
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceHydroselenation of olefins: elucidating the β-selenium effect†
Gabriel S.
Phun‡
 b,
Hannah S.
Slocumb‡
b,
Kirsten J.
Ruud
b,
Shaozhen
Nie
c,
Cheyenne
Antonio
d,
Filipp
Furche
*b,
Vy M.
Dong
*b and
Xiao-Hui
Yang
*ae
b,
Hannah S.
Slocumb‡
b,
Kirsten J.
Ruud
b,
Shaozhen
Nie
c,
Cheyenne
Antonio
d,
Filipp
Furche
*b,
Vy M.
Dong
*b and
Xiao-Hui
Yang
*ae
aAdvanced Research Institute of Multidisciplinary Science, School of Chemistry and Chemical Engineering, Key Laboratory of Medical Molecule Science and Pharmaceutical Engineering, Ministry of Industry and Information Technology, Beijing Institute of Technology, Beijing, 100081, P. R. China. E-mail: xhyang@bit.edu.cn
bDepartment of Chemistry, University of California, Irvine, California 92697, USA. E-mail: filipp.furche@uci.edu; dongv@uci.edu
cDepartment of Medicinal Chemistry, Glaxo-Smith-Kline, Collegeville, Pennsylvania 19426, USA
dDepartment of Chemistry, University of California, San Francisco, California 94143, USA
eState Key Laboratory of Elemento-Organic Chemistry, Nankai University, Tianjin, 300071, P. R. China
First published on 25th November 2024
Abstract
We report a light-promoted hydroselenation of alkenes with high anti-Markovnikov selectivity. Blue light activates an aryl diselenide to generate a seleno radical with subsequent addition into an alkene to form a β-seleno carbon radical. Hydrogen atom transfer (HAT) from the selenol to the carbon radical generates the linear selenide with high selectivity in preference to the branched isomer. These studies reveal a unique β-selenium effect, where a selenide β to a carbon radical imparts high anti-selectivity for radical addition through delocalization of the HAT transition state.
Introduction
While an element essential to life, selenium remains less investigated relative to earlier chalcogens (oxygen and sulfur). Proteins containing selenocysteine (selenoproteins) participate in essential antioxidant, redox, and metabolic processes. Selenoprotein thioredoxin reductase 1 catalyzes several antioxidant and redox processes via selenocysteine and cysteine residues through the formation and cleavage of a Se–S bond (Fig. 1A).1 Disulfide bonds between neighboring cysteines are generally conformationally unfavorable, but the use of selenocysteines makes the dichalcogen bond viable due to selenium's longer bond lengths.2 Selenoproteins act as biomarkers for diseases including cancer and diabetes.3 Recently, abnormal plasma levels of micronutrient selenium were found in patients suffering severe cases of COVID-19.4 Apart from its occurence in nature, selenides also appear in drug targets.5 Ethaselen is a potent thioredoxin reductase 1 inhibitor which has undergone phase I clinical trials (Fig. 1A).6 Selenides show promise as both ligands and catalysts; the Zhao lab established a series of aryl selenide organocatalysts (Fig. 1A) for stereoselective difunctionalizations.7 Selenides serve as useful alkyl radical precursors;8 they exhibit higher stability in comparison to other radical precursors (e.g., halides)9 and thus can be carried through multi-step syntheses.10 Inoue and coworkers employed phenyl selenide (Fig. 1A) to generate an alkyl radical to achieve a three-component couplingin the construction of resiniferatoxin.10a However, this selenide required multi-step preparation from a different radical precursor. These compelling applications for Se motivate the development of new methods to prepare organoselenides, with hydroselenation being an especially attractive strategy. | ||
| Fig. 1 (A) Selenium in nature (catalytic residues in selenoprotein thioredoxin reductase 1) and synthesis (Inoue seleno radical precursor, ethaselen, and chiral catalyst). (B) Regiodivergent hydroselenation of alkenes via Rh (previous work) or light (proposed work). | ||
Hydroselenation is an atom economical approach to add a Se–H bond across a C–C π-bond.11 Several metal-catalyzed hydroselenations have been developed, mainly using activated alkenes (such as heterobicyclic alkenes,12a allenes,12bN-vinyl lactams,12c or α,β-unsaturated thioamides12d) or alkynes.12 Ogawa reported a light-promoted hydroselenation of alkynes, proposing an alkenyl radical intermediate.13 A number of other light-promoted reactions with selenides are known, including difunctionalizations,14 couplings,15 and cyclizations.16 Our lab communicated the Rh-catalyzed enantioselective hydroselenation of styrenes where we were able to access the Markovnikov-addition products with high stereoselectivity (Fig. 1B).12e In this follow up article, we aim to develop a complementary radical-mediated hydroselenation to form the anti-Markovnikov product. Several strategies to achieve a radical hydrofunctionalization are known. In the metal–hydride hydrogen atom transfer (MHAT) approach, a metal hydride adds to an alkene to generate a carbon radical, which is intercepted with a reactive carbon center or heteroatom.17,18 An alternative strategy involves the addition of a heteroatom radical to an alkene, then delivery of an H-atom via an H-atom donor such as a silane or thiol. One classic example of this is the thiol-ene click reaction, where an alkene undergoes hydrothiolation with a thiol in the presence of a radical initiator.19 Within this strategy, the addition of a selenide radical to alkenes has had reports in difunctionalizations featuring the addition of a selenide and C-, N-, O-, S-, F- coupling partners.14b–d,20 These strategies propose the intermediacy of a β-selenide radical, and either subsequent interception or oxidation. These previous functionalizations often occur with high anti-selectivity, yet the origins of this phenomenon remains unexplained. Despite numerous works proposing a β-selenide carbon radical14,17 and the infamy of the radical thiol-ene reaction, the analogous selenol-ene proceeding through a β-selenide carbon radical has yet to be explored. Herein, we imagined a light-generated seleno radical that would add to the terminal position of an alkene to generate the more stable internal radical, which could then undergo HAT to give the anti-Markovnikov selenide (Fig. 1B). If successful, this approach would allow us to (1) access a complementary anti-Markovnikov motif, and (2) study the reactivity of the β-seleno radical by both experiment and theory.
Results and discussion
To access the anti-Markovnikov product by hydroselenation, we chose feedstock chemical styrene (1a) and commercially available benzeneselenol (2a) as our model system. The benzeneselenol sample contained 3 mol% diphenyl diselenide (3a) due to spontaneous selenol oxidative dimerization.21 We observed no reactivity when the experiment was performed in the dark (Table 1, entry 1), however under ambient light we found that the hydroselenation can occur to give the anti-Markovnikov isomer 4aa in 15% yield and >20![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 rr (Table 1, entry 2). By shining blue LEDs on the reaction mixture and monitoring by TLC, we observed reaction completion and nearly quantitative yield of 4aa within 20 minutes (Table 1, entry 3). We suspected that the trace diphenyl diselenide 3a could be the photoactive species promoting reactivity.13 Indeed, TLC monitoring indicated the reaction time could be shortened to five minutes by adding an additional 20 mol% of diphenyl diselenide (3a) (Table 1, entry 4). Together, these results support the critical role of blue light and catalytic diselenide in this coupling.
