHI2–I. It is apparent that the time delay of 40 ps is too early for
is observed, accompanied by an increase in the concentration of iso-CHI2–I. It is apparent that the time delay of 40 ps is too early for  and I˙ to undergo a non-geminate bimolecular recombination.26–28 Therefore, it can be assigned that during this time domain,
and I˙ to undergo a non-geminate bimolecular recombination.26–28 Therefore, it can be assigned that during this time domain, 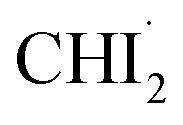 and I˙ radicals geminately recombined. An important observation here is that the molar amount of
and I˙ radicals geminately recombined. An important observation here is that the molar amount of  reduced during this time range is not equivalent to the molar amount of generated iso-CHI2–I. In Fig. 2d, it is evident that the sum of the concentrations of
reduced during this time range is not equivalent to the molar amount of generated iso-CHI2–I. In Fig. 2d, it is evident that the sum of the concentrations of 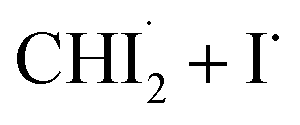 and iso-CHI2–I, plotted alongside the concentrations of each species, decreases. This result indicates that not all
and iso-CHI2–I, plotted alongside the concentrations of each species, decreases. This result indicates that not all  are converted into iso-CHI2–I; only a certain fraction undergoes conversion, while the remaining portion either recovers into the parent species, CHI3, or remains unchanged. As the time domain of ∼40 ps is too early for a non-geminate recombination, as mentioned earlier, the corresponding recombination of
are converted into iso-CHI2–I; only a certain fraction undergoes conversion, while the remaining portion either recovers into the parent species, CHI3, or remains unchanged. As the time domain of ∼40 ps is too early for a non-geminate recombination, as mentioned earlier, the corresponding recombination of  and I˙ to CHI3 should also be a geminate recombination process.
and I˙ to CHI3 should also be a geminate recombination process.
In brief, the concentration changes of each species, as determined through the LCF analysis, indicate the presence of the following two reaction pathways: (1)  and (2)
and (2)  . Before initiating the two pathways,
. Before initiating the two pathways,  undergoes a concentration-maintaining period referred to as an induction period. In this phase, the dissociated I radical remains unreactive until it undergoes geminate recombination. Fig. S2† illustrates the comparison between kinetic models with and without the induction period. Incorporating the induction period into the kinetic model yields time constants of (1) 14.5 ps and (2) 26.0 ps, providing a more accurate depiction of the experimental data than that without the induction period. In contrast, when the induction period is ignored, the kinetic model extends the time constants to account for the persistent nature of radical concentrations in the early time domain (<1.8 ps), leading to much larger time constants of 22.6 ps and 39.3 ps for the pathways (1) and (2), respectively. The absence of the induction period in the kinetic model exacerbates discrepancies, particularly in the 1.8 ps to 10 ps region (see Fig. S2c and d†). Based on these, we established the optimal kinetic model for the photoinduced reaction dynamics of CHI3, comprising (1) the amount of initially generated radicals from photoexcited CHI3, (2) the induction period of radicals, (3) the fraction of radicals involved in secondary geminate recombination, and (4, 5) two time constants τ1 and τ2 for each pathway described earlier, in total of five parameters.
undergoes a concentration-maintaining period referred to as an induction period. In this phase, the dissociated I radical remains unreactive until it undergoes geminate recombination. Fig. S2† illustrates the comparison between kinetic models with and without the induction period. Incorporating the induction period into the kinetic model yields time constants of (1) 14.5 ps and (2) 26.0 ps, providing a more accurate depiction of the experimental data than that without the induction period. In contrast, when the induction period is ignored, the kinetic model extends the time constants to account for the persistent nature of radical concentrations in the early time domain (<1.8 ps), leading to much larger time constants of 22.6 ps and 39.3 ps for the pathways (1) and (2), respectively. The absence of the induction period in the kinetic model exacerbates discrepancies, particularly in the 1.8 ps to 10 ps region (see Fig. S2c and d†). Based on these, we established the optimal kinetic model for the photoinduced reaction dynamics of CHI3, comprising (1) the amount of initially generated radicals from photoexcited CHI3, (2) the induction period of radicals, (3) the fraction of radicals involved in secondary geminate recombination, and (4, 5) two time constants τ1 and τ2 for each pathway described earlier, in total of five parameters.
