
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Visible light-induced ruthenium(II)-catalyzed hydroarylation of unactivated olefins†
Sven
Trienes‡
 ab,
Stéphane
Golling‡
a,
Matthew H.
Gieuw
a,
Marco
Di Matteo
a and
Lutz
Ackermann
*ab
ab,
Stéphane
Golling‡
a,
Matthew H.
Gieuw
a,
Marco
Di Matteo
a and
Lutz
Ackermann
*ab
aWöhler Research Institute for Sustainable Chemistry (WISCh), Georg-August-Universität, Tammannstraße 2, 37077 Göttingen, Germany. E-mail: Lutz.Ackermann@chemie.uni-goettingen.de
bDZHK (German Centre for Cardiovascular Research), Potsdamer Straße 58, 10875 Berlin, Germany
First published on 23rd October 2024
Abstract
Hydroarylation reactions have emerged as a valuable tool for the direct functionalization of C–H bonds with ideal atom economy. However, common catalytic variants for these transformations largely require harsh reaction conditions, which often translate into reduced selectivites. In contrast, we herein report on a photo-induced hydroarylation of unactivated olefins at room temperature employing a readily available ruthenium(II) catalyst. Our findings include high position- and regio-selectivity and remarkable tolerance of a wide range of functional groups, which further enabled the late-stage diversification.
Introduction
During the last decades, C–H activation has surfaced as a powerful and sustainable tool in modern organic synthesis.1–6 In this context, ruthenium has emerged as a privileged metal for this type of transformation.7,8 Particularly, the direct aryl addition to C–C double or triple bonds via hydroarylation turned out to be an attractive and highly atom-economic approach for the direct functionalization of aromatic C(sp2)–H bonds. Different approaches were developed to realize the transition metal-catalyzed anti-Markovnikov hydroarylation of unactivated olefins. Prominent transition metal-catalysts for this transformation9 are primarily based on ruthenium10–15 – with pioneering contributions by Lewis16 and Murai17 – rhodium18–23 or iridium.24–29 However, these processes are of limited utility for late-stage functionalization, as they require elevated reaction temperatures. For the same reason, the degree of C–H bond functionalization can be difficult to control, as overfunctionalization is observed in some cases. To overcome this challenge, Yoshikai30–32 reported on the cobalt-catalyzed hydroarylation of olefins at room temperature. Although those reactions were performed at reduced temperature and therefore prevented the formation of the bis-alkylated product, they required a substoichiometric amount of Grignard reagents which translates into reduced functional group tolerance, and hence potentially limits viable late-stage transformations of complex molecules (Scheme 1A). | ||
| Scheme 1 (A) Previous work: metal-catalyzed anti-Markovnikov hydroarylation of olefins; (B) this work: photo-induced ruthenium-catalyzed hydroarylation of non-activated olefins at room temperature. | ||
To circumvent the harsh reaction conditions represented either by high reaction temperatures or Grignard reagents, we envisioned that a photochemical approach could realize the desired transformation under significantly milder conditions. Additionally, visible light irradiation appears as sustainable and easily tunable driver for chemical reactions, comparable to electrochemistry,33–36 that has proven to be highly efficient in organic chemistry.37–43 Among a variety of different photochemical approaches, dual-catalytic systems with a transition metal-catalyst and an exogeneous photosensitizer, often based on ruthenium or iridium, showed remarkable efficiency. Alternatively, in some cases the transition metal-catalyst itself can show interesting photophysical properties, thereby superseding the addition of a separate photocatalyst. In the context of photochemical C–H functionalization, both with and without an exogenous photocatalyst, transformations utilizing transition metals,44,45 such as palladium,46–51 rhodium,52,53 copper54–56 or iron57 have been exploited. Another metal with remarkable synergistic effects of merging photochemistry and C–H activation is ruthenium.58 Within our program on photoinduced, ruthenium-catalyzed C–H functionalizations,59–63 we wondered whether a photo-induced ruthenium-catalyzed hydroarylation of non-activated olefins would indeed be viable at room temperature (Scheme 1B).