:1 rr (Table 1, entry 2). By shining blue LEDs on the reaction mixture and monitoring by TLC, we observed reaction completion and nearly quantitative yield of 4aa within 20 minutes (Table 1, entry 3). We suspected that the trace diphenyl diselenide 3a could be the photoactive species promoting reactivity.13 Indeed, TLC monitoring indicated the reaction time could be shortened to five minutes by adding an additional 20 mol% of diphenyl diselenide (3a) (Table 1, entry 4). Together, these results support the critical role of blue light and catalytic diselenide in this coupling.
|
|
||||
|---|---|---|---|---|
| Entry | Conditions | 3a mol% | Time | Yield |
| 1 | Dark | 3 | 24 h | ND |
| 2 | Ambient light | 3 | 24 h | 15% |
| 3 | Blue LEDs | 3 | 20 min | 98% |
| 4 | Blue LEDs | 23 | 5 min | 98% |

A condition-based sensitivity screen was performed as outlined by the Glorius group (Fig. 2).22 Increased oxygen content lowered the yield by 26%. While high intensity of light showed similar results as the optimized conditions, low intensity afforded diminished yield (83%). Slightly lower efficiency is observed at a larger reaction scale (20× optimized conditions, 69% yield).
 | ||
| Fig. 2 Scope of light-catalyzed hydroselenation for (A) alkenes and (B) selenols. Reaction conditions for alkene scope: 1 (1.0 equiv.), 2 (1.5 equiv.), 1,2-dichloroethane (0.25 M). Reaction conditions for selenol scope: 1 (1.0 equiv.), 3 (1.5 equiv.), Ph2POH (1.5 equiv.), 1,2-dichloroethane (0.25 M). Reactions are run in a light box under blue LEDs and monitored by TLC for reaction completion (20 min to 24 h). Temperature in the box rises to 50 °C in 20 minutes due to LEDs and maintains that temperature over time. Regioselectivity determined by 1H NMR analysis of the unpurified reaction mixture. | ||
Synthetic scope
We examined the light-promoted hydroselenation of twenty-eight different alkenes with benzeneselenol (2a) to generate the corresponding linear selenides (Fig. 2). Olefin partners were less hazardous and more readily available, so we chose to explore this scope more widely than the selenols. High reactivity and regioselectivity (>20:1 rr) are obtained with mono-substituted alkenes (4ab–4av, 44–98% yield). Wide functional group tolerance is shown, including halides (4ab, 4ad, 4ae, 4ah), acids (4ak, 4ao), and heterocycles (4ar–4au). This process can occur with unactivated alkenes (4av), in contrast to the activated alkenes required in previous hydroselenations.12a–e,23 Both 1,1-disubstituted (4aw–4ay, 90–94% yield) and 1,2-disubstituted alkenes (4az–4aab, 79–93% yield) undergo addition. A conjugated diene undergoes the selenol-ene to afford the homoallylic selenide (4aac) with 87% yield and >20:1 rr. We applied this hydroselenation to prepare a Se-analogue of eletriptan, a migraine medication, which has an –SO2Ph group instead of the SePh group in 4aad. The reaction gave a Se-analogue 4aad in a 31% yield. Several selenols (generated in situ from the diselenide and Ph2POH) provide anti-Markovnikov products with high regioselectivity. A more electron-rich aryl selenol (4ba, 68% yield) shows higher reactivity compared to an electron-poor aryl selenol (4ca, 46% yield). A bulkier, less aromatic selenol had diminished reactivity (4da, 33% yield). We hypothesize the lower yielding reactions to form 4ca and 4da are due to the heterogeneous character of the solutions, resulting from the precipitation of diphenylphosphinic acid and resulting diphenylphosphinyl selenide byproducts in the dichloroethane (DCE) solvent, which led to low absorption of light. Overall, this light promoted selenol-ene occurs under mild conditions to form linear organoselenides with excellent regiocontrol.
Mechanistic hypotheses
Initially, we investigated the photoactive species of the transformation. UV-visible spectra showed that absorbance in the blue light region corresponds to diphenyl diselenide (Fig. S3†). Ultra-fast transient absorption spectra also showed identical behavior between diphenyl diselenide (3a), the benzeneselenol sample (2a), and the reaction mixture (see ESI 5.2 for details†). Based on these spectroscopic results, we concluded that the photoactive species is diselenide (3), which is formed by selenols when exposed to light or oxygen.21 Considering Ogawa's light-promoted hydroselenation of alkynes,13 we were interested whether this selenol-ene involves radicals. We found that azobisisobutyronitrile (AIBN) could be used as an initiator, giving 54% yield (Fig. 3A). The background reaction at 70 °C gave only 15% yield, highlighting that AIBN effectively promotes hydroselenation. Additionally, a mixture of two distinct diselenides in DCE gave mixed diselenide product when exposed to blue LEDs (Fig. 3B). Thus, AIBN-mediated reactivity and crossover studies indicate the importance of seleno radicals in the reaction. A light–dark study (Fig. S6†) revealed that light is necessary for reaction progress; when the light is off, the reactivity halts, but resumes when re-exposed to light. This observation is consistent with a previous report of high rates of recombination for seleno radicals.20f | ||
| Fig. 3 (A) Reaction run in the dark using AIBN as an initiator; (B) diselenide crossover experiment; (C) anti-selective addition of a deuterated selenol to a cyclic styrene; (D) anti-selective addition of selenol to a trisubstituted styrene; (E) anti-selective addition of a dueterated thiol to a cyclic styrene. | ||
Diselenide excitation is supported computationally (see ESI 6.2).† Excitation energies of 1a, 2a, 3a and an optimized S1 state geometry of 3a were computed using time-dependent density functional theory (TDDFT) to support the feasibility of photoabsorption and photoinduced homolytic cleavage. The excitation energies are consistent with the experimental UV-visible spectra, with 3a being the only species with a S1 excitation within range of the blue LEDs. The S1 state of 3a has Se–Se σ* character. Upon S1 state geometry optimization, the Se–Se bond lengthens by 0.47 Å. Moreover, the computed Se–Se bond dissociation energy changes from energetically unfavorable (35.7 kcal mol−1) to favorable (−15.3 kcal mol−1) upon S1 excitation.
Notably, simply using thiophenol with blue LEDs does not promote reactivity. Likely the reagent does not contain any species with absorptions in the blue LED range. However, by spiking the solution with diphenyl diselenide, we were able to obtain the sulfide product in 81% yield, with 4% selenide product as well (see ESI 5.6).† Deuterium labeling studies revealed high anti-selectivity for cyclic selenide 4aab (Fig. 3C). Similarly, hydroselenation of a tri-substituted acyclic alkene gave high selectivity for a single diastereomer for selenide 4aae (Fig. 3D). Additionally, deuterium labeling studies with thiophenol revealed high anti-selectivity for cyclic sulfide 8aab (Fig. 3E). High diastereoselectivity is relatively uncommon in radical reactions, so we investigated several potential mechanisms to determine which reflected the observed selectivity.
The importance of diphenyl diselenide and light in the reaction suggests activation of the diselenide 3 by light (Fig. 4). Excited state diselenide 3* can undergo homolytic bond cleavage to form seleno radical Ia. Based on literature precedence,13 we propose that the radical could then add to alkene 1ab to form C-radical IIab. To investigate the origin of anti-selectivity from this β-seleno radical, we proposed three diverging mechanisms: M1, a diastereoselective HAT; M2, a seleniranium process; and M3, a radical selenirane process. Two other mechanisms, involving a Dexter Energy Transfer (DET) event or formation of a styrene radical cation were also investigated, but these mechanisms favored syn-addition and were therefore ruled out (see ESI for details).† We found that M1 (Fig. 4) is the most likely mechanism, and we herein detail our experimental and computational results which support or refute M1, M2, and M3.