In the best-fit results, 5.2 ± 0.6 mM of CHI3 out of 20 mM is initially excited by the pump pulse. Of this, 1.6 ± 0.2 mM of CHI3 relaxes back to the ground state, while the remaining 3.6 ± 0.4 mM undergoes C–I bond cleavage, generating 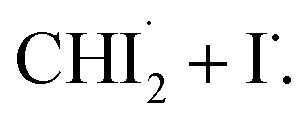 Among the initially generated 3.6 mM of
Among the initially generated 3.6 mM of  51 ± 1.1% (1.8 mM) undergo geminate recombination after a 1.8 ps of induction period, while the remaining 49 ± 1.1% (1.8 mM) persist unchanged until 100 ps. The reactive radicals (mentioned earlier, the 51%) are further subdivided, with 64 ± 2.3% (1.2 mM) undergoing isomerization to produce iso-CHI2–I with a time constant, τ1 = 14.5 ± 1.0 ps, and the remaining 36 ± 2.3% (0.65 mM) relaxing back to their parent molecule, CHI3, with a time constant, τ2 = 26.0 ± 1.9 ps. The ratio of recombination to the isomer and to the parent molecule, 64:36, can be further supported by employing the random collision model between
51 ± 1.1% (1.8 mM) undergo geminate recombination after a 1.8 ps of induction period, while the remaining 49 ± 1.1% (1.8 mM) persist unchanged until 100 ps. The reactive radicals (mentioned earlier, the 51%) are further subdivided, with 64 ± 2.3% (1.2 mM) undergoing isomerization to produce iso-CHI2–I with a time constant, τ1 = 14.5 ± 1.0 ps, and the remaining 36 ± 2.3% (0.65 mM) relaxing back to their parent molecule, CHI3, with a time constant, τ2 = 26.0 ± 1.9 ps. The ratio of recombination to the isomer and to the parent molecule, 64:36, can be further supported by employing the random collision model between  and I˙. In the model, the collision of
and I˙. In the model, the collision of  and
and 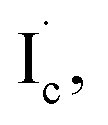 where the subscript c is used to indicate the dissociated I atom among three I atoms of CHI3, can result in the production of iso-CHI2–I with two targets (Ia and Ib, two iodine atoms in
where the subscript c is used to indicate the dissociated I atom among three I atoms of CHI3, can result in the production of iso-CHI2–I with two targets (Ia and Ib, two iodine atoms in  ) or CHI3 with one target (the carbon atom), in total of three targets (Ia, Ib, and C). The ratio between the newly formed iso-CHI2–I vs. CHI3 in this simplified model is 2:1 = 67:33, which is almost identical to the one obtained from the kinetic model (64:36). The two time constants, τ1 = 14.5 ps and τ2 = 26.0 ps, yield an apparent time constant, τ = 9.3 ± 1.1 ps, which aligns closely with the time constant, τ = 9.6 ± 0.6 ps, obtained from the results of the SVD analysis (see Fig. S3d†). Here, an “apparent” time constant refers to a simplified kinetic behavior that is observed in the data and represents the combined effect of two independent processes, each with distinct time constants τ1 and τ2. When these two processes contribute simultaneously to the observed kinetics, the apparent time constant can be calculated using the relationship: 1/τ = 1/τ1 + 1/τ2. This equation is derived from the principle that the overall rate of change (1/τ) is the sum of the rates (1/τ1 and 1/τ2) of the two independent processes. The combined result of these processes can be expressed through the apparent time constant, which provides a simplified but meaningful representation of the system's overall kinetics.
) or CHI3 with one target (the carbon atom), in total of three targets (Ia, Ib, and C). The ratio between the newly formed iso-CHI2–I vs. CHI3 in this simplified model is 2:1 = 67:33, which is almost identical to the one obtained from the kinetic model (64:36). The two time constants, τ1 = 14.5 ps and τ2 = 26.0 ps, yield an apparent time constant, τ = 9.3 ± 1.1 ps, which aligns closely with the time constant, τ = 9.6 ± 0.6 ps, obtained from the results of the SVD analysis (see Fig. S3d†). Here, an “apparent” time constant refers to a simplified kinetic behavior that is observed in the data and represents the combined effect of two independent processes, each with distinct time constants τ1 and τ2. When these two processes contribute simultaneously to the observed kinetics, the apparent time constant can be calculated using the relationship: 1/τ = 1/τ1 + 1/τ2. This equation is derived from the principle that the overall rate of change (1/τ) is the sum of the rates (1/τ1 and 1/τ2) of the two independent processes. The combined result of these processes can be expressed through the apparent time constant, which provides a simplified but meaningful representation of the system's overall kinetics.
The unchanged, remaining radicals (the 49%) undergo slow, non-geminate recombination after 100 ps, as already revealed in the previous TRXL study. The ratio between the reactive radicals and the remaining radicals in CHI3 (51:49) is similar to that observed in other triiodides, I3− (58:42)29 and BiI3 (60:40),8 but slightly smaller. The ratio is influenced by the characteristics of the solvent and solute system; future experiments in different solvent systems, such as CHI3 in methanol, could provide further confirmation.
The kinetic analysis of the TRXL data shows that the recombination of CHI2 and I radicals occurs over a timescale of several to tens of picoseconds, with specific time constants of 14.5 ps and 26.0 ps. Based on these time constants, we conclude that this can be interpreted as geminate recombination. More specifically, when compared to other systems containing two or three iodine atoms, such as HgI230 and I3−,31 this process likely corresponds to what is described as secondary geminate recombination in studies reporting the kinetics of those systems. In this process, geminate radicals that escaped the first solvation shell but remained in close proximity eventually recombine. While the lifetime of this radical pair may appear unusually long, similar instances of radical pairs remaining in close proximity for tens to hundreds of picoseconds have been reported in other systems, such as CH2I2.32
It should be noted that the accurate reaction pathways could be determined because the relative concentrations of both species were quantitatively determined via TRXL. Achieving such quantitative determination in time-resolved spectroscopic measurements is not trivial. In contrast, TRXL enables these quantitative measurements thanks to the quantitative nature of its signal. In other words, the TRXL signal—from any species—can be accurately simulated, capturing not only the shape but also the precise, absolute amplitude. This quantitative nature of the TRXL signal allows for precise determination of the relative branching ratios of any two species of interest (see Fig. S10†). Such quantitative determination via time-resolved spectroscopic signals requires prior knowledge of accurate transition cross-sections for the species of interest. Since such information is not readily available, it is generally estimated through quantum or TD-DFT calculations. However, the oscillator strengths derived from these calculations are often not as accurate as the scattering signals, which can be easily calculated from even roughly predicted molecular structures. This significant advantage of scattering over spectroscopy is frequently overlooked.
Ultrafast structural dynamics of photoexcited CHI3 during early time domain
For the data at time delays earlier than 500 fs, however, the results of the LCF analysis do not match well with the experimental data. This discrepancy is evident in the residuals shown in Fig. S1c.† The shape of the residual continuously changes in q-space over time. If the residual originated from a novel, short-lived intermediate species, we would expect the residuals to have a consistent shape, with the amplitude of the residual transiently increasing and decreasing. However, the residuals from the LCF analysis do not exhibit such behavior. This suggests that the residual likely does not originate from the formation and decay of an unknown species, besides and iso-CHI2–I, having a fixed molecular structure, but rather from time-dependent, continuous changes of the molecular structures of the involved species, i.e., a specific wavepacket trajectory. To describe the wavepacket trajectory, we modeled the time-dependent changes of the key structural parameters involved in the process of C–I bond cleavage in CHI3, leading to the formation of
and iso-CHI2–I, having a fixed molecular structure, but rather from time-dependent, continuous changes of the molecular structures of the involved species, i.e., a specific wavepacket trajectory. To describe the wavepacket trajectory, we modeled the time-dependent changes of the key structural parameters involved in the process of C–I bond cleavage in CHI3, leading to the formation of  These key parameters were selected based on the sensitivity plot of CHI3. For detailed information, refer to Fig. S4 and ESI.† The resulting wavepacket trajectory, depicted through changes in the representative structural parameters over time delays, is shown in Fig. S5.† The result confirms that the origin of the residual is the ultrafast, continuous structural changes in photoexcited CHI3 occurring during C–I bond dissociation process in the early time domain.
