
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceLigand cross-links as a design element in oligo- and polyMOFs†
Debobroto
Sensharma
 and
Seth M.
Cohen
*
and
Seth M.
Cohen
*
Department of Chemistry and Biochemistry, University of California San Diego, La Jolla, California 92093, USA. E-mail: scohen@ucsd.edu
First published on 11th November 2024
Abstract
Metal–Organic Frameworks (MOFs) constructed using cross-linked oligomeric or polymeric ligands (oligoMOFs and polyMOFs respectively) have so far relied on a handful of canonical structural blueprints, in which the cross-links have not played a significant role in determining structure. In this study, we show that cross-links between terephthalate ligands in dabco-based Zn-MOFs (DMOFs) can exert control over the overall phase landscape of resulting oligo- and polyMOFs. We find that cross-links can direct the overall topology of the resulting MOF (pcuvs.kag) based on their length or rigidity, and can influence the phase transformation behavior of the pcu network. We also show the first example of tethered ligand dimers adopting a different MOF structure to the analogous trimer and polymer. Understanding the influence of cross-links on the formation of these MOFs will help guide the design of future MOF–polymer hybrid materials.
Introduction
The modular nature of metal–organic frameworks (MOFs) has been one of the main motivations behind the broad research interest in this class of solids in the past several decades.1–5 MOFs can be constructed with predetermined architectures by judiciously selecting molecular building blocks, i.e., organic ligands and metal atoms or clusters of appropriate geometries. This approach, known as reticular chemistry, has been demonstrated powerfully by the construction of large families of isoreticular MOFs in which a single building block is substituted by others of the same connectivity and geometry, ultimately resulting in structures that share the same network topology, but show predictable differences in pore size and functionality.6–9 However, in some cases, MOFs adopt different network topologies even though they are constructed using identical molecular building blocks. This phenomenon is known as framework isomerism10–12 and is exemplified by groups of MOFs such as MIL-101 (mtn network) and MIL-88B (acs network),13,14UiO-66 (fcu) and EHU-30 (hex),15 and MOF-177-mNH2 (qom), MOF-155 (pyr), and MOF-156 (rtl).16Typically, distinct framework isomers are favored under different crystallization conditions, and these differences are utilized in synthesizing phase pure isomeric frameworks. In the case of the pcu and kag framework isomers of the archetypal “pillar-layered” MOF, DMOF-1, the kag structure is favored under kinetically controlled conditions, while the pcu structure is favored under thermodynamically forcing conditions. Following the discovery of each phase by Kim et al. and Chun et al.,17,18 work by Kitagawa et al. has shown that the formation of the triangular structural subunits of the kag network is initially favored by the constituent Zn(II) paddlewheels and terephthalate (bdc2−) ligands over the pcu network due to steric considerations (Fig. 1(a–d)).19 Under kinetically controlled conditions, these triangular subunits act as nuclei for the growth of the kag framework. However, the extended pcu structure is energetically favored over kag, and is obtained upon the provision of sufficient thermal energy and reaction time. These differences were utilized by the groups of Verpoort and Walton to develop a rapid room temperature synthetic route to kag-DMOF-1, while also observing that the use of certain solvents instead favored the formation of pcu-DMOF-1 under the same conditions.20,21 Additionally, Verpoort et al. have shown that solvents such as MeOH can mediate the transformation of kag-DMOF-1 to the thermodynamically favored pcu-DMOF-1 by simply soaking in the solvent at room temperature.22 Besides these isomeric structures, DMOF-1 also exhibits distinct phases of the pcu isomer through flexibility. These phases involve distortions of the framework induced by interactions with various guest molecules such as DMF, benzene, and isopropanol. The relationships between these phases and topologies is summarized in Fig. 1(e).
 | ||
| Fig. 1 Crystal structures of (a and b) pcu-DMOF-1 and (c and d) kag-DMOF-1; (e) schematic representation of the phases of DMOF-1 upon use of different synthetic conditions or other stimuli. | ||
Numerous studies have explored the effect of introducing substituent groups to the terephthalic acid linker in DMOF analogues, especially with respect to modulation of the adsorption-induced phase change behavior of the resulting frameworks.23–30 However, despite the variety of functional variants that have been made, the introduction of additional substituents to the H2bdc linker has not been reported to yield the kag structure, which Hungerford and Walton attribute to steric constraints.21
Recent work from the laboratories of Xiao, Johnson, He, Zhou, Cohen, and others, has explored the outcomes of incorporating flexible tethering groups between conventional ligands, resulting in oligomeric (oligoMOF) or polymeric (polyMOF) materials.31–42 These tethers are sufficiently flexible to allow the retention of the overall network structure of the analogous untethered “parent” MOF, but can, in principle, impose constraints on the relative distance and orientation between the tethered bdc2− ligands. OligoMOF analogues of materials like IRMOF-1 (MOF-5), NOTT-101, and MOF-74 have been synthesized using this approach, which show modified properties owing to the incorporation of the tethering alkyl chains, such as modified sorbate uptake or exploitable surface functionality.31,32,41,43 Notably, Xiao et al. showed that tether incorporation in an expanded MIL-53 analogue could modulate its phase change behavior. The tether stabilized the large-pore phase of the material, which is unfavored at low guest loadings in the untethered “parent” MOF.37
The use of ligands linked into polymers through repeating cross-links in a similar manner yields polyMOFs, which also typically adopt canonical MOF structures. Studies on systems such as IRMOF-1 and UiO-66 emphasize the versatility of these systems with respect to the polymer chains incorporated, without altering the overall crystalline lattice.31,32,44 In the examples reported so far, dimeric, trimeric, and polymeric tethers have adopted the same framework structure when combined with metal precursors under fixed conditions, and the number of repeating units has not been a factor in determining the structure adopted.
Despite some indications in previous studies that tether incorporation can result in unidentified phases, and that tether length can influence framework ordering relative to a single parent structure, the potential of tethers to influence the selection of competing structures has not been studied yet. In this work, we study the effects of joining terephthalic acid linkers with flexible alkyl chains and rigid xylyl spacers, in place of unmodified terephthalic acid in the synthesis of DMOF-1. We also study the impact of changing the number of repeating units on structure selection. The observed effect of ligand cross-linking on topology selection and phase transformations shows that flexible tethers may act as hitherto unstudied crystal engineering elements in the design and synthesis of MOFs, opening up a new chemical space for MOF discovery.