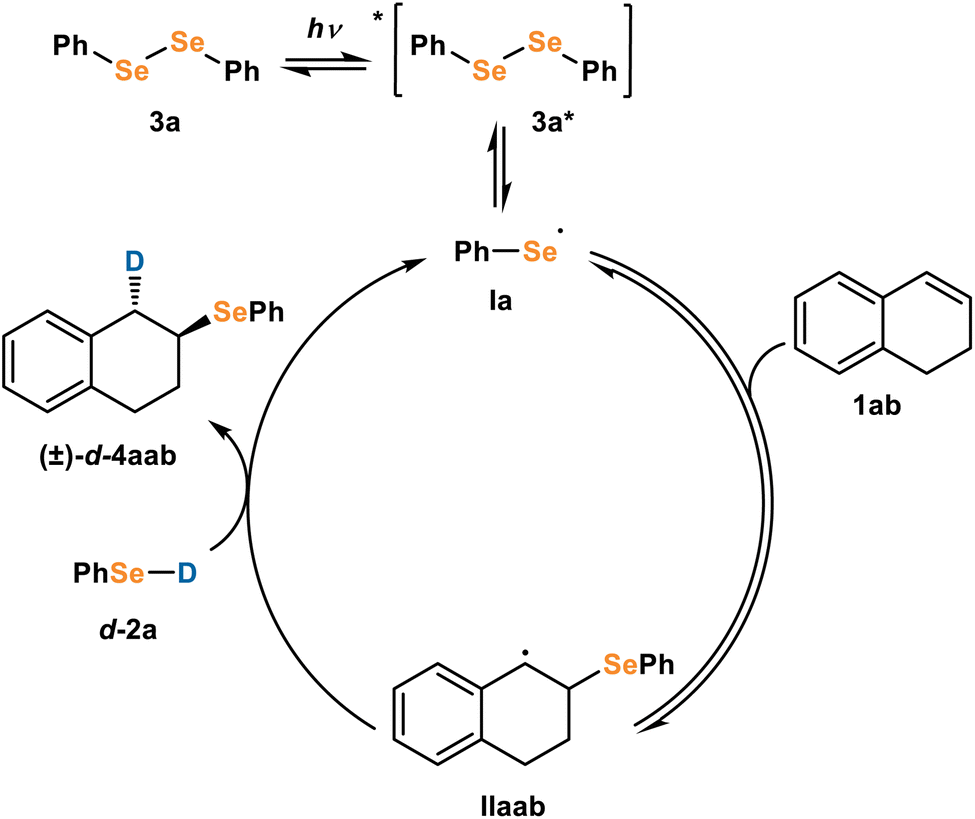 | ||
| Fig. 4 Favored mechanism for diastereoselective formation of d-4aab. | ||
Selective HAT mechanism (M1)
For M1 (Fig. 4), seleno radical Ia adds to the alkene 1ab to form alkyl radical IIaab. We propose a subsequent HAT with selenol d-2a provides product 4aab. Concerted addition of radical la and HAT with selenol d-2a to alkene 1ab would be a termolecular reaction which is kinetically unlikely. Ogawa has proposed a similar mechanism where the homolytic bond cleavage of diselenides generates seleno radicals, which undergo addition to alkynes.13 This mechanism is in accordance with the experimental observation of seleno radicals, but to probe whether the predicted diastereoselectivity reflected the experimental results we turned to computational studies.Computational studies of M1
We focused our computational studies on analyzing the predicted selectivity for cyclic styrene substrate 1ab. For M1, the diastereo-determining step will be the HAT step; hence we focused on the calculations for the intermediate (IIaab), transition state (TS2) and product (4aab) for that step.Relative free energies of transition states and intermediates were computed with DFT to predict the diastereomeric ratios (dr) (Fig. 5). It is likely that there is interconversion between the anti- and syn-isomers of IIaab, due to the low barrier of inversion for tetrahedral C-radicals. This profile is the condition for Curtin–Hammett control which results in a dr that only depends on the transition state free energies rather than the activation energies. The computed anti- to syn-product ratio is >99:1 dr, which corresponds well with the experimentally observed >20:1 dr. In the less likely scenario where there is not facile interconversion of anti- and syn-IIaab, the reaction would be under kinetic control and therefore depend upon the ΔΔG between anti- and syn-IIaab. In this case, calculations also indicate a >99:1 dr, and still support experimental results.
 | ||
| Fig. 5 Energies of the intermediate IIaab and HAT transition state (TS2) for syn- and anti-addition. Energies are: anti-IIaab, 0 kcal mol−1; syn-IIaab, 1.51 kcal mol−1; anti-TS2, 14.48 kcal mol−1; syn-TS2, 23.01 kcal mol−1; anti-4aab, −13.60 kcal mol−1; syn-4aab, −13.30 kcal mol−1. | ||
Comparing the singly occupied molecular orbitals (SOMOs) of the HAT step transition state (TS2) reveals a qualitative difference between the syn- and anti-TS2 electron densities (Fig. 6). The SOMO of the anti-TS2 is delocalized a greater distance because selenium atoms are more linear. The SOMO of both transition state structures has weak C–H bonding and Se–H antibonding character (Fig. 6). However, the almost linear (133°) Se–C–C configuration over 5.5 Å in the anti-TS2 structure affords additional stabilization of the SOMO by hyperconjugation with the σ* orbital of the adjacent C–Se bond, whereas the 77° Se–C–C angle reduces the length scale of delocalization (3.74 Å) in the syn-TS2 structure. The anti-TS2 SOMO delocalization shows some antibonding character and results in a small lengthening in the anti-TS2 β-Se–C bond (2.005 Å) compared to the syn-TS2 β-Se–C bond (2.003 Å). Based on these results, it is likely that electronic effects contribute greatly to give the high anti-selectivity. Sterics may also have an impact,20l however steric effects alone would not explain the observed bond lengthening and extended delocalization.
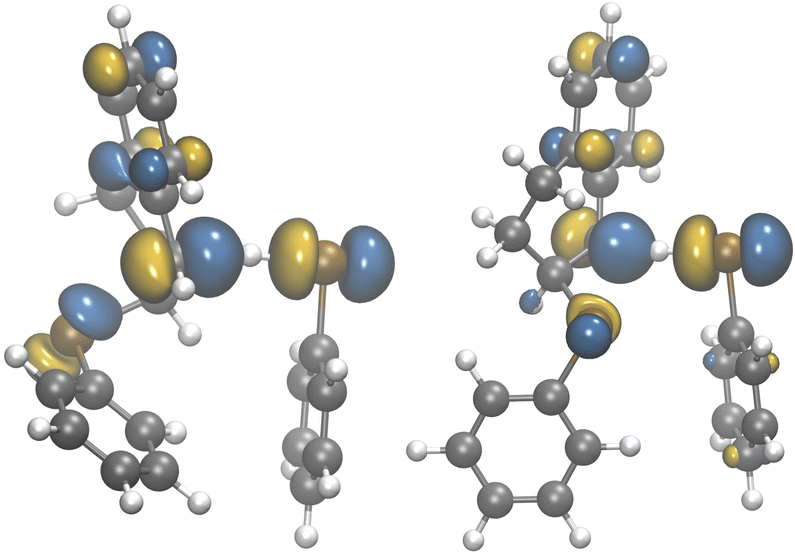 | ||
| Fig. 6 SOMO of the anti- and syn-HAT transition states (TS2). | ||
We were interested in the high diastereoselectivity observed for acyclic product 4aae. DFT calculations were performed on the entire reaction coordinate (see ESI).† Calculations indicated a high anti-selectivity in this case as well. However, both (E)- and (Z)-1ae give the same diastereomer experimentally, which was found to be the product of anti-addition to (E)-1ae. Depletion of (E)-1ae occurs more quickly in the reaction, and isomerization between the two alkenes under reaction conditions is also observed. Thus, it is possible that there is a pre-equilibrium between the alkenes followed by a faster anti-addition reaction to (E)-1ae. The calculations do not fully describe a significantly faster reaction with (E)-1ae, but do support high anti-selectivity in a similar fashion to the cyclic system.