Results and discussion
We commenced our studies by probing reaction conditions for the envisioned photo-enabled, room-temperature ruthenium-catalyzed hydroarylation of olefins with ketimine 1a as model substrate. [Ru(OAc)2(p-cymene)] turned out to be the catalyst of choice for this transformation, affording the corresponding alkylated ketone 2a in 85% yield after hydrolysis (Table 1, entry 1). Other ruthenium(II) catalysts, such as [RuCl2(p-cymene)]2 or [Ru(O2CMes)2(p-cymene)], allowed the formation of the desired product 2a, albeit with lower efficacy (Table 1, entries 2 and 3). Several other arene-containing ruthenium-complexes were also employed giving comparable efficacy.64 The biscationic ruthenium-aqua complex65 [Ru(H2O)(t-BuCN)5][BF4]2 gave low yields, also in the presence of KOAc (Table 1, entry 4). Likewise, the arene-free, biscationic complex66 [Ru(MeCN)6][BF4]2 failed to furnish the desired product 2a in the absence of light, highlighting the unique features of the metallaphotocatalysis (Table 1, entry 5). RuCl3·3H2O and Ru3(CO)12 failed to give the desired product (Table 1, entries 6, 7). Additional carboxylate sources67 did not improve the catalytic performance (Table 1, entries 8, 9). Finally, a similar result was obtained when the p-methoxyphenyl (PMP) moiety was substituted by a 3,4,5-trimethoxyphenyl group (TMP) (Table 1, entry 10). Control experiments were conducted, leading to a complete inhibition of reactivity in the dark or without the ruthenium catalyst, mirroring their essential role in the catalysis (Table 1, entry 11).|
|
||
|---|---|---|
| Entry | Deviation from standard conditions | Yield 2ab (%) |
| a Reaction conditions: 1a (0.20 mmol), 1-octene (0.60 mmol), [Ru(OAc)2(p-cymene)] (10 mol%), 1,4-dioxane (1.0 mL), room temperature (30–35 °C). Subsequent hydrolysis with 1 M HCl (3.0 mL). Detailed experimental procedures are shown in the ESI. b Yields of isolated product. Yields in parentheses determined by 1H NMR using an internal standard. c 24 h, 30 °C, in the dark. d Using [RuCl2(p-cymene)]2 as catalyst. PMP = p-methoxyphenyl. | ||
| 1 | No deviation | 85 |
| 2 | [RuCl2(p-cymene)]2 | 32 |
| 3 | [Ru(O2CMes)2(p-cymene)] | 69 |
| 4 | [Ru(H2O)(t-BuCN)5][BF4]2 + KOAc (20 mol%) | (6) |
| 5c | [Ru(MeCN)6][BF4]2 + KOAc (20 mol%) | 0 |
| 6 | RuCl3·3H2O | 0 |
| 7 | Ru3(CO)12 | 0 |
| 8d | 1-Cyclohexenecarboxylic acid as additive | 85 |
| 9d | Boc-Leu-OH as additive | (30) |
| 10 | 3,4,5-Trimethoxyphenyl instead of p-methoxyphenyl | 68 |
| 11 | Without light or without [Ru] | 0 |
Substrate scope evaluation
With the optimized conditions in hand, we explored the versatility of the direct hydroarylation of 1-octene with various ketimines 1 (Scheme 2A). Initially, the synthetic utility was successfully demonstrated by upscaling our standard reaction on 1.2 mmol scale yielding the alkylated compound 2a in 90% yield. Subsequently, a wide range of substituted ketimines 1 were subjected to our reaction conditions to evaluate the functional group compatibility. Noteworthy, numerous functionalities were well tolerated, and the mono-alkylated product was selectively obtained in all cases. Different alkyl substituents in the para-position (2b–2e) or a phenyl-group (2f) afforded the desired compounds in excellent yields. Additionally, the use of various electron donating groups such as methoxy (2g), thioether (2h), dimethylamine (2i), 1,3-dioxole (2j) or mesylate (2k) led to the targeted products in high yields. It is noteworthy that only one isomer was observed in the case of 1,3-dioxole derivative (2j). Other interesting functionalities, such as methyl ester (2l), p-fluoro (2m), p-chloro (2n), p-trifluoromethyl (2o) or even p-mesyl (2p) were well tolerated. Subsequently, the use of ketimines bearing a heterocycle in para-position were also smoothly transformed into the corresponding hydroarylation products with yields up to 95%, demonstrating the excellent functional group tolerance of this approach (2q–2s). Finally, substituents in the meta-position of the aromatic ring were also tolerated, and only one constitutional isomer was obtained (2t, 2u) in 59% and 74% yield, respectively. | ||
| Scheme 2 Substrate scope for various ketimines 1 with 1-octene. Reaction conditions: 1 (0.20 mmol), 1-octene (0.60 mmol), [Ru(OAc)2(p-cymene)] (10 mol%), 1,4-dioxane (1.0 mL), room temperature (30–35 °C). Detailed experimental procedures are shown in the ESI.† (a) Subsequent hydrolysis with 1 M HCl (3.0 mL). (b) 96 h reaction time. (c) 1.6 equiv. of 1-octene. | ||
Then, the effect of substituents on the alkene was explored (Scheme 2B). A longer alkyl chain did not impact the reactivity as ketone 2v was obtained in 95% yield. The use of an alkene bearing a phenyl ring led to an excellent yield of 98% (2w). A sensitive bromide derivative (2x) and a free alcohol (2y) were also compatible and afforded the corresponding compounds in good yields. Different silyl groups (2z-ab) proved to be suitable as well. Similarly, a boronic acid masking group (MIDA) was well tolerated (2ac). Finally, terminal alkenes incorporated in important biorelevant compounds were successfully transformed into the desired hydroarylation products. Therefore, the reaction was performed in the presence of ibuprofen (2ad) and cholesterol (2ae) derivatives giving the targeted products in acceptable to excellent yields.