Results and discussion
Tethered oligomeric and polymeric H2bdc ligands were prepared according to reported procedures (see ESI†). These ligands feature H2bdc groups tethered using ether linkages by flexible n-alkyl chains of varying length, butyl(bdc)2, pentyl(bdc)2, hexyl(bdc)2, and heptyl(bdc)2; relatively rigid xylyl groups with similar numbers of carbon atoms between ether oxygens: o-xylyl(bdc)2, m-xylyl(bdc)2, and p-xylyl(bdc)2; a trimeric linker, pentyl2(bdc)3; and polymeric linkers with n-pentyl and n-heptyl spacers: pbdc-5a and pbdc-7a (Fig. 2). We use the above terms to refer to cross-linked ligands in both protonated and fully deprotonated states. Polymer formation in the cases of pbdc-5a and pbdc-7a was confirmed by MALDI-TOF MS (Fig. S1 and S2†). Oligomeric ligands were combined with zinc nitrate hexahydrate and 1,4-diazabicyclo[2.2.2]octane (dabco) in DMF and heated under solvothermal conditions using a procedure optimized for the synthesis of the parent pcu-DMOF-1 (ESI†). Previously reported synthetic difficulties associated with the synthesis of polyDMOFs were overcome by modifying the synthetic procedure, allowing a precipitate to form upon mixing reactants, and subjecting the suspension to solvothermal conditions without filtering the solids out.45 The parent kag-DMOF-1 was synthesized using the “rapid” procedure reported by Hungerford and Walton.21 In order to keep the proportion of Zn2+, dabco, and bdc2− units identical across reactions with various tethered ligands, the number of moles of dimeric ligands (which contain two H2bdc units per formula mass) was halved relative to the procedure using untethered H2bdc, and the number of moles of pentyl2(bdc)3 was reduced by a factor of three. The number of moles of polymer ligands was not changed, since they contain one bdc unit per formula unit. | ||
| Fig. 2 The cross-linked terephthalic acid ligands chosen for the construction of oligo- and poly-DMOFs. | ||
Polycrystalline products were obtained from these reactions and were characterized by powder X-ray diffraction (PXRD), N2 physisorption (at 77 K), and 1H NMR (after digestion of the solid in acid). Upon comparison of the PXRD patterns obtained from the alkyl-tethered DMOFs, we find that although these reactions were carried out under identical conditions, the crystal structures adopted are clearly distinct (Fig. 3). The patterns shown by butyl(bdc)2-DMOF-1 and pentyl(bdc)2-DMOF-1 closely match the calculated pattern for kag-DMOF-1 with a characteristic 2θ peak at ca. 4.7°, whereas that shown by heptyl(bdc)2-DMOF-1 matches the calculated pattern for pcu-DMOF-1, with a characteristic 2θ peak at ca. 8.1°. The adoption of a phase pure kag structure by butyl(bdc)2-DMOF-1 and pentyl(bdc)2-DMOF-1, under conditions that yield the phase pure pcu structure using unfunctionalized terephthalic acid implies that the butyl and pentyl cross-links direct the adoption of the kag structure. This shows that the formation of the kag structure using functionalized bdc2− units is possible, but more importantly, that the cross-link between bdc2− units exerts a structure-directing influence under conditions that typically result in the pcu structure. The PXRD pattern of the putative hexyl(bdc)2-DMOF-1 does not correspond to either of these phases. The possibility of additional phases in the DMOF system was noted by Kitagawa et al. in their study of framework isomerism in DMOF-1, and this material too may represent a novel structural arrangement.19
 | ||
| Fig. 3 PXRD patterns of alkyl-tethered DMOFs in comparison with pcu-DMOF-1 and kag-DMOF-1. | ||
PXRD patterns obtained for o-xylyl(bdc)2-DMOF-1, m-xylyl(bdc)2-DMOF-1, and p-xylyl(bdc)2-DMOF-1 all match the calculated pcu-DMOF-1 pattern (Fig. 4). This is notable because of the similarity in the number of carbon atoms between ether oxygens in each linker: four in o-xylyl(bdc)2 and butyl(bdc)2, five in m-xylyl(bdc)2 and pentyl(bdc)2, and six in p-xylyl(bdc)2 and hexyl(bdc)2. None of the flexible alkyl-tethered linkers of corresponding lengths yielded pcu structures, implying that the rigidity of the xylyl tethers impose geometrical constraints incompatible with the formation of the kag structure.
 | ||
| Fig. 4 PXRD patterns of xylyl-tethered DMOFs in comparison with pcu-DMOF-1 and kag-DMOF-1. | ||
The trimer-based pentyl2(bdc)3-DMOF-1 and polymeric pbdc-5a-DMOF-1 also adopted the pcu structure, in contrast to the dimeric pentyl(bdc)2-DMOF-1 based on the same spacer (Fig. 5). This is the first observation of corresponding dimeric and trimeric oligoMOFs adopting distinct, isomeric framework structures, as well as the first example of isomerism between the framework structures of an oligoMOF and its exact polyMOF analogue. These results show the remarkable sensitivity of the DMOF system to subtle changes in the length, flexibility, and number of repeat units of tethered H2bdc ligands. Despite facile data collection on a laboratory source, Pawley refinement of the PXRD patterns of these MOFs showed good whole pattern fits, high phase purity, and minimal deviation of unit cell parameters from the reported single crystal structures of the parent pcu-DMOF-1 and kag-DMOF-1 (Table S1 and Fig. S3–S11†).