Based on our observed diastereoselectivity and computational investigations, we propose a novel “β-selenium effect” for radical additions due to delocalization of the transition state SOMO. This effect is an exciting parallel to the β-silicon effect.24 Further, our present results suggest that the β-selenium effect arises from delocalization rather than a seleniranium intermediate.25
We were interested in whether the β-selenium effect extends to other radical additions, so we investigated whether the C-radical can be trapped using H2O2. This type of reactivity is analogous to work in hydrodesulfurization.26 We found that a mixture of alkene 1ab, 3a, and H2O2 gave sole anti-addition (Scheme 1).
 | ||
| Scheme 1 Use of H2O2 as a radical trap reagent. | ||
From these studies, we conclude that the β-selenium effect is applicable to trapping of C-radicals β to a selenide with high stereocontrol.
Seleniranium mechanism (M2)
Mechanisms involving seleniraniums are well precedented.27 The addition of an electrophilic selenium moiety to an alkene forms a seleniranium, which can subsequently be opened by a range of nucleophiles. Addition of the nucleophile occurs with anti-selectivity. This reactivity includes applications such as polyene cyclizations,27b selenoetherifications28 and selenolactonizations.29 Strong evidence for seleniraniums has been observed, as the seleniranium ion structure has recently been thoroughly characterized.30 Based on this literature precedence and the observed anti-selectivity, we were curious whether our selenol-ene occurs via a seleniranium intermediate. In this proposed mechanism (Fig. 7), C-radical intermediate IIaab can be oxidized to form seleniranium intermediate IIIaab. Subsequent HAT and reduction (order is unknown so they are drawn as a single step) can give the product 4aab and regenerate seleno radical Ia. To probe this mechanism, we imagined independently synthesizing a seleniranium and subjecting it to the reaction conditions, but the instability of seleniraniums precluded their use at reaction temperature.28b,30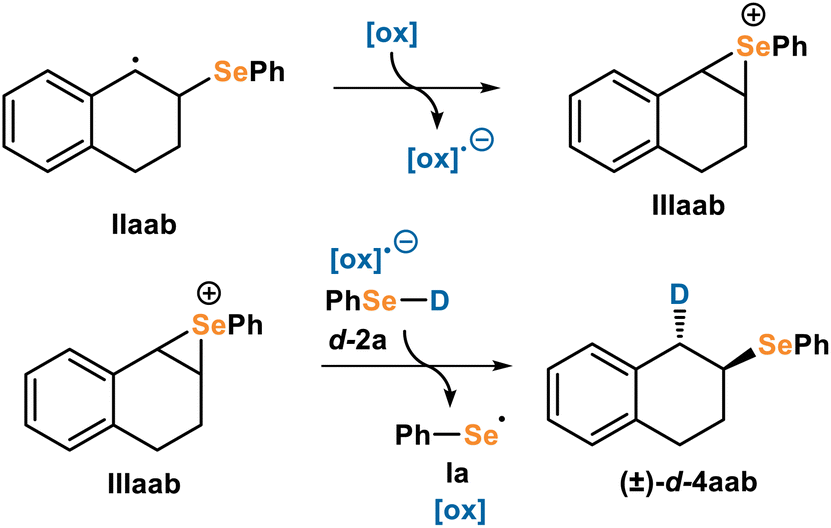 | ||
| Fig. 7 Proposed seleniranium mechanism (M2) for formation of anti-d-4aab starting from carbon radical IIaab. | ||
To form the seleniranium, oxidation is required and so, we investigated the presence of oxidants in the reaction. There are two possible mechanisms for oxidation: first, C-radical II can be oxidized to form a carbocation followed by ring closing to form III (Fig. 8A), or ring closing can occur first to form the radical selenirane IV followed by oxidation to form III (Fig. 8B).
 | ||
| Fig. 8 (A) Oxidation pathway involving carbocation; (B) oxidation pathway involving selenirane; (C) investigated oxidation of C-radical. | ||
Based upon previous redox studies of benzeneselenol, styrene, and diphenyl diselenide, we realized that the ground state species of these reagents were unable to facilitate oxidation.31 Therefore, we calculated the redox potentials of various excited-state species in the reaction. The 1e− reduction potential for seleno radical Ia is calculated to be −4.46 eV. For the carbocation pathway (Fig. 8A), the oxidation of the C-radical intermediate IIaa is +5.13 eV (Table 2). Based on these calculations, the redox event using a seleno radical Ia as an oxidant for IIaa is energetically uphill and therefore unlikely (Fig. 8C). DFT studies of the neutral selenirane radical indicated that this was not an energetically feasible intermediate (vide infra).
| Ia → Ia˙− | IIaa → IIaa+ | |
|---|---|---|
| Adiabatic electronic energy difference | −4.46 eV | 5.13 eV |
To investigate whether oxygen could be the oxidant, parallel experiments were run: one under inert conditions (<5.0 ppm O2), one under brief exposure to the atmosphere, and one under an O2 balloon. All three reactions proceeded similarly, and the reaction occurred in the absence of O2; therefore, we concluded that O2 is unlikely to affect reactivity (Table 3).
| Inert | Air | O2 balloon | |
|---|---|---|---|
| Yield (μmol) | 72.1 ± 4.5 | 73.2 ± 4.6 | 74.4 ± 4.6 |
Further, we investigated the impact of solvent dielectric constant on reaction rate. Based on Marcus theory,32 if redox reactivity occurred to form a seleniranium, the reaction should be faster in solvents with higher dielectric constants. A higher solvent dielectric constant allows for rapid solvent reorganization upon charged species formation. However, we observed no correlation between solvent dielectric constant and yield at an early time point in the reaction (Fig. 9). In fact, acetonitrile, which has the highest dielectric constant and smallest radius, led to one of the lowest yields early in the reaction. Therefore, it is unlikely that charged species formation occurs during this reaction.
 | ||
| Fig. 9 Observed lack of trend between dielectric constant and product formation early in the reaction. Tested solvents included heptane, 1,2-dichloroethane, α,α,α-trifluorotoluene, tetrahydrofuran, and acetonitrile. Reaction conditions: 1a (0.1 mmol), 2a (0.15 mmol), solvent (0.25 M), blue LED irradiation for five minutes. Yield measured using GC-FID analysis with trimethoxybenzene as internal standard. | ||
Based on these mechanistic studies, we were able to conclude that the presence of oxidants and ionized intermediates is unlikely. Therefore, a seleniranium mechanism for this reaction is improbable.