These key parameters were selected based on the sensitivity plot of CHI3. For detailed information, refer to Fig. S4 and ESI.† The resulting wavepacket trajectory, depicted through changes in the representative structural parameters over time delays, is shown in Fig. S5.† The result confirms that the origin of the residual is the ultrafast, continuous structural changes in photoexcited CHI3 occurring during C–I bond dissociation process in the early time domain.
Comparative analysis of triiodine systems with and without roaming-mediated isomer formation
Polyhalomethanes and their relatives, including CHBr3 and BiI3, exhibit roaming-mediated isomer formation within 200 fs.7,8 Notably, BiI3, even though it is also one of the triiodides like CHI3, demonstrates significant differences compared to CHI3 in (1) the presence of early isomer formation through a roaming reaction and (2) the mechanism of (late) isomer generation. Specifically, photolysis of CHI3 exclusively yields radicals immediately after the dissociation of I, without involving a roaming reaction. In contrast, in the case of BiI3, both roaming-mediated early isomer formation and radical generation through the dissociation of I occur in a 50:50 ratio. The rapid formation of the early isomer through roaming reaction, within <200 fs, is known to be solvent-independent7 and relies on the interaction between the roaming iodine and the remaining fragment, representing a characteristic of the solute. Therefore, the presence or absence of roaming reaction is predicted to be based on the difference in the central moiety between CHI3 and BiI3.
radicals immediately after the dissociation of I, without involving a roaming reaction. In contrast, in the case of BiI3, both roaming-mediated early isomer formation and radical generation through the dissociation of I occur in a 50:50 ratio. The rapid formation of the early isomer through roaming reaction, within <200 fs, is known to be solvent-independent7 and relies on the interaction between the roaming iodine and the remaining fragment, representing a characteristic of the solute. Therefore, the presence or absence of roaming reaction is predicted to be based on the difference in the central moiety between CHI3 and BiI3.
For isomer formation, CHI3 exclusively undergoes secondary geminate recombination of radicals to form isomer, while BiI3 generates the late isomer through the relaxation of the early isomer only. Considering that both cases occur in the fast time regime of a few picoseconds, it is conceivable that the late isomer formation through secondary geminate recombination of radicals could also occur in the BiI3 system. However, the absence of such pathway in the previous study is attributed to differences in the central moiety (CH vs. Bi) and the solvent (cyclohexane vs. acetonitrile) between the two systems, leading to variations in the energy levels of radicals and (late) isomers.
Through further comparison of polyhalomethanes, we observed that halogen identity (for example, Br and I) plays a critical role in influencing roaming dynamics. CH2I2,32 like CHI3, contains iodine atoms and does not exhibit roaming-based isomer formation within ∼1 ps, similar to CHI3. The structural and behavioral similarities between these iodine-containing systems suggest that the heavier iodine atoms may hinder early-time isomer formation, in contrast to bromine-containing systems like CHBr3.7 This highlights the significant role that halogen atoms play in determining the dynamics of roaming and recombination in systems with similar central moieties.
Kinetics of anisotropic signals
As previously mentioned, we aimed to analyze the anisotropic signal isolated from the experimental data in a manner similar to the isotropic signal analysis to confirm the kinetics of the anisotropy. Fig. 3a presents a contour plot of qΔS2(q, t). For time delays less than 500 fs, the ultrafast structural dynamics during C–I bond cleavage induced a peak-shifting feature, as observed in the isotropic signal. This feature was well described by the theoretical anisotropic signal calculated for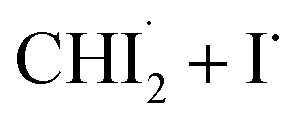 formation via C–I dissociation process, which aligns with the structural dynamics derived from the isotropic signal analysis. For time delays beyond 500 fs, a single major decaying component with a time constant of τ = 3.0 ± 0.2 ps was identified through an SVD analysis (see Fig. S6† and 3d). As described in the previous section, the time constants for the radicals returning to either the isomer or CHI3 are 14.5 ps and 26.0 ps, respectively, indicating a negligible amount of isomer formation around the 3 ps time range. Therefore, during the time period relevant to the observed anisotropic time constant, the primary contributors to the anisotropic signal are
formation via C–I dissociation process, which aligns with the structural dynamics derived from the isotropic signal analysis. For time delays beyond 500 fs, a single major decaying component with a time constant of τ = 3.0 ± 0.2 ps was identified through an SVD analysis (see Fig. S6† and 3d). As described in the previous section, the time constants for the radicals returning to either the isomer or CHI3 are 14.5 ps and 26.0 ps, respectively, indicating a negligible amount of isomer formation around the 3 ps time range. Therefore, during the time period relevant to the observed anisotropic time constant, the primary contributors to the anisotropic signal are  and depleted CHI3 (hole). Based on these considerations, the origin of the anisotropic signal can be explained as follows. Before irradiation, the transition dipole moments of CHI3 in its ground state are evenly distributed. Linearly polarized light selectively excites CHI3 molecules whose transition dipole moments align well with the polarization direction of the light, with the excitation probability proportional to cos2θ where θ is the angle between the laser polarization (E) and the transition dipole moment. As a result, the excited CHI3 and the radicals that subsequently form exhibit a specific directionality according to the transition dipole moment. After orientation-selective photoexcitation, the remaining unexcited CHI3 molecules exhibit an uneven distribution of transition dipole moments, as the molecules whose transition dipole moments matched the polarization direction of the light have been selectively excited, leaving an uneven distribution among the unexcited population. This leads to an uneven distribution of transition dipole moments in the ground-state holes and particles, which is depicted in Fig. 4g. Through subsequent rotational dephasing, the ground-state holes and particles gradually return to an even distribution, which is observed as a decaying anisotropic signal over several picoseconds. In this study, we based our fitting on the assumption that the anisotropic signal arises from ground-state holes and excited CHI3 particles, which subsequently dissociate into CHI2 and I radicals. We tested three possible models for the origin of the anisotropic signal: (1) 1:1 combination of radicals and ground-state holes, (2) radicals only, and (3) ground-state holes only. Among the models, the 1:1 combination model provided the best fit to the experimental data (Fig. 4). Fig. 3e illustrates the rotational dephasing dynamics of the CHI3 system during the photodissociation process. The decay time constants of rotational dephasing for the particle and hole, calculated under the assumption of ellipsoidal molecules,33 are 2.3 ps and 4.0 ps, respectively. These values are consistent with the experimental decay time constant of τ = 3.0 ± 0.2 ps, further supporting the result.