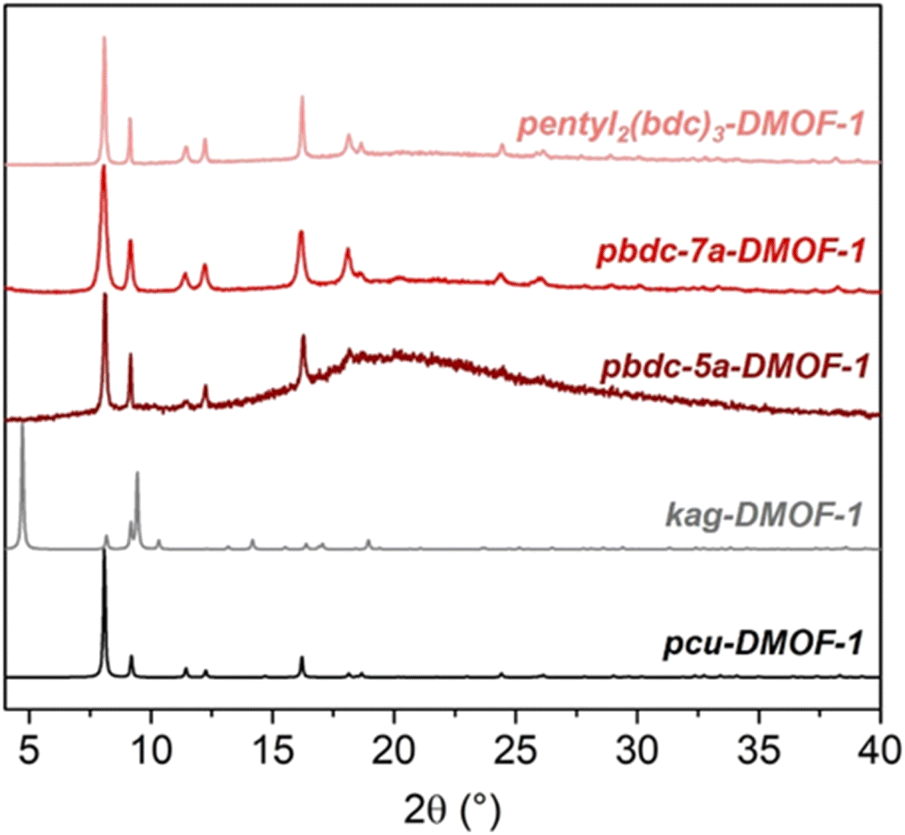 | ||
| Fig. 5 PXRD patterns of trimer- and polymer-based DMOFs in comparison with pcu-DMOF-1 and kag-DMOF-1. | ||
To better understand the role of the alkyl cross-link in directing the formation of isomeric networks, we conducted further experiments on pentyl(bdc)2-DMOF-1 and heptyl(bdc)2-DMOF-1. When synthetic conditions were changed to the rapid method outlined by Hungerford and Walton, in which unfunctionalized terephthalate forms the kag network in the presence of triethylamine and DMF (see ESI Fig. S12†), the PXRD pattern of the product formed using pentyl(bdc)2 is well-defined and corresponds to the phase pure kag product. However, under the same conditions, the heptyl(bdc)2 product shows poor crystallinity, only retaining the major peaks associated with the pcu structure. Therefore, although conditions that promote the formation of the kinetically favored kag structure do influence the reaction, the effect of the pentyl and heptyl cross-links remains the same as under solvothermal conditions. In addition, this observation shows that the heptyl cross-link also exerts its own structure directing effect on the product.
To investigate whether the pentyl cross-link permits the formation of the pcu structure under thermodynamically forcing conditions, we allowed solvothermal syntheses using pentyl(bdc)2 to proceed for up to two weeks. PXRD patterns taken after one week and two weeks under these conditions showed the exclusive formation of the kag phase of pentyl(bdc)2-DMOF-1 with no discernible decrease of crystallinity, suggesting that the modulation of the landscape of available structures by the pentyl cross-link is indeed of a thermodynamic nature (Fig. S13†). This is consistent with observations from solvent soaking experiments, in which we observe that pentyl(bdc)2-DMOF-1 shows no change from its kag structure or decrease in crystallinity upon soaking for up to a week in solvents including MeOH, whereas unfunctionalized kag-DMOF-1 transforms to the pcu isomer in less than 72 hours under the same conditions, in agreement with Verpoort et al. (Fig. S14†).22
N2 physisorption experiments carried out on the tethered DMOFs showed the retention of appreciable BET surface areas in most materials, with characteristic Type I isotherms. Among alkyl-tethered DMOFs, BET surface area values varied from 841 ± 60 and 1150 ± 4 m2 g−1 for butyl(bdc)2-DMOF-1 and pentyl(bdc)2-DMOF-1, respectively to 962 ± 1 m2 g−1 for heptyl(bdc)2-DMOF-1 (Fig. 6). These values are reduced relative to the values obtained for the parent pcu-DMOF-1 and kag-DMOF-1, 1779 ± 58 m2 g−1 and 1813 ± 22 m2 g−1, due to partial occlusion of pores by the flexible tethering moieties. Pore volumes were found to be 0.429 cm3 g−1 and 0.578 cm3 g−1 for butyl and pentyl tethered kag DMOFs, compared to 0.863 cm3 g−1 in the parent kag-DMOF-1. The pore volume of heptyl(bdc)-DMOF-1 was found to be 0.443 cm3 g−1, compared to 0.732 cm3 g−1 in pcu-DMOF-1. These values confirm that large fractions of the parent pore volume are retained in oligo-DMOFs adopting both kag and pcu structures, despite the incorporation of tethering groups.
 | ||
| Fig. 6 N2 sorption isotherms (77 K) of alkyl-tethered DMOFs in comparison with pcu-DMOF-1 and kag-DMOF-1. Closed symbols represent adsorption and open symbols represent desorption. | ||
MOFs constructed using the xylyl-tethered H2bdc linkers showed BET surface areas of 1093 ± 84 m2 g−1 for o-xylyl(bdc)2-DMOF-1, 892 ± 1 m2 g−1 for m-xylyl(bdc)2-DMOF-1, and 892 ± 1 m2 g−1 for p-xylyl(bdc)2-DMOF-1 (Fig. S15†). These three MOFs adopt the pcu structure, and the surface area available varies systematically with the increased centrality of the bulky phenyl ring in the tethered ligand molecule. This trend is also observed in pore volumes. The trimer-based pentyl2(bdc)3-DMOF-1 shows reduced porosity compared to its parent or dimer-based counterparts, and has a BET surface area of 448 ± 88 m2 g−1. The pentyl-spaced polyMOF, pbdc-5a-DMOF-1 shows minimal microporosity with a BET surface area of just 100 ± 6 m2 g−1. The heptyl-spaced polyMOF, pbdc-7a-DMOF-1, retains more of its microporosity in comparison, with a BET surface area of 251 ± 5 m2 g−1 (Fig. S16†). The reduced porosity observed in polyDMOFs compared to oligoDMOFs is consistent with the increased density of tethering units in polyDMOFs – 1 tethering unit per bdc unit in polymeric linkers, versus 0.5 tethering unit per bdc unit in dimer linkers – resulting in greatly diminished accessible void space in the ultramicroporous MOF structure.