Additionally, the ruthenium-catalyzed hydroarylation proved viable for different directing groups (Scheme 2C). The use of a bis(p-methoxyphenyl)imine led to the corresponding mono-alkylated product 3a in a satisfactory yield of 59%. A good yield of 58% was observed for bis(diethylamino phenyl)imine 3b. Moreover, a cyclic imine was employed to furnish the corresponding dihydroisoquinoline 3c in moderate yield. Furthermore, heteroarenes also enabled the photo-induced hydroarylation. Therefore, 2-phenylpyridine gave access to the alkylated product 3d in 80% yield. The mild conditions of the photo-induced C–H activation translated into improved mono-selectivities as compared to thermal mode of action.10,64 A reduced efficacy was observed for a pyrimidyl coordinating group (3e) and an aldimine (3f).
Mechanistic investigations
To gain insights into the working mode of the metallaphotocatalysis, mechanistic studies were conducted. Initially, the role of the blue light irradiation was investigated in detail with an on-off experiment (Scheme 3A). Here, the essential role of the light was confirmed. Next, the reaction was monitored via1H NMR spectroscopy, with free p-cymene being observed.59 The addition of a substoichiometric amount of p-cymene to the reaction mixture inhibited catalysis.64 Additionally, a quantum yield64 of 1.2% was in good agreement. Finally, UV-spectroscopy allowed us to identify that both the ruthenium complex and ruthenacycle Ru-1 show a strong absorbance of light in the range between 380–460 nm. Analysis of the reaction mixture before and after irradiation with blue light also show the formation of new species (Scheme 3C).64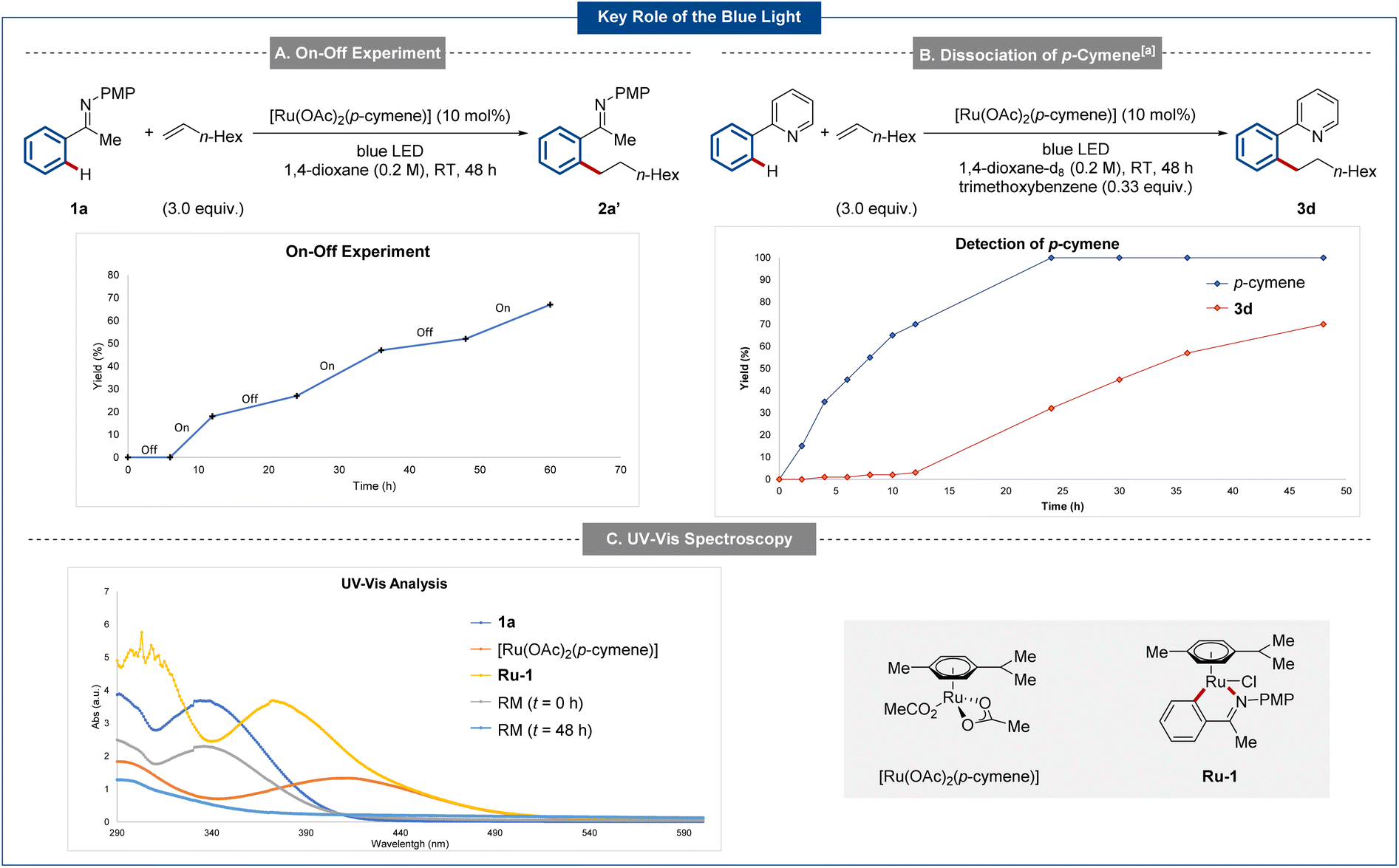 | ||
| Scheme 3 Key mechanistic findings on the role of the blue light. Detailed experimental procedures are shown in ESI.† (a) Relative yields referring to the initial amount of phenylpyridine (0.2 mmol) or [Ru(OAc)2(p-cymene)] (0.02 mmol). RM = reaction mixture. | ||
To gain further insights into the potential mode of action, an intermolecular competition experiment was performed between electron-rich ketimine 1b and electron-deficient ketimine 1o (Scheme 4A). After 24 h, the trifluoromethyl-substituted compound 2o was obtained in 30% yield against 5% for the methyl-substituted product 2b. Consequently, the C–H activation may occur preferentially on electron-poor arenes with weaker C–H bonds indicating a carboxylate-assisted cycloruthenation. This tendency was also observed during the evaluation of the substrate scope of the ketimine 1, as slightly higher yields were obtained in the case of electron-poor ketimines (Scheme 2). To pursue our studies, the independently synthesized ruthenacycle Ru-1 was employed as catalyst under our standard conditions. Notably, this complex was only effective in the presence of a catalytic amount of potassium acetate (20 mol%), further substantiating that a carboxylate-assisted ruthenation is involved in the catalytic cycle (Scheme 4B). Interestingly, product formation was not observed in the dark with complex Ru-1 as catalyst, being suggestive of light-independent cyclometallation, along