and depleted CHI3 (hole). Based on these considerations, the origin of the anisotropic signal can be explained as follows. Before irradiation, the transition dipole moments of CHI3 in its ground state are evenly distributed. Linearly polarized light selectively excites CHI3 molecules whose transition dipole moments align well with the polarization direction of the light, with the excitation probability proportional to cos2θ where θ is the angle between the laser polarization (E) and the transition dipole moment. As a result, the excited CHI3 and the radicals that subsequently form exhibit a specific directionality according to the transition dipole moment. After orientation-selective photoexcitation, the remaining unexcited CHI3 molecules exhibit an uneven distribution of transition dipole moments, as the molecules whose transition dipole moments matched the polarization direction of the light have been selectively excited, leaving an uneven distribution among the unexcited population. This leads to an uneven distribution of transition dipole moments in the ground-state holes and particles, which is depicted in Fig. 4g. Through subsequent rotational dephasing, the ground-state holes and particles gradually return to an even distribution, which is observed as a decaying anisotropic signal over several picoseconds. In this study, we based our fitting on the assumption that the anisotropic signal arises from ground-state holes and excited CHI3 particles, which subsequently dissociate into CHI2 and I radicals. We tested three possible models for the origin of the anisotropic signal: (1) 1:1 combination of radicals and ground-state holes, (2) radicals only, and (3) ground-state holes only. Among the models, the 1:1 combination model provided the best fit to the experimental data (Fig. 4). Fig. 3e illustrates the rotational dephasing dynamics of the CHI3 system during the photodissociation process. The decay time constants of rotational dephasing for the particle and hole, calculated under the assumption of ellipsoidal molecules,33 are 2.3 ps and 4.0 ps, respectively. These values are consistent with the experimental decay time constant of τ = 3.0 ± 0.2 ps, further supporting the result.
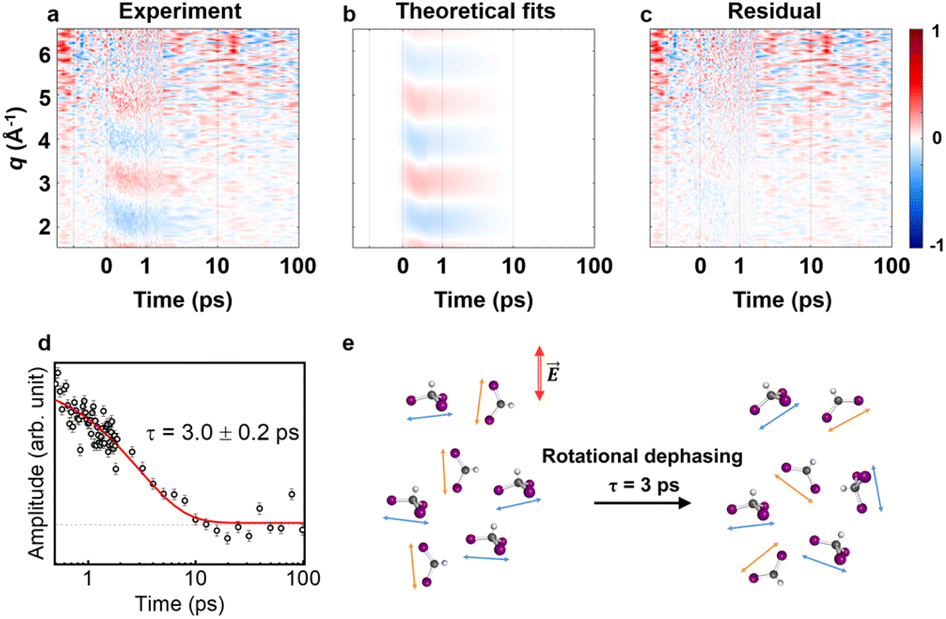 | ||
Fig. 3 Anisotropic signals aligned with theoretical fit results. (a) Experimental data qΔS2(q, t) plotted in a contour map with time (ps) and q (Å−1) as x- and y-axis, respectively. (b) Corresponding theoretical fit results derived from the structural analysis and the exponential kinetics model. (c) Residual obtained by subtracting theoretical fit results from the corresponding experimental data. All panels share a common color scale representing the amplitude of the signal in arbitrary unit on the very right side. (d) The first RSV was fitted by a single exponential function, giving τ = 3.0 ± 0.2 ps. (e) The rotational dephasing dynamics of CHI3 and 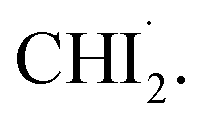 | ||
 | ||
Fig. 4 Contribution analysis of the anisotropic experimental data. (a–c) qΔS2(q) (black), corresponding to the time constant of 3.0 ps, compared to the theoretical anisotropic signals (red) derived from (a) 1:1 combination of radicals and ground-state holes, (b) radicals only, and (c) ground-state holes only. (d–f) Residuals of (a)–(c), respectively, obtained by subtracting theoretical anisotropic signals from corresponding qΔS2(q). The weighted R-factors (wR) are shown, with the relative ratios compared to the 1:1 combination model provided in the parentheses. The residual in (a) is the smallest, suggesting that both radicals and ground-state holes contribute to the anisotropic signals. (g) Schematic diagram illustrating the origin of the anisotropic signals and their kinetics. The blue and pale blue ellipses represent the distributions of the transition dipole moments of  and CHI3 (hole), respectively, which are perturbed by linearly polarized laser pulses depending on the angle (θ) between the laser polarization (E) and the transition dipole moment. The concentration distributions of and CHI3 (hole), respectively, which are perturbed by linearly polarized laser pulses depending on the angle (θ) between the laser polarization (E) and the transition dipole moment. The concentration distributions of  and CHI3 hole with respect to θ are plotted in black and red lines, respectively. The 1:1 combination model (a) encompasses both pathways, (i) rotational dephasing of the generated and CHI3 hole with respect to θ are plotted in black and red lines, respectively. The 1:1 combination model (a) encompasses both pathways, (i) rotational dephasing of the generated 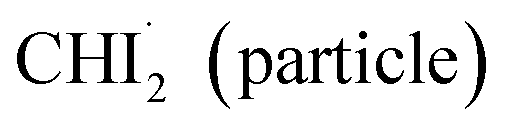 and (ii) rotational dephasing of depleted CHI3 (hole). The radicals-only model (b) involves only the pathway (i), and the ground-state hole-only model (c) involves only the pathway (ii). and (ii) rotational dephasing of depleted CHI3 (hole). The radicals-only model (b) involves only the pathway (i), and the ground-state hole-only model (c) involves only the pathway (ii). | ||
Methods
Experimental methods and sample preparation