The observation of varying degrees of crystallinity between oligo- and polyDMOFs bearing different tethering groups is found to correlate with the observation of varying degrees of macropore N2 condensation in the high-pressure region of the respective isotherms. Materials with less crystalline PXRD patterns, such as p-xylyl(bdc)2-DMOF-1, pentyl2(bdc)3-DMOF-1, pbdc-5a-DMOF-1, and pbdc-7a-DMOF-1, show noticeably larger steps due to macropore condensation, suggesting a relationship between crystal attributes such as size, morphology, defectivity, and N2 uptake.
Pore size distributions calculated from these isotherms by the Horvath–Kawazoe (HK) method provided corroborating structural insights (Table S2†). The parent pcu-DMOF-1 shows a sharp unimodal distribution of pore widths with a maximum at ca. 6.1 Å, whereas the parent kag-DMOF-1 shows a bimodal distribution of pore widths due to the presence of narrow triangular (ca. 6.3 Å) and broad hexagonal (ca. 11.3 Å) microporous channels (Fig. S17–S27†). We observe that tethered DMOFs adopting the kag structure, butyl(bdc)2-DMOF-1 and pentyl(bdc)2-DMOF-1, also show bimodal pore size distributions, while those that adopt the pcu structure show unimodal distributions. In contrast to the small deviations from the parent structures found in unit cell parameters, deviations from the pore size distribution maxima in the tethered material were found to be as large as ca. 1.0 Å. Pore contraction due to the incorporation of tethering groups can account for many of these deviations, but in some cases, such as the increase of the HK plot maximum associated with the micropore in o-xylyl(bdc)2-DMOF-1 and p-xylyl(bdc)2-DMOF-1, and the triangular micropore in butyl(bdc)2-DMOF-1 and pentyl(bdc)2-DMOF-1, distortion of framework components (e.g. torsion of phenyl rings) is suggested.
1H NMR experiments carried out on activated and acid-digested MOFs confirmed that cross-links in each ligand were intact and provided information on the composition of each material, by integration of proton signals corresponding to tethered H2bdc ligands and the single peak corresponding to dabco (Table S3 and Fig. S28–S39†). This method confirms that the parent MOFs show a H2bdc-to-dabco mole ratio of 2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1, corresponding to the expected formula of [Zn2(bdc)2(dabco)]. The dimeric oligoDMOFs in this study conform to a general formula of [Zn2(tether(bdc)2)(dabco)], and the trimer-based pentyl2(bdc)3-DMOF-1 conforms to a formula of [Zn3(pentyl2(bdc)3)(dabco)1.5]. The polyDMOFs have an expected H2bdc-to-dabco mole ratio of 2:1. However while pbdc-7a-DMOF-1 agrees with this ratio, the ratio found for pbdc-5a-DMOF-1 is nearly 3:1. Since 1H NMR conducted on thoroughly washed but unactivated pbdc-5a-DMOF-1 (Fig. S40†) shows the expected H2bdc-to-dabco mole ratio of 2:1, the loss of dabco linkers can be attributed to degradation of the framework structure during activation. Finally, 1H NMR of the as-synthesized hexyl(bdc)2-DMOF-1 material indeed conformed to the expected bdc:dabco mole ratio of 2:1, and may indicate a novel framework isomer of DMOF. Work to prepare single crystalline samples of hexyl(bdc)2-DMOF is ongoing.
:1, corresponding to the expected formula of [Zn2(bdc)2(dabco)]. The dimeric oligoDMOFs in this study conform to a general formula of [Zn2(tether(bdc)2)(dabco)], and the trimer-based pentyl2(bdc)3-DMOF-1 conforms to a formula of [Zn3(pentyl2(bdc)3)(dabco)1.5]. The polyDMOFs have an expected H2bdc-to-dabco mole ratio of 2:1. However while pbdc-7a-DMOF-1 agrees with this ratio, the ratio found for pbdc-5a-DMOF-1 is nearly 3:1. Since 1H NMR conducted on thoroughly washed but unactivated pbdc-5a-DMOF-1 (Fig. S40†) shows the expected H2bdc-to-dabco mole ratio of 2:1, the loss of dabco linkers can be attributed to degradation of the framework structure during activation. Finally, 1H NMR of the as-synthesized hexyl(bdc)2-DMOF-1 material indeed conformed to the expected bdc:dabco mole ratio of 2:1, and may indicate a novel framework isomer of DMOF. Work to prepare single crystalline samples of hexyl(bdc)2-DMOF is ongoing.
FTIR spectra of the activated oligoDMOFs (Fig. S41–S49†) do not show any observable peaks in the region between 1720 cm−1 and 1680 cm−1, which is associated with the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O stretching vibration (νs CO) of the free carboxylate groups in the respective tethered ligands. Instead, strong peaks associated with the asymmetric –COO− stretching mode (νas COO−) of coordinated carboxylates are observed in each case between 1640 cm−1 and 1630 cm−1. This implies that uncoordinated carboxylate groups are not present in the oligo-MOFs in significant quantities. However, small shoulders are observed at ca. 1720 cm−1 in the FTIR spectra of the polyDMOFs (Fig. S48 and S49†), suggesting that although the peak intensity and area corresponding to the νs CO stretch are greatly reduced, a notable fraction of carboxylate groups remain uncoordinated unlike in the oligoDMOFs, and that the additional constraints imposed by multiple repeating units hinder the efficient binding of all carboxylate groups in the pcu structure.