with a light-induced activation – arguably through decoordination of the p-cymene ligand.64 Next, deuterium-labelling experiments were carried out. When ketimine 1a-d5 was treated with [Ru(OAc)2(p-cymene)] under blue light irradiation, a significant H/D exchange was observed (Scheme 4C). This result points to the reversibility of the cleavage of the ortho C–H bond. The hydroarylation reaction between 1a-d5 and dimethyl(phenyl)(vinyl)silane was also examined. Deuterium incorporation was only observed at the β-position of the hydroarylation product 2ac-d5 while no deuterium was introduced at the α-position (Scheme 4C). This observation is in contrast to rhodium(I)-catalyzed alkylations of aromatic amides reported by Chatani whose mechanistic proposal was considered to undergo a carbene mechanism which was confirmed by deuterium labelling experiments and DFT calculations.68 The carbene mechanism was therefore considered unlikely for the photo-induced hydroarylation.
 | ||
| Scheme 4 Key mechanistic findings. | ||
Based on our experiments and literature precedents,59–61 a plausible mechanism for the photo-enabled hydroarylation of olefins involves an initial C–H ruthenation of ketimine 1 to generate ruthenacycle A (Scheme 5). After light-induced p-cymene dissociation, the catalytic active species B is formed. Subsequently, the coordination of the olefin via intermediate C, along with migratory insertion into the Ru–C bond forms intermediate D. Then, acetic acid enables proto-demetallation to generate the species E. Finally, ligand exchange with another equivalent of the starting material releases the desired product 2a′, while regenerating catalyst B through cyclometalation.
 | ||
| Scheme 5 Proposed catalytic cycle. | ||
Conclusions
In summary, we reported on versatile C–H alkylations enabled by photo-induced ruthenium(II)-catalyzed hydroarylation of non-activated alkenes. In sharp contrast to established catalytic systems, the carboxylate-assisted ruthenium(II) catalysis proved efficient under exceedingly mild reaction conditions, namely at ambient temperature, enabled by visible light irradiation. The ruthenaphotocatalysis was easily conducted on gram-scale and its robustness was mirrored by a broad functional group tolerance.Data availability
The data supporting this article have been uploaded as part of the ESI.†Author contributions
Conceptualization, L. A.; funding acquisition, L. A.; investigation S. T., S. G., M. H. G., M. D. M., methodology, S. T., S. G.; resources, L. A., supervision, L. A.; writing – original draft, S. T., S. G., L. A., writing – reviewing & editing, all authors.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors gratefully acknowledge generous support by the DZHK, the DFG (Gottfried Wilhelm Leibniz award to L.A.) and the ERC Advanced Grant no. 101021358 (L.A.).Notes and references
- T. Rogge, N. Kaplaneris, N. Chatani, J. Kim, S. Chang, B. Punji, L. L. Schafer, D. G. Musaev, J. Wencel-Delord, C. A. Roberts, R. Sarpong, Z. E. Wilson, M. A. Brimble, M. J. Johansson and L. Ackermann, C–H activation, Nat. Rev. Methods Primers, 2021, 1, 43 CrossRef CAS.