The femtosecond time-resolved X-ray liquidography (fs-TRXL) experiment was conducted at the XSS beamline of PAL-XFEL (Pohang Accelerator Laboratory X-ray Free-Electron Laser).34,35 X-ray pulses, with an energy of 12.7 keV and a temporal width of <50 fs, were delivered at a repetition rate of 30 Hz and focused to a spot approximately 30 μm in diameter. Optical pump pulses had a wavelength of 267 nm, focused to a spot approximately 200 μm in diameter (full width at half maximum, FWHM), resulting in a laser fluence of ∼2.0 mJ mm−2 and a temporal width of ∼100 fs at the sample position. A 20 mM CHI3 solution in cyclohexane served as a sample solution, alongside an 8.5 mM 4-bromo-4′-(N,N-diethylamino)-azobenzene solution in cyclohexane for measuring the solvent heating signals. The sample solution was pumped through a quartz capillary nozzle, generating a 100 μm cylindrical jet vertically. Scattering intensities from the two-dimensional patterns were captured by a charge-coupled-device detector (Rayonix MX225-HS, 5760 × 5760 pixels, 39 × 39 μm2 per pixel, 4 × 4 binning mode) with a sample-to-detector distance of ∼34 mm. To capture the CHI3 photolysis, the scattering images were collected across a wide-range of pump-probe time delays, spanning from −450 fs to 100 ps, with a total of 108 time delays. More details are described in ESI.†Data processing and data reduction
The time-resolved difference scattering images were obtained by subtracting the laser-off scattering images from their corresponding laser-on images of the sample solution. To resolve the X-ray intensity jitter issues that often arise at XFEL facilities, we first paired the laser-off and laser-on scattering images with similar intensities. These paired images were then subtracted to obtain difference scattering images, effectively compensating for the X-ray intensity jitter. Subsequently, these difference images were decomposed into isotropic (ΔS0(q, t)) and anisotropic (ΔS2(q, t)) components and further transformed into one-dimensional difference scattering curves. The solvent heating signals were then subtracted from the sample solution data via the projection to extract the perpendicular component (PEPC) method, thereby removing solvent heating contributions and experimental artifacts from the dataset. The PEPC-treated data underwent further analysis through the linear combination fitting (LCF) and the singular value decomposition (SVD) to exclusively assess the kinetic contributions of solute. More details are described in ESI.†Anisotropic scattering curve calculation
The anisotropic scattering curves of the ground-state holes and particles were calculated using the method described by Biasin et al.24 and later applied to a triiodide system by Heo et al.29 In our analysis, we utilized this method, despite it being specifically designed for symmetric top molecules. The relevant equations used in these studies are not inherently applicable to molecules that do not exhibit symmetric top geometry, such as the photoproduct CHI2. The deviation from symmetric top geometry in CHI2 may introduce minor inaccuracies in retrieving the anisotropic component of the experimental scattering signal and in calculating the theoretical scattering curves corresponding to the radical species. However, we believe these effects are unlikely to substantially impact our results because the extent of inherent discrepancy between the experimental and simulated anisotropic signals obtained through this approach is expected to be negligible, without interfering with the analysis of dephasing kinetics. Additionally, we did not refine the molecular structure using the anisotropic signal; instead, we used the structural information obtained from the isotropic signal analysis directly in the anisotropic signal analysis. This approach was employed to assign the identity of the species contributing to the anisotropic signal. The small expected distortions in the anisotropic signal are not large enough to affect the identification of these contributing species. Heo et al.29 successfully used this method for a similar non-symmetric triiodide system. Nevertheless, further simulations or derivations may be necessary to fully validate its application to CHI2.Details of density functional theory calculation
The initial structures and charges of the reactant and candidate intermediates were obtained from density functional theory (DFT) calculations. We used the ωB97X36 functional as the DFT exchange–correlation functional, and the calculations were done using the Gaussian 16 package,37 including the NBO program38 for the charge calculations. For carbon and hydrogen atoms, aug-cc-pVTZ (AVTZ) all-electron basis sets were used. For iodine atoms, dhf-TZVPP small-core relativistic effective core potential (RECP) was used39 to consider the scalar relativistic effects. Solvent effects of cyclohexane were implicitly included by applying an integral equation formalism polarizable continuum model (IEFPCM). DFT calculations were performed to obtain the atomic charge distributions required for the MD simulations, with the MD simulation results subsequently used to calculate the cage terms. We followed the procedures outlined in a previous study22 for both DFT calculations and MD simulations.Weighted R-factor
In Fig. 4, the agreement between the experimental anisotropic signal (qΔS2(q)) and those derived from the three theoretical models (qΔS2,theo(q)s) was evaluated by the weighted R-factor (wR): | (1) |
Conclusions
In summary, we observed the ultrafast structural dynamics of the photodissociation of CHI3 in cyclohexane via fs-TRXL. Following the iodine dissociation accompanied by structural changes, both iso-CHI2–I and CHI3 are competitively formed through secondary geminate recombination of and I˙ radicals after the induction period. The CHI3 system, in contrast to the BiI3 system where a roaming-mediated early isomer is generated, only exhibits the formation of radicals, followed by subsequent isomer formation through geminate recombination of them. This disparity between the two systems is attributed to the differences between the two systems, the central moieties (CH vs. Bi) and the solvents (cyclohexane vs. acetonitrile). Through this paper, we aim to provide insights into whether isomer formation in polyhalomethanes and similar systems is based on geminate recombination or roaming reaction, and further provide clues for controlling the process.