O stretching vibration (νs CO) of the free carboxylate groups in the respective tethered ligands. Instead, strong peaks associated with the asymmetric –COO− stretching mode (νas COO−) of coordinated carboxylates are observed in each case between 1640 cm−1 and 1630 cm−1. This implies that uncoordinated carboxylate groups are not present in the oligo-MOFs in significant quantities. However, small shoulders are observed at ca. 1720 cm−1 in the FTIR spectra of the polyDMOFs (Fig. S48 and S49†), suggesting that although the peak intensity and area corresponding to the νs CO stretch are greatly reduced, a notable fraction of carboxylate groups remain uncoordinated unlike in the oligoDMOFs, and that the additional constraints imposed by multiple repeating units hinder the efficient binding of all carboxylate groups in the pcu structure.
Examination of PXRD patterns of these materials before and after activation (Fig. 7) shows that the dimeric oligoDMOFs adopting pcu networks do not show noticeable structural changes upon activation, unlike pcu-DMOF-1 which is known to undergo a phase transformation. Although cross-linked DMOFs adopt a pcu network, the incorporation of the heptyl cross-link results in clear phase differences from unfunctionalized DMOF-1. Whereas the flexibility of DMOF-1 results in the adoption of a narrow pore DMF-loaded phase upon solvothermal synthesis, the structures of as synthesized heptyl(bdc)2-DMOF-1 and pbdc-7a-DMOF-1 closely resemble the large pore phase that corresponds to the guest-free DMOF-1 structure.17 This difference between phases is illustrated by a shift of the (100) peak from 8.25° (2θ) in the narrow pore phase to 8.10° (2θ) in the large pore phase.
 | ||
| Fig. 7 PXRD patterns of as synthesized and activated DMOFs in comparison with pcu-DMOF-1. The red region highlights the (100) peak. | ||
Xylyl-tethered oligoDMOFs, pentyl2(bdc)3-DMOF-1, and pbdc-5a-DMOF-1 which all adopt pcu networks similarly form the large pore phase directly upon synthesis in DMF. The assignment of phases in this manner is corroborated by Pawley refinement of PXRD data for all pcu oligoDMOFs against the large pore parent pcu-DMOF-1 structure. We postulate this is due to the occupation of a large fraction of the pore volume of the structure by the cross-links, which may act as de facto guests and prevent pore contraction. Therefore, cross-links effectively “lock” each pcu structure into its large pore phase (Fig. S50†). To our knowledge, this is the second case of large pore phase stabilization upon cross-link incorporation, following the report by Xiao et al.37 However, the pcu structures obtained using pentyl cross-links, pentyl2(bdc)3-DMOF-1 and pbdc-5a-DMOF-1, do show significant losses in crystallinity upon solvent removal at elevated temperature. This, taken together with the dabco linker vacancies seen in the NMR spectrum of digested pbdc-5a-DMOF-1, indicates that although increasing the number of repeating H2bdc units to three or greater directs the formation of pcu structures, these structures may be more strained and less robust than other pcu oligo- or polyDMOFs with better matches in cross-link length and framework dimensions.
Further insight into the stability of these materials is obtained from thermogravimetric analysis (TGA). After showing initial mass loss due to vaporization of interparticle and pore DMF below 200 °C, the kag oligoDMOFs based on butyl and pentyl cross-links show mass loss steps due to decomposition, with onsets at approximately 310 °C and 305 °C respectively, close to the reported value of 300 °C for the parent kag-DMOF-1 (Fig. S51 and S52†). Similarly, heptyl and xylyl-tethered pcu oligoDMOFs show decomposition steps between 325 °C and 335 °C, in agreement with reported values for pcu-DMOF-1 (Fig. S53–S56†). However, while pbdc-7a-DMOF-1 shows a similar decomposition temperature to the pcu oligo-DMOFs (325 °C), pentyl2(bdc)3-DMOF-1 and pbdc-5a-DMOF-1 show significantly lower decomposition temperatures (315 and 300 °C respectively), supporting the hypothesis that pentyl-tethered structures based on multiple repeating units show diminished stability (Fig. S57–S59†). We note that values for decomposition temperature are approximate due to the gradual slope of the mass loss steps, but provide helpful points of comparison between the materials in this study.
Scanning electron microscopy (SEM) conducted on selected tethered DMOFs reveals mixtures of discrete and intergrown crystals. Strikingly, the crystals of pentyl(bdc)2-DMOF-1 show a distinct hexagonal morphology (Fig. 8(a)). In contrast, pentyl2(bdc)3-DMOF-1 forms cuboidal rod-like crystals (Fig. 8(b) and S60†), and pbdc-5a-DMOF-1 forms tapered cuboidal crystals together with smaller, intergrown nanocrystals. Heptyl(bdc)2-DMOF-1 and m-xylyl(bdc)2-DMOF-1 form mixtures of cuboidal and irregular block-shaped crystals (Fig. 8(c) and S61†), and pbdc-7a-DMOF forms a mixture of aggregated needle crystals and larger cuboidal crystals (Fig. 8(d)). Therefore, the kag structure of pentyl(bdc)2-DMOF-1 results in the adoption of crystals with hexagonal symmetry, whereas the cross-linked pcu DMOFs adopt morphologies with square cross-sections. These morphologies are consistent with the layer structure in each set of materials and with the morphologies shown by crystals of the parent MOFs.17,18 Unlike some previously reported polyMOFs, pbdc-5a-DMOF-1 and pbdc-7a-DMOF-1 do not show significant hierarchical structuring of individual crystallites.46 While we cannot rule out the formation of amorphous polymer-only domains, the combination of FTIR, SEM, and digestion NMR data strongly suggests that such domains – implying larger fractions of uncoordinated carboxylate groups and ligand to dabco ratios than those observed – do not comprise more than ca. 10% of the final polyDMOF materials.
 | ||
| Fig. 8 Scanning Electron Microscopy (SEM) images of (a) pentyl(bdc)2-DMOF-1, (b) pbdc-5a-DMOF-1, (c) heptyl(bdc)2-DMOF-1, and (d) pbdc-7a-DMOF-1. | ||
Observations from the various experiments detailed above can be rationalized from analysis of the parent kag- and pcu-DMOF-1 structures. Both structures are based on pillared layers, with dinuclear Zn(II) paddlewheel SBUs providing their equatorial sites for layer formation through four ditopic bdc2− ligands, and their axial sites for pillaring through neutral ditopic dabco ligands. The key difference between the structures is that the pcu network is constructed by the pillaring of sql sheets in which bdc2− ligands make angles of ca. 90° with the paddlewheel axis, while the kag network consists of pillared kgm sheets in which bdc2− ligands make angles of ca. 60° and ca. 120° with the paddlewheel axis (Fig. 9).