- S. Rej, Y. Ano and N. Chatani, Bidentate Directing Groups: An Efficient Tool in C–H Bond Functionalization Chemistry for the Expedient Construction of C–C Bonds, Chem. Rev., 2020, 120, 1788–1887 CrossRef CAS PubMed.
- P. Gandeepan, T. Müller, D. Zell, G. Cera, S. Warratz and L. Ackermann, 3d Transition Metals for C–H Activation, Chem. Rev., 2019, 119, 2192–2452 CrossRef CAS PubMed.
- C. Sambiagio, D. Schönbauer, R. Blieck, T. Dao-Huy, G. Pototschnig, P. Schaaf, T. Wiesinger, M. F. Zia, J. Wencel-Delord, T. Besset, B. U. W. Maes and M. Schnürch, A comprehensive overview of directing groups applied in metal-catalysed C–H functionalisation chemistry, Chem. Soc. Rev., 2018, 47, 6603–6743 RSC.
- J. A. Leitch and C. G. Frost, Regioselective Transition-Metal-Catalyzed C–H Functionalization of Anilines, Synthesis, 2018, 50, 2693–2706 CrossRef CAS.
- J. Wencel-Delord and F. Glorius, C–H bond activation enables the rapid construction and late-stage diversification of functional molecules, Nat. Chem., 2013, 5, 369–375 CrossRef CAS PubMed.
- J. A. Leitch and C. G. Frost, Ruthenium-catalysed σ-activation for remote meta-selective C–H functionalisation, Chem. Soc. Rev., 2017, 46, 7145–7153 RSC.
- P. B. Arockiam, C. Bruneau and P. H. Dixneuf, Ruthenium(II)-Catalyzed C–H Bond Activation and Functionalization, Chem. Rev., 2012, 112, 5879–5918 CrossRef CAS PubMed.
- Z. Dong, Z. Ren, S. J. Thompson, Y. Xu and G. Dong, Transition-Metal-Catalyzed C–H Alkylation Using Alkenes, Chem. Rev., 2017, 117, 9333–9403 CrossRef CAS PubMed.
- M. Schinkel, I. Marek and L. Ackermann, Carboxylate-Assisted Ruthenium(II)-Catalyzed Hydroarylations of Unactivated Alkenes through C–H Cleavage, Angew. Chem., Int. Ed., 2013, 52, 3977–3980 CrossRef CAS PubMed.
- G. Rouquet and N. Chatani, Ruthenium-catalyzed ortho-C–H bond alkylation of aromatic amides with α,β-unsaturated ketones via bidentate-chelation assistance, Chem. Sci., 2013, 4, 2201–2208 RSC.
- L. Ackermann, S. I. Kozhushkov and D. S. Yufit, Ruthenium-Catalyzed Hydroarylation of Methylenecyclopropanes through C–H Bond Cleavage: Scope and Mechanism, Chem.–Eur. J., 2012, 18, 12068–12077 CrossRef CAS PubMed.
- M.-O. Simon, G. Ung and S. Darses, Tandem Catalysis: Alcohol Oxidation and C–C Bond Formation via C–H Bond Activation, Adv. Synth. Catal., 2011, 353, 1045–1048 CrossRef CAS.
- S. I. Kozhushkov, D. S. Yufit and L. Ackermann, Ruthenium-Catalyzed Hydroarylations of Methylenecyclopropanes: Mild C−H Bond Functionalizations with Conservation of Cyclopropane Rings, Org. Lett., 2008, 10, 3409–3412 CrossRef CAS PubMed.
- R. Martinez, R. Chevalier, S. Darses and J.-P. Genet, A Versatile Ruthenium Catalyst for C–C Bond Formation by C–H Bond Activation, Angew. Chem., Int. Ed., 2006, 45, 8232–8235 CrossRef CAS PubMed.
- L. N. Lewis and J. F. Smith, Catalytic carbon-carbon bond formation via ortho-metalated complexes, J. Am. Chem. Soc., 1986, 108, 2728–2735 CrossRef CAS.
- S. Murai, F. Kakiuchi, S. Sekine, Y. Tanaka, A. Kamatani, M. Sonoda and N. Chatani, Efficient catalytic addition of aromatic carbon-hydrogen bonds to olefins, Nature, 1993, 366, 529–531 CrossRef CAS.