and I˙ radicals after the induction period. The CHI3 system, in contrast to the BiI3 system where a roaming-mediated early isomer is generated, only exhibits the formation of radicals, followed by subsequent isomer formation through geminate recombination of them. This disparity between the two systems is attributed to the differences between the two systems, the central moieties (CH vs. Bi) and the solvents (cyclohexane vs. acetonitrile). Through this paper, we aim to provide insights into whether isomer formation in polyhalomethanes and similar systems is based on geminate recombination or roaming reaction, and further provide clues for controlling the process.
Data availability
The data supporting this article have been included as part of the ESI.†Author contributions
Conceptualization: H. K., H. I.; data curation: Y. C.; formal analysis: Y. C., H. K.; funding acquisition: H. I.; investigation: Y. L., S. L., J. K., J. H. L., J. K.; methodology: Y. C., H. K., H. I.; software: Y. C., H. K., D. I.; supervision: H. I.; visualization: Y. C.; writing – original draft: Y. C., H. K., H. I.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the Institute for Basic Science (IBS-R033). The experiment was performed at the XSS of PAL-XFEL (proposal no. 2019-1st-XSS-011). The authors thank Chi Woo Ahn, Eun Hyuk Choi, Jun Heo, Minseo Choi, Hanui Kim, Jong Goo Kim, Intae Eom, Minseok Kim, and Sae Hwan Chun for their dedicated support for the beamtime experiment.References
- J. H. Lee, T. K. Kim, J. Kim, Q. Kong, M. Cammarata, M. Lorenc, M. Wulff and H. Ihee, Capturing transient structures in the elimination reaction of haloalkane in solution by transient X-ray diffraction, J. Am. Chem. Soc., 2008, 130, 5834–5835 CrossRef CAS PubMed.
- A. N. Tarnovsky, J.-L. Alvarez, A. P. Yartsev, V. Sundström and E. Åkesson, Photodissociation dynamics of diiodomethane in solution, Chem. Phys. Lett., 1999, 312, 121–130 CrossRef CAS.
- A. N. Tarnovsky, I. Pascher and T. Pascher, Reactivity of iso-diiodomethane and iso-iodoform, isomers of CH2I2 and CHI3, toward the double bond of a variety of cycloalkenes, J. Phys. Chem. A, 2007, 111, 11814–11817 CrossRef CAS PubMed.
- X. Zheng and D. L. Phillips, Density functional theory and resonance Raman investigation of the ultraviolet electronic excited states of CF2I2, Chem. Phys. Lett., 2000, 316, 524–530 CrossRef CAS.
- J. Davidsson, J. Poulsen, M. Cammarata, P. Georgiou, R. Wouts, G. Katona, F. Jacobson, A. Plech, M. Wulff, G. Nyman and R. Neutze, Structural determination of a transient isomer of CH2I2 by picosecond X-ray diffraction, Phys. Rev. Lett., 2005, 94, 245503 CrossRef.
- T. J. Preston, M. A. Shaloski and F. F. Crim, Probing the photoisomerization of CHBr3 and CHI3 in solution with transient vibrational and electronic spectroscopy, J. Phys. Chem. A, 2013, 117, 2899–2907 CrossRef CAS PubMed.
- A. S. Mereshchenko, E. V. Butaeva, V. A. Borin, A. Eyzips and A. N. Tarnovsky, Roaming-mediated ultrafast isomerization of geminal tri-bromides in the gas and liquid phases, Nat. Chem., 2015, 7, 562–568 CrossRef CAS PubMed.
- E. H. Choi, J. G. Kim, J. Kim, H. Ki, Y. Lee, S. Lee, K. Yoon, J. Kim, J. Kim and H. Ihee, Filming ultrafast roaming-mediated isomerization of bismuth triiodide in solution, Nat. Commun., 2021, 12, 4732 CrossRef CAS PubMed.
- T. Marolewski and N. C. Yang, Photochemical addition of polyhalogenomethanes to olefins, Chem. Commun., 1967, 1225–1226, 10.1039/C19670001225.
- N. C. Yang and T. A. Marolewski, The addition of halomethylene to 1,2-dimethylcyclobutene, a methylene-olefin reaction involving a novel rearrangement, J. Am. Chem. Soc., 1968, 90, 5644–5646 CrossRef CAS.
- J. H. Lee, J. Kim, M. Cammarata, Q. Kong, K. H. Kim, J. Choi, T. K. Kim, M. Wulff and H. Ihee, Transient X-ray diffraction reveals global and major reaction pathways for the photolysis of iodoform in solution, Angew. Chem., Int. Ed., 2008, 47, 1047–1050 CrossRef CAS PubMed.
- X. Zheng and D. L. Phillips, Solvation effects on the iodoform ultraviolet direct photodissociation reaction: opening the photoisomerization channel, Chem. Phys. Lett., 2000, 324, 175–182 CrossRef CAS.
- Y.-L. Li, D. M. Chen, D. Wang and D. L. Phillips, Time-resolved resonance Raman and density functional theory investigation of iodocyclopropanation and addition reactions with alkenes after ultraviolet photolysis of iodoform, J. Org. Chem., 2002, 67, 4228–4235 CrossRef CAS PubMed.