 | ||
| Fig. 9 Perspective views of the crystal structures of (a) pcu-DMOF-1 and (b) kag-DMOF-1, with highlighted inter-ligand distances and angles. | ||
This results in structures that have approximately equal interlayer distances, governed by the size of the pillaring dabco ligand, but distinct pore architectures. pcu-DMOF-1 has uniform channels with a square cross section bounded by four bdc2− ligands. In contrast, kag-DMOF-1 has two types of channels, one narrow and triangular, and the other wide and hexagonal. Considering that tethering groups are bound to ether substituents on the bdc2− ligand, distances measured between aryl protons in the crystal structures of the parent frameworks provide an approximate measure of the space available for tether incorporation between pairs of bdc linkers. The interlayer distances in pcu-DMOF-1 (7.5 Å) and kag-DMOF-1 (7.4 Å) are very similar and do not provide a basis for structure directing effects upon tether incorporation. However, the distances between perpendicular (6.7 Å) and parallel (8.7 Å) pairs of bdc2− ligands in pcu-DMOF-1, and the narrow triangular channel in kag-DMOF-1 (5.9 Å) are in the appropriate range for the tethers studied here to exert an influence over framework formation.
Histograms were generated from the crystal structures deposited in the Cambridge Structural Database (CSD, see ESI for details†) of O⋯O distances between oxygen atoms bridged by alkyl and xylyl groups that are used as tethers in our study (Table S4 and Fig. S62†), to illustrate the typical range of distances shown by each tethering group in crystal structures.47 It was found that most butyl chains and a significant fraction of pentyl chains in reported structures adopt conformations resulting in O⋯O distance below the 5.9 Å value associated with the narrow pore in kag-DMOF-1. In contrast, histograms for the hexyl and heptyl tethers show distributions with no distances below 6.0 Å, which provides an empirical justification for the structure directing behavior observed. m-Xylyl and p-xylyl groups show distributions centered around ca. 6.5 Å and 7 Å respectively, compatible with the pcu structure they adopt. However, o-xylyl groups show distances only in the 4–6 Å range yet prefer the pcu structure. This suggests that additional factors such as tether rigidity play a role in structure selection. The narrow triangular pore in the kag structure may be accessible to the butyl tether of comparable length due to its flexibility and ability to contort into the confined space presented by the pore, while remaining inaccessible to the rigid o-xylyl tether.
Similar factors can explain the formation of the kag network by the dimeric pentyl-tethered ligand, and the pcu network by its trimeric and polymeric counterparts. Although the pentyl tether is short and flexible enough to bridge two bdc2− groups in the triangular pore of the kag structure, any additional pentyl tether from the same bdc2− unit would be required to traverse the hexagonal pore, in which parallel bdc2− ligands are 19.9 Å apart, and adjacent ligands are 6.7 Å apart at a very obtuse angle. In these circumstances, we propose that the formation of a strained pcu structure becomes favored over kag, as seen in pentyl2(bdc)3-DMOF-1 and pbdc-5a-DMOF-1. The modified relationships between phases and topologies upon tether incorporation are summarized in Fig. 10.
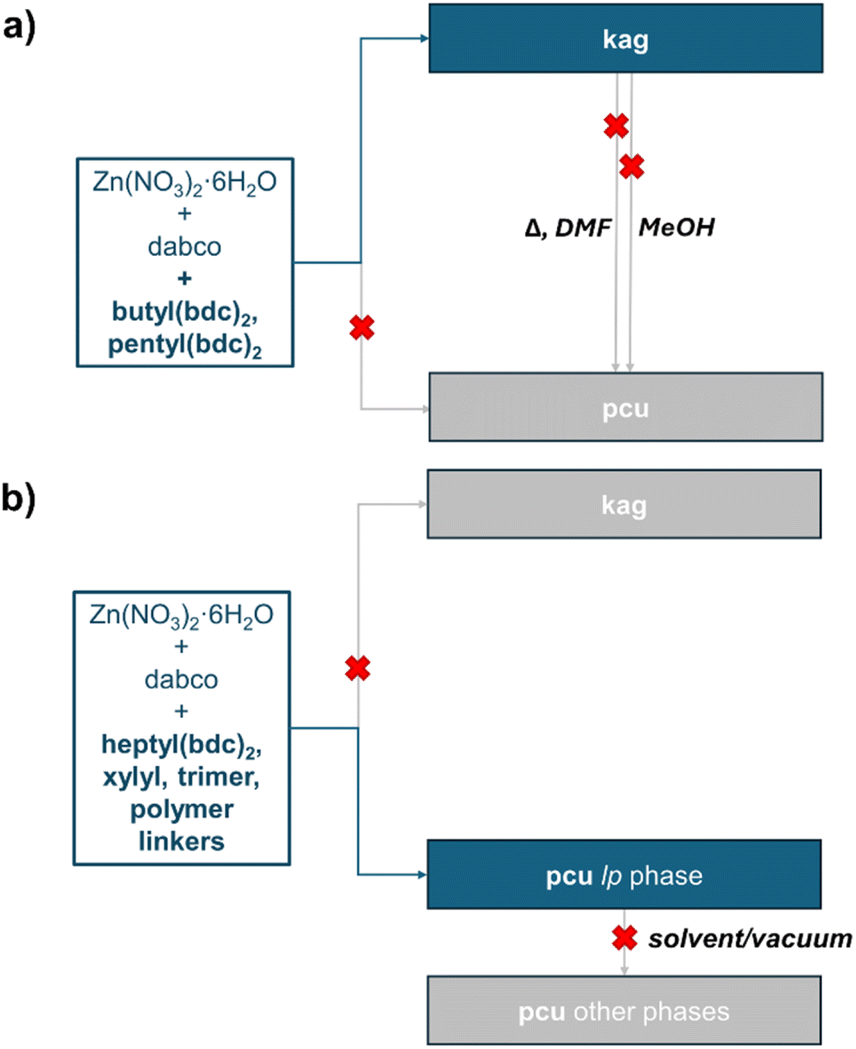 | ||
| Fig. 10 Schematic representation of the DMOF phases accessible to (a) butyl- and pentyl-tethered dimeric bdc ligands; (b) heptyl-tethered dimeric, pentyl-tethered dimeric, and polymeric bdc ligands. | ||
Conclusions
In this work, we have synthesized and characterized several new DMOFs based on oligomeric and polymeric linkers, with varying cross-link length, flexibility, and number of repeating ligand units. Our results show that although oligomeric and polymeric terephthalate-based ligands with dabco co-ligands adopt the canonical pcu-DMOF-1 structure in most cases, the ultimate structures are dictated by the length and flexibility of the tethering group. Specifically, we find that butyl- and pentyl-tethered dimers adopt an isomeric kag structure even under thermodynamically forcing conditions, whereas more rigid xylyl tethers of comparable length, or longer heptyl tethers adopt the parent