- K. Shibata, T. Yamaguchi and N. Chatani, Rhodium-Catalyzed Alkylation of C–H Bonds in Aromatic Amides with Styrenes via Bidentate–Chelation Assistance, Org. Lett., 2015, 17, 3584–3587 CrossRef CAS PubMed.
- J. Kwak, Y. Ohk, Y. Jung and S. Chang, Rollover Cyclometalation Pathway in Rhodium Catalysis: Dramatic NHC Effects in the C–H Bond Functionalization, J. Am. Chem. Soc., 2012, 134, 17778–17788 CrossRef CAS PubMed.
- C.-H. Jun, C. W. Moon, J.-B. Hong, S.-G. Lim, K.-Y. Chung and Y.-H. Kim, Chelation-Assisted RhI-Catalyzed ortho-Alkylation of Aromatic Ketimines or Ketones with Olefins, Chem.–Eur. J., 2002, 8, 485–492 CrossRef CAS PubMed.
- C.-H. Jun, J.-B. Hong, Y.-H. Kim and K.-Y. Chung, The Catalytic Alkylation of Aromatic Imines by Wilkinson's Complex: The Domino Reaction of Hydroacylation and ortho-Alkylation, Angew. Chem., Int. Ed., 2000, 39, 3440–3442 CrossRef CAS PubMed.
- Y.-G. Lim, J.-B. Kang and Y. H. Kim, Regioselective alkylation of 2-phenylpyridines with terminal alkenes via C–H bond activation by a rhodium catalyst, J. Chem. Soc., Perkin Trans. 1, 1996, 2201–2206 RSC.
- Y.-G. Lim, Y. H. Kim and J.-B. Kang, Rhodium-catalysed regioselective alkylation of the phenyl ring of 2-phenylpyridines with olefins, J. Chem. Soc., Chem. Commun., 1994, 2267–2268 RSC.
- J. Kim, S.-W. Park, M.-H. Baik and S. Chang, Complete Switch of Selectivity in the C–H Alkenylation and Hydroarylation Catalyzed by Iridium: The Role of Directing Groups, J. Am. Chem. Soc., 2015, 137, 13448–13451 CrossRef CAS PubMed.
- T. Shibata and T. Shizuno, Iridium-Catalyzed Enantioselective C–H Alkylation of Ferrocenes with Alkenes Using Chiral Diene Ligands, Angew. Chem., Int. Ed., 2014, 53, 5410–5413 CrossRef CAS PubMed.
- S. Pan, N. Ryu and T. Shibata, Iridium(I)-Catalyzed Direct C–H Bond Alkylation of the C-7 Position of Indolines with Alkenes, Adv. Synth. Catal., 2014, 356, 929–933 CrossRef CAS.
- S. Pan, N. Ryu and T. Shibata, Ir(I)-Catalyzed C–H Bond Alkylation of C2-Position of Indole with Alkenes: Selective Synthesis of Linear or Branched 2-Alkylindoles, J. Am. Chem. Soc., 2012, 134, 17474–17477 CrossRef CAS PubMed.
- K. Tsuchikama, M. Kasagawa, Y.-K. Hashimoto, K. Endo and T. Shibata, Cationic iridium–BINAP complex-catalyzed addition of aryl ketones to alkynes and alkenes via directed C–H bond cleavage, J. Organomet. Chem., 2008, 693, 3939–3942 CrossRef CAS.
- R. Dorta and A. Togni, Addition of the ortho-C–H bonds of phenol across an olefin catalysed by a chiral iridium(I) diphosphine complex, Chem. Commun., 2003, 760–761 RSC.
- W. Xu and N. Yoshikai, Highly Linear Selective Cobalt-Catalyzed Addition of Aryl Imines to Styrenes: Reversing Intrinsic Regioselectivity by Ligand Elaboration, Angew. Chem., Int. Ed., 2014, 53, 14166–14170 CrossRef CAS PubMed.
- K. Gao and N. Yoshikai, Regioselectivity-Switchable Hydroarylation of Styrenes, J. Am. Chem. Soc., 2011, 133, 400–402 CrossRef CAS PubMed.
- K. Gao and N. Yoshikai, Cobalt–Phenanthroline Catalysts for the ortho Alkylation of Aromatic Imines under Mild Reaction Conditions, Angew. Chem., Int. Ed., 2011, 50, 6888–6892 CrossRef CAS PubMed.
- Y.-K. Xing, Z.-H. Wang, P. Fang, C. Ma and T.-S. Mei, Divergent synthesis of aryl amines and dihydroquinazolinones via electrochemistry-enabled rhodium-catalyzed C–H functionalization, Sci. China Chem., 2023, 66, 2863–2870 CrossRef CAS.
- Y. Zhang, Z. Cai, S. Warratz, C. Ma and L. Ackermann, Recent advances in electrooxidative radical transformations of alkynes, Sci. China Chem., 2023, 66, 703–724 CrossRef CAS.