- M. Wall, A. N. Tarnovsky, T. Pascher, V. Sundström and E. Åkesson, Photodissociation dynamics of iodoform in solution, J. Phys. Chem. A, 2003, 107, 211–217 CrossRef CAS.
- P. Z. El-Khoury, W. M. Kwok, X. Guan, C. Ma, D. L. Phillips and A. N. Tarnovsky, Photochemistry of iodoform in methanol: formation and fate of the iso-CHI2-I photoproduct, ChemPhysChem, 2009, 10, 1895–1900 CrossRef CAS PubMed.
- Q. Kong, J. H. Lee, K. H. Kim, J. Kim, M. Wulff, H. Ihee and M. H. J. Koch, Ultrafast X-ray solution scattering reveals different reaction pathways in the photolysis of triruthenium dodecacarbonyl (Ru3(CO)12) after ultraviolet and visible excitation, J. Am. Chem. Soc., 2010, 132, 2600–2607 CrossRef CAS PubMed.
- K. H. Kim, J. H. Lee, J. Kim, S. Nozawa, T. Sato, A. Tomita, K. Ichiyanagi, H. Ki, J. Kim, S.-i. Adachi and H. Ihee, Solvent-dependent molecular structure of ionic species directly measured by ultrafast X-ray solution scattering, Phys. Rev. Lett., 2013, 110, 165505 CrossRef PubMed.
- J. H. Lee, M. Wulff, S. Bratos, J. Petersen, L. Guerin, J.-C. Leicknam, M. Cammarata, Q. Kong, J. Kim, K. B. Møller and H. Ihee, Filming the birth of molecules and accompanying solvent rearrangement, J. Am. Chem. Soc., 2013, 135, 3255–3261 CrossRef CAS PubMed.
- J. G. Kim, T. W. Kim, J. Kim and H. Ihee, Protein structural dynamics revealed by time-resolved X-ray solution scattering, Acc. Chem. Res., 2015, 48, 2200–2208 CrossRef CAS PubMed.
- D. J. Hsu, D. Leshchev, D. Rimmerman, J. Hong, M. S. Kelley, I. Kosheleva, X. Zhang and L. X. Chen, X-ray snapshots reveal conformational influence on active site ligation during metalloprotein folding, Chem. Sci., 2019, 10, 9788–9800 RSC.
- S. Westenhoff, E. Malmerberg, D. Arnlund, L. Johansson, E. Nazarenko, M. Cammarata, J. Davidsson, V. Chaptal, J. Abramson, G. Katona, A. Menzel and R. Neutze, Rapid readout detector captures protein time-resolved WAXS, Nat. Methods, 2010, 7, 775–776 CrossRef CAS PubMed.
- C. W. Ahn, H. Ki, J. Kim, J. Kim, S. Park, Y. Lee, K. H. Kim, Q. Kong, J. Moon, M. N. Pedersen, M. Wulff and H. Ihee, Direct observation of a transiently formed isomer during iodoform photolysis in solution by time-resolved X-ray liquidography, J. Phys. Chem. Lett., 2018, 9, 647–653 CrossRef CAS PubMed.
- U. Lorenz, K. B. Møller and N. E. Henriksen, On the interpretation of time-resolved anisotropic diffraction patterns, New J. Phys., 2010, 12, 113022 CrossRef.
- E. Biasin, T. B. van Driel, G. Levi, M. G. Laursen, A. O. Dohn, A. Moltke, P. Vester, F. B. K. Hansen, K. S. Kjaer, T. Harlang, R. Hartsock, M. Christensen, K. J. Gaffney, N. E. Henriksen, K. B. Møller, K. Haldrup and M. M. Nielsen, Anisotropy enhanced X-ray scattering from solvated transition metal complexes, J. Synchrotron Radiat., 2018, 25, 306–315 CrossRef CAS PubMed.
- H. Ki, J. Gu, Y. Cha, K. W. Lee and H. Ihee, Projection to extract the perpendicular component (PEPC) method for extracting kinetics from time-resolved data, Struct. Dyn., 2023, 10, 034103 CrossRef CAS PubMed.
- H. Vennekate, D. Schwarzer, J. Torres-Alacan, O. Krahe, A. C. Filippou, F. Neese and P. Vöhringer, Ultrafast primary processes of an iron-(iii) azido complex in solution induced with 266 nm light, Phys. Chem. Chem. Phys., 2012, 14, 6165–6172 RSC.
- C. F. Sailer and E. Riedle, Photogeneration and reactions of benzhydryl cations and radicals: a complex sequence of mechanisms from femtoseconds to microseconds, Pure Appl. Chem., 2013, 85, 1487–1498 CrossRef CAS.
- K. H. Kim, H. Ki, K. Y. Oang, S. Nozawa, T. Sato, J. Kim, T. K. Kim, J. Kim, S.-i. Adachi and H. Ihee, Global reaction pathways in the photodissociation of I3− ions in solution at 267 and 400 nm studied by picosecond X-ray liquidography, ChemPhysChem, 2013, 14, 3687–3697 CrossRef CAS PubMed.
- J. Heo, J. G. Kim, E. H. Choi, H. Ki, D.-S. Ahn, J. Kim, S. Lee and H. Ihee, Determining the charge distribution and the direction of bond cleavage with femtosecond anisotropic x-ray liquidography, Nat. Commun., 2022, 13, 522 CrossRef CAS PubMed.
- D. Leshchev, D. Khakhulin, G. Newby, H. Ki, H. Ihee and M. Wulff, Sub-nanosecond secondary geminate recombination in mercury halides HgX2 (X = I, Br) investigated by time-resolved x-ray scattering, J. Chem. Phys., 2019, 151, 054310 Search PubMed.
- A. Nimmrich, M. R. Panman, O. Berntsson, E. Biasin, S. Niebling, J. Petersson, M. Hoernke, A. Björling, E. Gustavsson, T. B. van Driel, A. O. Dohn, M. Laursen, D. B. Zederkof, K. Tono, T. Katayama, S. Owada, M. M. Nielsen, J. Davidsson, J. Uhlig, J. S. Hub, K. Haldrup and S. Westenhoff, Solvent-dependent structural dynamics in the ultrafast photodissociation reaction of triiodide observed with time-resolved X-ray solution scattering, J. Am. Chem. Soc., 2023, 145, 15754–15765 CrossRef CAS PubMed.