pcu structure. Upon linking three or more terephthalate groups using pentyl spacers, we have found that ligands become incompatible with the kag structure and revert to pcu. Therefore, oligoMOFs based on dimeric ligands can form distinct structures from those based on trimers, or the corresponding polyMOFs under identical conditions, depending on the specific framework structure. These results are the first example of flexible inter-ligand tethers acting as structure-directing elements in MOFs and show that the influences of these tethers on overall structure are more nuanced than previously realized.Furthermore, we have shown that the phase transformations shown by the parent pcu-DMOF-1 are altered upon cross-link incorporation, and the large pore phase is stabilized. The solvent-mediated transformation of kag-DMOF-1 to pcu-DMOF-1 is also rendered inaccessible upon cross-link incorporation. Therefore, the phase and topological landscapes of the kag- and pcu-DMOF-1 structures have been shown to be highly sensitive to manipulation of the constraints over ligand geometry imposed by means of cross-linking moieties. The use of hexyl-tethered dimeric ligands further resulted in the formation of a possible new isomeric phase of DMOFs, showing that cross-links may also provide a route to novel structures. Understanding the impact of ligand cross-links as design elements represents a step towards enhanced understanding of MOF-polymer compatibility.
Data availability
The data supporting this article have been included as part of the ESI.†Author contributions
D. S. and S. M. C designed the materials and experimental strategy; D. S. conducted the experiments; D. S. and S. M. C wrote the manuscript. All authors have given approval to the final version of the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the Department of Energy, Office of Basic Energy Sciences, Division of Materials Science and Engineering (under Award No. DE-FG02-08ER46519). Preliminary funding was provided by the Defense Threat Reduction Agency (under Award No. HDTRA-12210037). We thank Dr Yongxuan Su for mass spectrometry at the Molecular Mass Spectrometry Facility at UC San Diego. We also thank Dr Minjung Kang for SEM images collected at the San Diego Nano-Technology Infrastructure (SDNI) of UC San Diego, a member of the National Nanotechnology Coordinated Infrastructure, which is supported by the National Science Foundation (ECCS-1542148). This work also made use of facilities and instrumentation at the UC San Diego Materials Research Science and Engineering Center (UCSD MRSEC, grant #DMR-2011924, supported by NSF), and the UC San Diego Department of Nanoengineering Materials Research Center (NE-MRC).References
- O. M. Yaghi, H. Li, C. Davis, D. Richardson and T. L. Groy, Acc. Chem. Res., 1998, 31, 474–484 CrossRef CAS
.
- B. F. Hoskins and R. Robson, J. Am. Chem. Soc., 1989, 111, 5962–5964 Search PubMed
- S. Kitagawa, R. Kitaura and S.-i. Noro, Angew. Chem., Int. Ed., 2004, 43, 2334–2375 CrossRef CAS PubMed
- G. Férey, J. Solid State Chem., 2000, 152, 37–48 Search PubMed
- B. Moulton and M. J. Zaworotko, Chem. Rev., 2001, 101, 1629–1658 Search PubMed
- H. Furukawa, Y. B. Go, N. Ko, Y. K. Park, F. J. Uribe-Romo, J. Kim, M. O'Keeffe and O. M. Yaghi, Inorg. Chem., 2011, 50, 9147–9152 CrossRef CAS PubMed
- H. Deng, S. Grunder, K. E. Cordova, C. Valente, H. Furukawa, M. Hmadeh, F. Gándara, A. C. Whalley, Z. Liu, S. Asahina, H. Kazumori, M. O'Keeffe, O. Terasaki, J. F. Stoddart and O. M. Yaghi, Science, 2012, 336, 1018–1023 CrossRef CAS PubMed
- M. Eddaoudi, J. Kim, N. Rosi, D. Vodak, J. Wachter, M. O'Keeffe and O. M. Yaghi, Science, 2002, 295, 469–472 CrossRef CAS PubMed
- R. Freund, S. Canossa, S. M. Cohen, W. Yan, H. Deng, V. Guillerm, M. Eddaoudi, D. G. Madden, D. Fairen-Jimenez, H. Lyu, L. K. Macreadie, Z. Ji. Y. Zhang, B. Wang, F. Haase, C. Woll, O. Zaremba, J. Andrea, S. Wuttke and C. S. Diercks, Angew. Chem., Int. Ed., 2021, 60, 23946–23974 CrossRef CAS PubMed
- T. L. Hennigar, D. C. MacQuarrie, P. Losier, R. D. Rogers and M. J. Zaworotko, Angew. Chem., Int. Ed., 1997, 36, 972–973 Search PubMed
- A. Karmakar, A. Paul and A. J. L. Pombeiro, CrystEngComm, 2017, 19, 4666–4695 RSC
- N. Zhu, D. Sensharma, P. Wix, M. J. Lennox, T. Düren, W. Y. Wong and W. Schmitt, Eur. J. Inorg. Chem., 2016, 1943–1947 Search PubMed