Selenirane mechanism (M3)
After establishing M2 is unlikely, we imagined a similar mechanism involving a neutral selenirane (Fig. 10).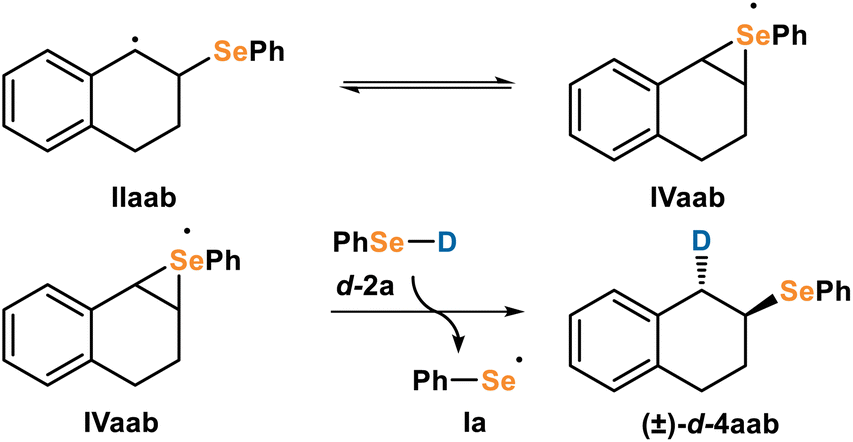 | ||
| Fig. 10 Proposed radical selenirane mechanism (M3) for formation of anti-d-4aab starting from carbon radical IIaab. | ||
In this mechanism, C-centered radical IIaab could be in equilibrium with the Se-centered radical selenirane IVaab, and could undergo HAT with selenol 2a to give the product 4aab and regenerate seleno radical Ia. This pathway would similarly have high anti-selectivity but would not require redox activity. A similar structure has been proposed by Beckwith in thiol-oxygen cooxidation reactions.26g We investigated M3 computationally to determine the energetic feasibility. A potential energy surface scan of IV (Fig. 11) varying the Se–C1 and Se–C2 bond lengths showed no local minimum that corresponded to the selenirane ring and no saddle points that would correspond to a transition state (Fig. 11). The lack of energetically feasible intermediates or transition states for a radical selenirane indicates that M3 is unlikely to be the operative mechanism.
 | ||
| Fig. 11 Potential energy surface scan of selenirane mechanism. No minima (intermediates) or saddle points (transition states) were found corresponding to a selenirane. | ||
Conclusions
The generation of carbon radicals from alkenes is an exciting approach to hydrofunctionalization. This strategy can be achieved through (1) metal–hydride hydrogen atom transfer (MHAT);30 (2) generation of radical cations from alkenes;31 or (3) the addition of a heteroatom radical to alkenes.32 We report a novel hydroselenation falling under this third approach, where a selenide radical adds to an alkene. Using blue LEDs, we were able to access novel selenides. The transformation proceeds with a variety of substrates, including unactivated alkenes and drug-like molecules. The exploration of several potential mechanisms (Table 4) led us to identify a mechanism involving a C-radical as the most likely pathway (M1).| M1 | M2 | M3 | ||
|---|---|---|---|---|
| Experimental evidence | anti-Addition | ✓ | ✓ | ✓ |
| Radical evidence | ✓ | ✓ | ✓ | |
| No rate correlation with polarity of solvent | ✓ | ✗ | ✓ | |
| Computational evidence | Energetically feasible | ✓ | ✗ | ✓ |
| Found transition states | ✓ | N/A | ✗ | |
| Favors anti-addition | ✓ | ✓ | ✓ |
Through computational studies, we have been able to identify a novel β-selenium effect which leads to high anti-selectivity for trapping C-radicals β to a selenide. In previous studies, sterics have been proposed to impact selectivity of addition to β-carbon radicals; however, through computational studies we have been able to elucidate additional electronic factors which play a significant role in selectivity. This relatively strong stereoelectronic effect appears to be caused by the much greater energetic accessibility of C–Se σ* orbitals compared to other C–X σ bonds with second-row elements. We hope that the application of the β-selenium effect can aid in future stereoselective syntheses, and we expect insights from this study will help to guide future work in hydroselenation and functionalization of selenides.
Methods
Computational methods
All calculations were performed using Turbomole.33 The molecular geometries were optimized using the DFT functional PBE0,34 the triple ζ quality def2-TZVP basis set,35 resolution-of-the-identity approximation (RI),36 D3 dispersion corrections,37 and COSMO with a dielectric constant of 10.45 corresponding to DCE.38 GKS-RPA39 single point calculations using the DFT functional PBE were then done on these optimized geometries and adiabatic electronic energies using the resolution-of-identity random phase approximation (RI-RPA),40 60 frequency quadrature points, triple ζ quality def2-TZVP basis set, an SCF convergence threshold of 10−8 Hartree, and grids of size 6 for functional integrations. Finally, the free energy corrections were computed using the “Thermo submodule” of xTB.41Experimental methods
Author contributions
G. S. P.: investigation, writing – original draft. H. S. S.: investigation, writing – original draft. K. J. R.: investigation, writing – review, editing, and reviewer response. S. N.: conceptualization, investigation. C. A.: investigation. F. F.: funding acquisition, supervision, writing – original draft. V. M. D.: funding acquisition, supervision, writing – original draft. X.-H. Y.: conceptualization, investigation, supervision, writing – original draft.Data availability
Details regarding experimental procedures and spectral data for all new compounds (PDF) are available in the ESI.†Conflicts of interest
Principal Investigator Filipp Furche has an equity interest in TURBOMOLE GmbH. The terms of this arrangement have been reviewed and approved by the University of California, Irvine, in accordance with its conflict of interest policies.Acknowledgements
We thank Dr Dmitry Fishman for his work on the transient absorption spectroscopy. We also thank the UCI mass spectroscopy facility for their work on the high resolution mass spectroscopy data. We thank the Analysis and Testing Center of Beijing Institute of Technology. X.-H. Y. thanks the National Natural Science Foundation of China (No. 22371016 and 22201018), Beijing Natural Science Foundation (No. 2222024), and the National R&D Program of China (No. 2021YFA1401200) for funding. V. M. D. thanks the U.S. National Science Foundation (No. CHE-2247923) and National Institutes of Health (2R35GM127071) for funding. H. S. S. thanks the National Science Foundation Graduate Research Fellowship (No. DGE-1839285). F. F. thanks the U.S. Department of Energy, Office of Basic Energy Sciences (DE-SC0025405).Notes and references
- (a) Q. Cheng, T. Sandalova, Y. Lindqvist and E. S. J. Arnér, J. Biol. Chem., 2009, 284, 3998–4008 CrossRef CAS; (b) Q. Cheng, T. Sandalova, Y. Lindqvist and E. S. J. Arnér, Crystal structure of recombinant rat selenoprotein thioredoxin reductase 1 with oxidized C-terminal tail, The Research Collaboratory for Structural Bioinformatics Protein Data Bank, 2008, accessed March 2024, https://www.rcsb.org/structure/3EAO Search PubMed.
- S. Gromer, L. Johansson, H. Bauer, L. D. Arscott, S. Rauch, D. P. Ballou, C. H. Williams, R. H. Schirmer and E. S. J. Arnér, Proc. Natl. Acad. Sci. U. S. A., 2003, 100, 12618–12623 CrossRef CAS PubMed.
- M. Roman, P. Jitaru and C. Barbante, Metallomics, 2013, 6, 25–54 Search PubMed.