- M. R. Panman, E. Biasin, O. Berntsson, M. Hermann, S. Niebling, A. J. Hughes, J. Kübel, K. Atkovska, E. Gustavsson, A. Nimmrich, A. O. Dohn, M. Laursen, D. B. Zederkof, A. Honarfar, K. Tono, T. Katayama, S. Owada, T. B. van Driel, K. Kjaer, M. M. Nielsen, J. Davidsson, J. Uhlig, K. Haldrup, J. S. Hub and S. Westenhoff, Observing the structural evolution in the photodissociation of diiodomethane with femtosecond solution X-ray scattering, Phys. Rev. Lett., 2020, 125, 226001 CrossRef CAS PubMed.
- F. Perrin, Mouvement brownien d'un ellipsoide - I. dispersion diélectrique pour des molécules ellipsoidales, J. Phys. Radium, 1934, 5, 497–511 CrossRef CAS.
- I. S. Ko, H.-S. Kang, H. Heo, C. Kim, G. Kim, C.-K. Min, H. Yang, S. Y. Baek, H.-J. Choi, G. Mun, B. R. Park, Y. J. Suh, D. C. Shin, J. Hu, J. Hong, S. Jung, S.-H. Kim, K. Kim, D. Na, S. S. Park, Y. J. Park, Y. G. Jung, S. H. Jeong, H. G. Lee, S. Lee, S. Lee, B. Oh, H. S. Suh, J.-H. Han, M. H. Kim, N.-S. Jung, Y.-C. Kim, M.-S. Lee, B.-H. Lee, C.-W. Sung, I.-S. Mok, J.-M. Yang, Y. W. Parc, W.-W. Lee, C.-S. Lee, H. Shin, J. H. Kim, Y. Kim, J. H. Lee, S.-Y. Park, J. Kim, J. Park, I. Eom, S. Rah, S. Kim, K. H. Nam, J. Park, J. Park, S. Kim, S. Kwon, R. An, S. H. Park, K. S. Kim, H. Hyun, S. N. Kim, S. Kim, C.-J. Yu, B.-S. Kim, T.-H. Kang, K.-W. Kim, S.-H. Kim, H.-S. Lee, H.-S. Lee, K.-H. Park, T.-Y. Koo, D.-E. Kim and K. B. Lee, Construction and commissioning of PAL-XFEL facility, Appl. Sci., 2017, 7, 479 CrossRef.
- H.-S. Kang, C.-K. Min, H. Heo, C. Kim, H. Yang, G. Kim, I. Nam, S. Y. Baek, H.-J. Choi, G. Mun, B. R. Park, Y. J. Suh, D. C. Shin, J. Hu, J. Hong, S. Jung, S.-H. Kim, K. Kim, D. Na, S. S. Park, Y. J. Park, J.-H. Han, Y. G. Jung, S. H. Jeong, H. G. Lee, S. Lee, S. Lee, W.-W. Lee, B. Oh, H. S. Suh, Y. W. Parc, S.-J. Park, M. H. Kim, N.-S. Jung, Y.-C. Kim, M.-S. Lee, B.-H. Lee, C.-W. Sung, I.-S. Mok, J.-M. Yang, C.-S. Lee, H. Shin, J. H. Kim, Y. Kim, J. H. Lee, S.-Y. Park, J. Kim, J. Park, I. Eom, S. Rah, S. Kim, K. H. Nam, J. Park, J. Park, S. Kim, S. Kwon, S. H. Park, K. S. Kim, H. Hyun, S. N. Kim, S. Kim, S.-m. Hwang, M. J. Kim, C.-y. Lim, C.-J. Yu, B.-S. Kim, T.-H. Kang, K.-W. Kim, S.-H. Kim, H.-S. Lee, H.-S. Lee, K.-H. Park, T.-Y. Koo, D.-E. Kim and I. S. Ko, Hard X-ray free-electron laser with femtosecond-scale timing jitter, Nat. Photonics, 2017, 11, 708–713 CrossRef CAS.
- J.-D. Chai and M. Head-Gordon, Systematic optimization of long-range corrected hybrid density functionals, J. Chem. Phys., 2008, 128, 084106 CrossRef PubMed.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, G. A. Petersson, H. Nakatsuji, X. Li, M. Caricato, A. V. Marenich, J. Bloino, B. G. Janesko, R. Gomperts, B. Mennucci, H. P. Hratchian, J. V. Ortiz, A. F. Izmaylov, J. L. Sonnenberg, D. Williams-Young, F. Ding, F. Lipparini, F. Egidi, J. Goings, B. Peng, A. Petrone, T. Henderson, D. Ranasinghe, V. G. Zakrzewski, J. Gao, N. Rega, G. Zheng, W. Liang, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, K. Throssell, J. A. Montgomery Jr, J. E. Peralta, F. Ogliaro, M. J. Bearpark, J. J. Heyd, E. N. Brothers, K. N. Kudin, V. N. Staroverov, T. A. Keith, R. Kobayashi, J. Normand, K. Raghavachari, A. P. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, J. M. Millam, M. Klene, C. Adamo, R. Cammi, J. W. Ochterski, R. L. Martin, K. Morokuma, O. Farkas, J. B. Foresman and D. J. Fox, Gaussian 16, Revision C.01, Gaussian, Inc., Wallingford CT, 2016 Search PubMed.
- E. D. Glendening, A. E. Reed, J. E. Carpenter and F. Weinhold, NBO version 3.1, 1998 Search PubMed.
- F. Weigend and A. Baldes, Segmented contracted basis sets for one- and two-component Dirac–Fock effective core potentials, J. Chem. Phys., 2010, 133, 174102 CrossRef PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental details, data processing, figures and experimental data. See DOI: https://doi.org/10.1039/d4sc04604h |
| This journal is © The Royal Society of Chemistry 2024 |