- F. Carson, J. Su, A. E. Platero-Prats, W. Wan, Y. Yun, L. Samain and X. Zou, Cryst. Growth Des., 2013, 13, 5036–5044 CrossRef CAS
- T. Tanasaro, K. Adpakpang, S. Ittisanronnachai, K. Faungnawakij, T. Butburee, S. Wannapaiboon, M. Ogawa and S. Bureekaew, Cryst. Growth Des., 2018, 18, 16–21 CrossRef CAS
- M. Perfecto-Irigaray, G. Beobide, O. Castillo, I. da Silva, D. García-Lojo, A. Luque, A. Mendia and S. Pérez-Yáñez, Chem. Commun., 2019, 55, 5954–5957 RSC
- Y.-B. Zhang, H. Furukawa, N. Ko, W. Nie, H. J. Park, S. Okajima, K. E. Cordova, H. Deng, J. Kim and O. M. Yaghi, J. Am. Chem. Soc., 2015, 137, 2641–2650 CrossRef CAS PubMed
- D. N. Dybtsev, H. Chun and K. Kim, Angew. Chem., Int. Ed., 2004, 43, 5033–5036 Search PubMed
- H. Chun and J. Moon, Inorg. Chem., 2007, 46, 4371–4373 CrossRef CAS PubMed
- M. Kondo, Y. Takashima, J. Seo, S. Kitagawa and S. Furukawa, CrystEngComm, 2010, 12, 2350–2353 RSC
- K. Zhou, S. Chaemchuen, Z. Wu and F. Verpoort, Microporous Mesoporous Mater., 2017, 239, 28–33 CrossRef CAS
- J. Hungerford and K. S. Walton, Inorg. Chem., 2019, 58, 7690–7697 Search PubMed
- S. Chaemchuen, K. Zhou, M. S. Yusubov, P. S. Postnikov, N. Klomkliang and F. Verpoort, Microporous Mesoporous Mater., 2019, 278, 99–104 CrossRef CAS
- S. Henke, R. Schmid, J. D. Grunwaldt and R. A. Fischer, Chem. Eur. J., 2010, 16, 14296–14306 CrossRef CAS PubMed
- S. Henke, A. Schneemann, A. Wütscher and R. A. Fischer, J. Am. Chem. Soc., 2012, 134, 9464–9474 CrossRef CAS PubMed
- A. Schneemann, S. Henke, I. Schwedler and R. A. Fischer, ChemPhysChem, 2014, 15, 823–839 Search PubMed
- L. K. Cadman, J. K. Bristow, N. E. Stubbs, D. Tiana, M. F. Mahon, A. Walsh and A. D. Burrows, Dalton Trans., 2016, 45, 4316–4326 RSC
- H. Hahm, K. Yoo, H. Ha and M. Kim, Inorg. Chem., 2016, 55, 7576–7581 CrossRef CAS PubMed
- H. Ha, H. Hahm, D. G. Jwa, K. Yoo, M. H. Park, M. Yoon, Y. Kim and M. Kim, CrystEngComm, 2017, 19, 5361–5368 RSC
- M. Kim, J. A. Boissonnault, P. V. Dau and S. M. Cohen, Angew. Chem., Int. Ed., 2011, 51, 12193–12196 Search PubMed
- Z. Wang and S. M. Cohen, J. Am. Chem. Soc., 2009, 131, 16675–16677 Search PubMed
- H. Kim and S. M. Cohen, Inorg. Chem., 2024, 63, 1853–1857 Search PubMed
- R. A. Dodson, J. Park, J. Kim, M. J. Cliffe and S. M. Cohen, Inorg. Chem., 2022, 61, 12284–12292 CrossRef CAS PubMed
- C. A. Allen, J. A. Boissonnault, J. Cirera, R. Gulland, F. Paesani and S. M. Cohen, Chem. Commun., 2013, 49, 3200–3202 RSC
- C. Allen and S. Cohen, Inorg. Chem., 2014, 53, 7014–7019 CrossRef CAS PubMed
- Z. Zhang, H. T. H. Nguyen, S. A. Miller and S. M. Cohen, Angew. Chem., Int. Ed., 2015, 54, 6152–6157 CrossRef CAS PubMed
- P. G. Mileo, S. Yuan, S. Ayala Jr, P. Duan, R. Semino, S. M. Cohen, K. Schmidt-Rohr and G. Maurin, J. Am. Chem. Soc., 2020, 142, 10863–10868 CrossRef CAS PubMed
- D. S. Rollins, J. Geary, A. H. Wong and D. J. Xiao, Chem. Commun., 2022, 58, 12361–12364 Search PubMed
- J. Geary, J. P. Aalto and D. J. Xiao, Chem. Mater., 2024, 36, 3949–3956 CrossRef CAS
- Y. Gu, M. Huang, W. Zhang, M. A. Pearson and J. A. Johnson, Angew. Chem., Int. Ed., 2019, 58, 16676–16681 CrossRef CAS PubMed
- M. MacLeod and J. Johnson, Polym. Chem., 2017, 8, 4488–4493 RSC
- J. Jiao, H. Liu, D. Bai and Y. He, Inorg. Chem., 2016, 55, 3974–3979 CrossRef CAS PubMed
- L. Feng, K.-Y. Wang, X.-L. Lv, J. A. Powell, T.-H. Yan, J. Willman and H.-C. Zhou, J. Am. Chem. Soc., 2019, 141, 14524–14529 CrossRef CAS PubMed
- J. Geary, A. H. Wong and D. J. Xiao, J. Am. Chem. Soc., 2021, 143, 10317–10323 CrossRef CAS PubMed
- S. Ayala, K. C. Bentz and S. M. Cohen, Chem. Sci., 2019, 10, 1746–1753 RSC
- Z. Zhang, H. T. H. Nguyen, S. A. Miller, A. M. Ploskonka, J. B. DeCoste and S. M. Cohen, J. Am. Chem. Soc., 2016, 138, 920–925 Search PubMed
- S. Ayala, Z. Zhang and S. M. Cohen, Chem. Commun., 2017, 53, 3058–3061 Search PubMed
- C. R. Groom, I. J. Bruno, M. P. Lightfoot and S. C. Ward, Acta Crystallogr., Sect. B: Struct. Sci., Cryst. Eng. Mater., 2016, 72, 171–179 CrossRef CAS PubMed
Footnote |
| † Electronic supplementary information (ESI) available: General experimental details, synthetic procedures for all ligands and MOFs, adsorption data, Pawley refinement data, 1H digestion NMR data, Fig. S1–S60, and Tables S1–S4. See DOI: https://doi.org/10.1039/d4sc06109h |
| This journal is © The Royal Society of Chemistry 2024 |