- (a) A. Moghaddam, R. A. Heller, Q. Sun, J. Seelig, A. Cherkezov, L. Seibert, J. Hackler, P. Seemann, J. Diegmann, M. Pilz, M. Bachmann, W. B. Minich and L. Schomburg, Nutrients, 2020, 12, 2098 CrossRef CAS; (b) A. Alkattan, K. Alabdulkareem, A. Kamel, H. Abdelseed, Y. Almutairi and E. Alsalameen, Alexandria J. Med., 2021, 57, 21–27 Search PubMed.
- For select reviews, see: (a) H. Chuai, S. Q. Zhang, H. Bai, J. Li, Y. Wang, J. Sun, E. Wen, J. Zhang and M. Xin, Eur. J. Med. Chem., 2021, 223, 113621 CrossRef CAS; (b) D. Radomska, R. Czarnomysy, D. Radomski and K. Bielawski, Int. J. Mol. Sci., 2021, 22, 1009 Search PubMed; (c) W. Ali, R. Benedetti, J. Handzlik, C. Zwergel and C. Battistelli, Drug Discovery Today, 2021, 26, 256–263 CrossRef CAS; (d) V. Gandin, P. Khalkar, J. Braude and A. P. Fernandes, Free Radical Biol. Med., 2018, 127, 80–97 Search PubMed; (e) A. P. Fernandes and V. Gandin, Biochim. Biophys. Acta, 2015, 1850, 1642–1660 CrossRef CAS.
- (a) L. Wang, Z. Yang, J. Fu, H. Yin, K. Xiong, Q. Tan, H. Jin, J. Li, T. Wang, W. Tang, J. Yin, G. Cai, M. Liu, S. Kehr, K. Becker and H. Zeng, Free Radical Biol. Med., 2012, 52, 898–908 CrossRef CAS; (b) X. Zheng, Y. Chen, M. Bai, Y. Liu, B. Xu, R. Sun and H. Zeng, Free Radical Biol. Med., 2019, 131, 7–17 CrossRef CAS.
- For a review, see: (a) L. Liao and X. Zhao, Acc. Chem. Res., 2022, 55, 2439–2453 CrossRef CAS; for select examples, see: (b) X. Liu, Y. Liang, J. Ji, J. Luo and X. Zhao, J. Am. Chem. Soc., 2018, 140, 4782–4786 CrossRef CAS; (c) Y. Liang and X. Zhao, ACS Catal., 2019, 9, 6896–6902 CrossRef CAS; (d) Y. Zhang, Y. Liang and X. Zhao, ACS Catal., 2021, 11, 3755–3761 CrossRef CAS.
- (a) G. Pandey, K. S. Sesha, P. Rao and B. B. V Soma Sekhar, J. Chem. Soc., Chem. Commun., 1993, 21, 1636–1638 RSC; (b) G. Pandey, K. S. Sesha Poleswara Rao and K. V. Nageshwar Rao, J. Org. Chem., 1996, 61, 6799–6804 Search PubMed; (c) P. Renaud, Organoselenium Chemistry, Radical Reactions Using Selenium Precursors, Springer, Berlin, 2000, pp. 81–112 Search PubMed.
- W. R. Bowman, Organoselenium Chemistry: Synthesis and Reactions, 2011, pp. 111–146 Search PubMed.
- (a) S. Hashimoto, S. I. Katoh, T. Kato, D. Urabe and M. Inoue, J. Am. Chem. Soc., 2017, 139, 16420–16429 CrossRef CAS PubMed; (b) D. L. J. Clive, Y. Tao, A. Khodabocus, Y.-J. Wu, A. Gae’tan Angoh, S. M. Bennett, C. N. Boddy, L. Bordeleau, D. Kellner, G. Kleiner, D. S. Middleton, C. J. Nichols, S. R. Richardson and P. G. Vernon, J. Am. Chem. Soc., 1994, 116, 11275–11286 CrossRef CAS; (c) A. Hirose, A. Watanabe, K. Ogino, M. Nagatomo and M. Inoue, J. Am. Chem. Soc., 2021, 143, 12387–12396 CrossRef CAS PubMed; (d) P. Yuan and T. Gaich, Org. Lett., 2022, 24, 4717–4721 CrossRef CAS PubMed; (e) T. Asaba, Y. Katoh, D. Urabe and M. Inoue, Angew. Chem., Int. Ed., 2015, 54, 14457–14461 CrossRef CAS; (f) W. Zi, W. Xie and D. Ma, J. Am. Chem. Soc., 2012, 134, 9126–9129 CrossRef CAS PubMed.
- B. M. Trost, Science, 1991, 254, 1471–1477 CrossRef CAS PubMed.
- (a) S. Li, Q. Yang, Z. Bian and J. Wang, Org. Lett., 2020, 22, 2781–2785 CrossRef CAS; (b) A. Ogawa, A. Kudo and T. Hirao, Tetrahedron Lett., 1998, 39, 5213–5216 CrossRef CAS; (c) T. Tamai, M. Yoshikawa, S. Higashimae, A. Nomoto and A. Ogawa, J. Org. Chem., 2016, 81, 324–329 CrossRef CAS; (d) H. Tian, H. M. Zhang and L. Yin, Angew. Chem., Int. Ed., 2023, 62, e202301422 CrossRef CAS PubMed; (e) H. S. Slocumb, S. Nie, V. M. Dong and X. H. Yang, J. Am. Chem. Soc., 2022, 144, 18246–18250 CrossRef CAS; (f) S. I. Kawaguchi, M. Kotani, S. Atobe, A. Nomoto, M. Sonoda and A. Ogawa, Organometallics, 2011, 30, 6766–6769 CrossRef CAS; (g) V. P. Ananikov, D. A. Malyshev, I. P. Beletskaya, G. G. Aleksandrov and I. L. Eremenko, J. Organomet. Chem., 2003, 679, 162–172 CrossRef CAS ; for select reviews: ; (h) A. Ishii and N. Nakata, The Mechanism for Transition-Metal-Catalyzed Hydrochalcogenation of Unsaturated Organic Molecules, in Hydrofunctionalization, Springer, Berlin, 2011, pp. 21–50 Search PubMed; (i) A. Ogawa, Transition-Metal-Catalyzed S–H and Se–H Bonds Addition to Unsaturated Molecules, Hydrofunctionalization, Springer, Berlin, 2011, pp. 325–360 Search PubMed.
- Y. Kobiki, S. I. Kawaguchi and A. Ogawa, Tetrahedron Lett., 2013, 54, 5453–5456 CrossRef CAS.
- (a) A. Ogawa, H. Yokoyama, K. Yokoyama, T. Masawaki, N. Kambe and N. Sonoda, J. Org. Chem., 1991, 56, 5721–5723 CrossRef CAS; (b) D. V. Patil, Y. T. Hong, H. Y. Kim and K. Oh, Org. Lett., 2022, 24, 8465–8469 CrossRef CAS PubMed; (c) J. Chen, R. Chen, L. Mei, S. Yan, Y. Wu, Q. Li and B. Yuan, Asian J. Org. Chem., 2020, 9, 181–184 CrossRef CAS; (d) G. Q. Liu, C. F. Zhou, Y. Q. Zhang, W. Yi, P. F. Wang, J. Liu and Y. Ling, Green Chem., 2021, 23, 9968–9973 RSC.
- (a) Z. P. Ye, P. J. Xia, F. Liu, Y. Z. Hu, D. Song, J. A. Xiao, P. Huang, H. Y. Xiang, X. Q. Chen and H. Yang, J. Org. Chem., 2020, 85, 5670–5682 CrossRef CAS PubMed; (b) V. Rathore and S. Kumar, Green Chem., 2019, 21, 2670–2676 RSC; (c) Q. B. Zhang, Y. L. Ban, P. F. Yuan, S. J. Peng, J. G. Fang, L. Z. Wu and Q. Liu, Green Chem., 2017, 19, 5559–5563 RSC.