- M. Yan, Y. Kawamata and P. S. Baran, Synthetic Organic Electrochemical Methods Since 2000: On the Verge of a Renaissance, Chem. Rev., 2017, 117, 13230–13319 CrossRef CAS PubMed.
- Y. Wang, S. Dana, H. Long, Y. Xu, Y. Li, N. Kaplaneris and L. Ackermann, Electrochemical Late-Stage Functionalization, Chem. Rev., 2023, 123, 11269–11335 CrossRef CAS PubMed.
- P. Bellotti, H.-M. Huang, T. Faber and F. Glorius, Photocatalytic Late-Stage C–H Functionalization, Chem. Rev., 2023, 123, 4237–4352 CrossRef CAS PubMed.
- J. Großkopf, T. Kratz, T. Rigotti and T. Bach, Enantioselective Photochemical Reactions Enabled by Triplet Energy Transfer, Chem. Rev., 2022, 122, 1626–1653 CrossRef PubMed.
- C.-S. Wang, P. H. Dixneuf and J.-F. Soulé, Photoredox Catalysis for Building C–C Bonds from C(sp2)–H Bonds, Chem. Rev., 2018, 118, 7532–7585 CrossRef CAS PubMed.
- L. Marzo, S. K. Pagire, O. Reiser and B. König, Visible-Light Photocatalysis: Does It Make a Difference in Organic Synthesis?, Angew. Chem., Int. Ed., 2018, 57, 10034–10072 CrossRef CAS PubMed.
- J. Twilton, C. Le, P. Zhang, M. H. Shaw, R. W. Evans and D. W. C. MacMillan, The merger of transition metal and photocatalysis, Nat. Rev. Chem, 2017, 1, 0052 CrossRef CAS.
- N. A. Romero and D. A. Nicewicz, Organic Photoredox Catalysis, Chem. Rev., 2016, 116, 10075–10166 CrossRef CAS PubMed.
- J. M. R. Narayanam and C. R. J. Stephenson, Visible light photoredox catalysis: applications in organic synthesis, Chem. Soc. Rev., 2011, 40, 102–113 RSC.
- K. P. S. Cheung, S. Sarkar and V. Gevorgyan, Visible Light-Induced Transition Metal Catalysis, Chem. Rev., 2022, 122, 1543–1625 CrossRef PubMed.
- R. Kancherla, K. Muralirajan, A. Sagadevan and M. Rueping, Visible Light-Induced Excited-State Transition-Metal Catalysis, Trends Chem., 2019, 1, 510–523 CrossRef CAS.
- Y.-C. Hsu, V. C. C. Wang, K.-C. Au-Yeung, C.-Y. Tsai, C.-C. Chang, B.-C. Lin, Y.-T. Chan, C.-P. Hsu, G. P. A. Yap, T. Jurca and T.-G. Ong, One-Pot Tandem Photoredox and Cross-Coupling Catalysis with a Single Palladium Carbodicarbene Complex, Angew. Chem., Int. Ed., 2018, 57, 4622–4626 CrossRef CAS PubMed.
- W.-J. Zhou, G.-M. Cao, G. Shen, X.-Y. Zhu, Y.-Y. Gui, J.-H. Ye, L. Sun, L.-L. Liao, J. Li and D.-G. Yu, Visible-Light-Driven Palladium-Catalyzed Radical Alkylation of C−H Bonds with Unactivated Alkyl Bromides, Angew. Chem., Int. Ed., 2017, 56, 15683–15687 CrossRef CAS PubMed.
- D. Kalyani, K. B. McMurtrey, S. R. Neufeldt and M. S. Sanford, Room-Temperature C–H Arylation: Merger of Pd-Catalyzed C–H Functionalization and Visible-Light Photocatalysis, J. Am. Chem. Soc., 2011, 133, 18566–18569 CrossRef CAS PubMed.
- A. Saha, S. Guin, W. Ali, T. Bhattacharya, S. Sasmal, N. Goswami, G. Prakash, S. K. Sinha, H. B. Chandrashekar, S. Panda, S. S. Anjana and D. Maiti, Photoinduced Regioselective Olefination of Arenes at Proximal and Distal Sites, J. Am. Chem. Soc., 2022, 144, 1929–1940 CrossRef CAS PubMed.
- K. Pak Shing Cheung, J. Fang, K. Mukherjee, A. Mihranyan and V. Gevorgyan, Asymmetric intermolecular allylic C–H amination of alkenes with aliphatic amines, Science, 2022, 378, 1207–1213 CrossRef CAS PubMed.
- S. Sarkar, K. P. S. Cheung and V. Gevorgyan, Recent Advances in Visible Light Induced Palladium Catalysis, Angew. Chem., Int. Ed., 2024, 63, e202311972 CrossRef CAS PubMed.