- (a) X. J. Zhou, H. Y. Liu, Z. Y. Mo, X. L. Ma, Y. Y. Chen, H. T. Tang, Y. M. Pan and Y. L. Xu, Chem.–Asian J., 2020, 15, 1536–1539 CrossRef CAS PubMed; (b) H. Sahoo, G. S. Grandhi, I. Ramakrishna and M. Baidya, Org. Biomol. Chem., 2019, 17, 10163–10166 RSC; (c) X. L. Ma, Q. Wang, X. Y. Feng, Z. Y. Mo, Y. M. Pan, Y. Y. Chen, M. Xin and Y. L. Xu, Green Chem., 2019, 21, 3547–3551 RSC; (d) H. Hou, Y. Sun, Y. Pan, H. Yu, Y. Han, Y. Shi, C. Yan and S. Zhu, J. Org. Chem., 2021, 86, 1273–1280 CrossRef CAS PubMed; (e) C. C. Tran, S. I. Kawaguchi, F. Sato, A. Nomoto and A. Ogawa, J. Org. Chem., 2020, 85, 7258–7266 CrossRef CAS PubMed; (f) Q. Shi, P. Li, Y. Zhang and L. Wang, Org. Chem. Front., 2017, 4, 1322–1330 RSC; (g) P. Tan, L. Lu, S. Wang, J. Wang, J. Chen, Y. Zhang, L. Xie, S. Yang, J. Chen and Z. Zhang, J. Org. Chem., 2023, 88, 7245–7255 CrossRef CAS PubMed.
- For select reviews, see: (a) S. W. M. Crossley, C. Obradors, R. M. Martinez and R. A. Shenvi, Chem. Rev., 2016, 116, 8912–9000 CrossRef CAS PubMed; (b) S. L. Shevick, C. V. Wilson, S. Kotesova, D. Kim, P. L. Holland and R. A. Shenvi, Chem. Sci., 2020, 11, 12401–12422 Search PubMed; (c) R. A. Shenvi, J. L. M. Matos and S. A. Green, Organic Reactions, 2019, 100, 383–470 CAS; (d) For the seminal work on MHAT, see: J. C. Lo, J. Gui, Y. Yabe, C. M. Pan and P. S. Baran, Nature, 2014, 516, 343–348 CrossRef CAS PubMed.
- For a review, see: (a) K. A. Margrey and D. A. Nicewicz, Acc. Chem. Res., 2016, 49, 1997–2006 Search PubMed; for select examples, see: (b) D. J. Wilger, J. M. M. Grandjean, T. R. Lammert and D. A. Nicewicz, Nat. Chem., 2014, 6, 720–726 CrossRef CAS PubMed; (c) T. M. Nguyen and D. A. Nicewicz, J. Am. Chem. Soc., 2013, 135, 9588–9591 CrossRef CAS PubMed; (d) N. A. Romero and D. A. Nicewicz, J. Am. Chem. Soc., 2014, 136, 17024–17035 Search PubMed.
- For select reviews, see: (a) A. K. Sinha and D. Equbal, Asian J. Org. Chem., 2019, 8, 32–47 Search PubMed; (b) C. E. Hoyle, A. B. Lowe and C. N. Bowman, Chem. Soc. Rev., 2010, 39, 1355–1387 RSC; (c) T. Posner, Ber. Dtsch. Chem. Ges., 1905, 38, 646–657 CrossRef; (d) A. J. Musacchio, B. C. Lainhart, X. Zhang, S. G. Naguib, T. C. Sherwood and R. R. Knowles, Science, 2017, 355, 727–730 CrossRef CAS PubMed; (e) E. Y. Xu, J. Werth, C. B. Roos, A. J. Bendelsmith, M. S. Sigman and R. R. Knowles, J. Am. Chem. Soc., 2022, 144, 18948–18958 CrossRef CAS PubMed; (f) M. Teders, C. Henkel, L. Anhäuser, F. Strieth-Kalthoff, A. Gómez-Suárez, R. Kleinmans, A. Kahnt, A. Rentmeister, D. Guldi and F. Glorius, Nat. Chem., 2018, 10, 981–988 CrossRef CAS PubMed; (g) A. B. Lowe, Polym. Chem., 2010, 1, 17–36 RSC.
- (a) L. Sun, L. Wang, H. Alhumade, H. Yi, H. Cai and A. Lei, Org. Lett., 2021, 23, 7724–7729 CrossRef CAS PubMed; (b) L. Sun, Y. Yuan, M. Yao, H. Wang, D. Wang, M. Gao, Y. H. Chen and A. Lei, Org. Lett., 2019, 21, 1297–1300 Search PubMed; (c) S. F. Wu, Y. Yu, Y. Yuan, Z. Li and K. Y. Ye, Eur. J. Org. Chem., 2022, 2022, e202201032 CrossRef CAS; (d) G. Q. Liu, W. Yi, P. F. Wang, J. Liu, M. Ma, D. Y. Hao, L. Ming and Y. Ling, Green Chem., 2021, 23, 1840–1846 Search PubMed; (e) F. H. Cui, Y. Hua, Y. M. Lin, J. Fei, L. H. Gao, X. Zhao and H. Xia, J. Am. Chem. Soc., 2022, 144, 2301–2310 CrossRef CAS PubMed; (f) O. Ito, J. Am. Chem. Soc., 1983, 105, 850–853 Search PubMed; (g) B. Huang, Y. Li, C. Yang and W. Xia, Green Chem., 2020, 22, 2804–2809 RSC; (h) Y. Yin, C. Li, K. Sun, Y. Liu and X. Wang, Chin. J. Org. Chem., 2022, 42, 1431–1437 CrossRef CAS; (i) C. Xu, Z. He, X. Kang and Q. Zeng, Green Chem., 2021, 23, 7544–7548 RSC; (j) H. Chen, L. Chen, Z. He and Q. Zeng, Green Chem., 2021, 23, 2624–2627 RSC; (k) K. Sun, X. Wang, Y. Lv, G. Li, H. Jiao, C. Dai, Y. Li, C. Zhang and L. Liu, Chem. Commun., 2016, 52, 8471–8474 RSC; (l) B. Giese, Angew. Chem., Int. Ed., 1983, 22, 753–764 CrossRef.
- N. Sonoda, A. Ogawa and F. Recupero, in Encyclopedia of Reagents for Organic Synthesis, 2005, DOI:10.1002/047084289X.RB018.PUB2.
- L. Pitzer, F. Schäfers and F. Glorius, Angew. Chem., Int. Ed., 2019, 58, 8572–8576 CrossRef CAS PubMed.
- B. Srinivas, V. P. Kumar, R. Sridhar, V. P. Reddy, Y. V. D. Nageswar and K. R. Rao, Helv. Chim. Acta, 2009, 92, 1080–1084 CrossRef CAS.
- S. N. Ushakov and A. M. Itenberg, Zh. Obshch. Khim., 1937, 7, 2495–2498 CAS.
- For select reviews, see: (a) D. D. Roberts and M. G. McLaughlin, Adv. Synth. Catal., 2022, 364, 2307–2332 CrossRef CAS; (b) J. B. Lambert, Y. Zhao, R. W. Emblidge, L. A. Salvador, X. Liu, J. H. So and E. C. Chelius, Acc. Chem. Res., 1999, 32, 183–190 CrossRef CAS.
- (a) J. E. Hong, Y. Jung, D. Min, M. Jang, S. Kim, J. Park and Y. J. Park, J. Org. Chem., 2022, 87, 7378–7391 CrossRef CAS PubMed; (b) H. Ruan, L. G. Meng, L. Zhu and L. Wang, Adv. Synth. Catal., 2019, 361, 3217–3222 CrossRef CAS; (c) S. F. Zhou, X. Pan, Z. H. Zhou, A. Shoberu and J. P. Zou, J. Org. Chem., 2015, 80, 3682–3687 CrossRef CAS PubMed; (d) A. Das and K. R. J. Thomas, ACS Omega, 2023, 8, 18275–18289 CrossRef CAS PubMed; (e) J. Shi, X. W. Gao, Q. X. Tong and J. J. Zhong, J. Org. Chem., 2021, 86, 12922–12931 CrossRef CAS PubMed; (f) H. Wang, Q. Lu, C. Qian, C. Liu, W. Liu, K. Chen and A. Lei, Angew. Chem., Int. Ed., 2015, 55, 1094–1097 CrossRef PubMed; (g) A. L. J. Beckwith and R. D. Wagner, J. Org. Chem., 1981, 46, 3638–3645 CrossRef CAS; (h) M. S. Kang, J. Y. X. Khoo, Z. Jia and T. Loh, Green Synth. Catal., 2022, 3, 309–316 CrossRef; (i) G. Qin, R. Wang, Z. Cheng, Y. Zhang, B. Wang, Y. Xia, W. Jin and C. Liu, Green Synth. Catal., 2024, 5, 131–135 CrossRef CAS.
- For select reviews, see: (a) L. Lu, D. Huang, Z. Wang, X. Wang and X. Wu, Adv. Synth. Catal., 2023, 365, 2310–2331 CrossRef CAS; (b) B. Maji, Adv. Synth. Catal., 2019, 361, 3453–3489 Search PubMed; (c) V. A. Potapov, M. V. Musalov, M. V. Musalova and S. V. Amosova, Curr. Org. Chem., 2015, 20, 136–145 Search PubMed.
- (a) H. Zhang, S. Lin and E. N. Jacobsen, J. Am. Chem. Soc., 2014, 136, 16485–16488 CrossRef CAS PubMed; (b) S. E. Denmark, D. Kalyani and W. R. Collins, J. Am. Chem. Soc., 2010, 132, 15752–15765 CrossRef CAS PubMed; (c) J. Y. See, H. Yang, Y. Zhao, M. W. Wong, Z. Ke and Y. Y. Yeung, ACS Catal., 2018, 8, 850–858 CrossRef CAS.
- (a) D. L. J. Clive and G. Chittattu, J. Chem. Soc. Chem. Commun., 1977, 484–485 RSC; (b) K. C. Nicolaou and Z. Lysenko, J. Am. Chem. Soc., 1977, 99, 3185–3187 CrossRef CAS; (c) K. C. Nicolaou, S. P. Seitz, W. J. Sipio and J. F. Blount, J. Am. Chem. Soc., 1979, 101, 3884–3893 CrossRef CAS; (d) D. L. J. Clive, C. G. Russell, G. Chittattu and A. Singh, Tetrahedron, 1980, 36, 1399–1408 CrossRef CAS; (e) S. E. Denmark and M. G. Edwards, J. Org. Chem., 2006, 71, 7293–7306 CrossRef CAS PubMed; (f) W. Niu and Y. Y. Yeung, Org. Lett., 2015, 17, 1660–1663 CrossRef CAS PubMed.
- (a) S. E. Denmark, W. R. Collins and M. D. Cullen, J. Am. Chem. Soc., 2009, 131, 3490–3492 CrossRef CAS PubMed; (b) H. Poleschner and K. Seppelt, Chem.–Eur. J., 2018, 24, 17155–17161 CrossRef CAS PubMed; (c) J. Bock, C. G. Daniliuc, K. Bergander, C. Mück-Lichtenfeld and U. Hennecke, Org. Biomol. Chem., 2019, 17, 3181–3185 RSC.
- (a) X. Luo, X. Tang, J. Ni, B. Wu, C. Li, M. Shao and Z. Wei, Chem. Sci., 2023, 14, 1679–1686 RSC; (b) S. Y. Ren, Q. Zhou, H. Y. Zhou, L. W. Wang, O. M. Mulina, S. A. Paveliev, H. T. Tang, A. O. Terent'ev, Y. M. Pan and X. J. Meng, J. Org. Chem., 2023, 88, 5760–5771 CrossRef CAS PubMed.
- R. A. Marcus, J. Chem. Phys., 1956, 24, 966–978 CrossRef CAS.
- Y. J. Franzke, C. Holzer, J. H. Andersen, T. Begušić, F. Bruder, S. Coriani, F. Della Sala, E. Fabiano, D. A. Fedotov, S. Fürst, S. Gillhuber, R. Grotjahn, M. Kaupp, M. Kehry, M. Krstić, F. Mack, S. Majumdar, B. D. Nguyen, S. M. Parker, F. Pauly, A. Pausch, E. Perlt, G. S. Phun, A. Rajabi, D. Rappoport, B. Samal, T. Schrader, M. Sharma, E. Tapavicza, R. S. Treß, V. Voora, A. Wodyński, J. M. Yu, B. Zerulla, F. Furche, C. Hättig, M. Sierka, D. P. Tew and F. Weigend, J. Chem. Theory Comput., 2023, 19, 6859–6890 CrossRef CAS PubMed.
- (a) C. Adamo, G. E. Scuseria and V. Barone, J. Chem. Phys., 1999, 111, 2889–2899 CrossRef CAS; (b) J. P. Perdew, M. Ernzerhof and K. Burke, J. Chem. Phys., 1996, 105, 9982–9985 CrossRef CAS.
- F. Weigend and R. Ahlrichs, Phys. Chem. Chem. Phys., 2005, 7, 3297–3305 RSC.
- B. I. Dunlap, J. W. D. Connolly and J. R. Sabin, J. Chem. Phys., 1979, 71, 3396–3402 CrossRef CAS.
- S. Grimme, J. Antony, S. Ehrlich and H. Krieg, J. Chem. Phys., 2010, 132, 46 CrossRef PubMed.
- A. Klamt and G. Schüürmann, J. Chem. Soc., Perkin Trans. 2, 1993, 799–805 RSC.
- (a) G. P. Chen, V. K. Voora, M. M. Agee, G. Balasubramani and F. Furche, Annu. Rev. Phys. Chem., 2017, 68, 421–466 CrossRef CAS PubMed; (b) H. Eshuis, J. E. Bates and F. Furche, Theor. Chem. Acc., 2012, 131, 1–18 Search PubMed; (c) V. K. Voora, S. G. Balasubramani and F. Furche, Phys. Rev. A, 2019, 99, 012518 CrossRef CAS.
- H. Eshuis, J. Yarkony and F. Furche, J. Chem. Phys., 2010, 132, 37 CrossRef PubMed.
- C. Bannwarth, E. Caldeweyher, S. Ehlert, A. Hansen, P. Pracht, J. Seibert, S. Spicher and S. Grimme, Wiley Interdiscip. Rev.: Comput. Mol. Sci., 2021, 11, e1493 CAS.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4sc05766j |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2024 |