- J. Tanaka, Y. Nagashima, A. J. Araujo Dias and K. Tanaka, Photo-Induced ortho-C–H Borylation of Arenes through In Situ Generation of Rhodium(II) Ate Complexes, J. Am. Chem. Soc., 2021, 143, 11325–11331 CrossRef CAS PubMed.
- J. Thongpaen, R. Manguin, V. Dorcet, T. Vives, C. Duhayon, M. Mauduit and O. Baslé, Visible Light Induced Rhodium(I)-Catalyzed C−H Borylation, Angew. Chem., Int. Ed., 2019, 58, 15244–15248 CrossRef CAS PubMed.
- S. Trienes, J. Xu and L. Ackermann, Photoinduced C–H arylation of 1,3-azoles via copper/photoredox dual catalysis, Chem. Sci., 2024, 15, 7293–7299 RSC.
- C. Li, B. Chen, X. Ma, X. Mo and G. Zhang, Light-Promoted Copper-Catalyzed Enantioselective Alkylation of Azoles, Angew. Chem., Int. Ed., 2021, 60, 2130–2134 CrossRef CAS PubMed.
- F. Yang, J. Koeller and L. Ackermann, Photoinduced Copper-Catalyzed C−H Arylation at Room Temperature, Angew. Chem., Int. Ed., 2016, 55, 4759–4762 CrossRef CAS PubMed.
- A. M. Messinis, T. von Münchow, M. Surke and L. Ackermann, Room temperature photo-promoted iron-catalysed arene C–H alkenylation without Grignard reagents, Nat. Catal., 2024, 7, 273–284 CrossRef CAS.
- X. Wang, G. An and G. Li, Visible Light-Induced Ru-Catalyzed Directing-Group-Assisted C−H Activation of Arenes, ChemCatChem, 2024, 16, e202301523 CrossRef CAS.
- Y. Wang, S. Chen, X. Chen, A. Zangarelli and L. Ackermann, Photo-Induced Ruthenium-Catalyzed Double Remote C(sp2)−H/C(sp3)−H Functionalizations by Radical Relay, Angew. Chem., Int. Ed., 2022, 61, e202205562 CrossRef CAS PubMed.
- J. Struwe, K. Korvorapun, A. Zangarelli and L. Ackermann, Photo-Induced Ruthenium-Catalyzed C−H Benzylations and Allylations at Room Temperature, Chem.–Eur. J., 2021, 27, 16237–16241 CrossRef CAS PubMed.
- K. Korvorapun, J. Struwe, R. Kuniyil, A. Zangarelli, A. Casnati, M. Waeterschoot and L. Ackermann, Photo-Induced Ruthenium-Catalyzed C−H Arylations at Ambient Temperature, Angew. Chem., Int. Ed., 2020, 59, 18103–18109 CrossRef CAS PubMed.
- P. Gandeepan, J. Koeller, K. Korvorapun, J. Mohr and L. Ackermann, Visible-Light-Enabled Ruthenium-Catalyzed meta-C−H Alkylation at Room Temperature, Angew. Chem., Int. Ed., 2019, 58, 9820–9825 CrossRef CAS PubMed.
- T. Michiyuki, I. Maksso and L. Ackermann, Photo-Induced Ruthenium-Catalyzed C−H Arylation Polymerization at Ambient Temperature, Angew. Chem., Int. Ed., 2024, 63, e202400845 CrossRef CAS PubMed.
- For detailed information, see the ESI.†.
- G. McArthur, J. H. Docherty, M. D. Hareram, M. Simonetti, I. J. Vitorica-Yrezabal, J. J. Douglas and I. Larrosa, An air- and moisture-stable ruthenium precatalyst for diverse reactivity, Nat. Chem., 2024, 16, 1141–1150 CrossRef CAS PubMed.
- C. C. Underwood, B. S. Stadelman, M. L. Sleeper and J. L. Brumaghim, Synthesis and electrochemical characterization of [Ru(NCCH3)6]2+, tris(acetonitrile) tris(pyrazolyl)borate, and tris(acetonitrile) tris(pyrazolyl)methane ruthenium(II) complexes, Inorg. Chim. Acta, 2013, 405, 470–476 CrossRef CAS.
- L. Ackermann, Carboxylate-Assisted Transition-Metal-Catalyzed C−H Bond Functionalizations: Mechanism and Scope, Chem. Rev., 2011, 111, 1315–1345 CrossRef CAS PubMed.
- T. Yamaguchi, S. Natsui, K. Shibata, K. Yamazaki, S. Rej, Y. Ano and N. Chatani, Rhodium-Catalyzed Alkylation of C−H Bonds in Aromatic Amides with Non-activated 1-Alkenes: The Possible Generation of Carbene Intermediates from Alkenes, Chem.–Eur. J., 2019, 25, 6915–6919 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4sc06005a |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2024 |