
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSimplifying complexity: integrating color science for predictable full-color and on-demand persistent luminescence using industrial disperse dyes†
Guowei
Xiao‡
a,
Xiaoyan
Wang‡
a,
Xiaoyu
Fang
 b,
Jinmei
Du
a,
Yang
Jiang
a,
Dagang
Miao
a,
Dongpeng
Yan
*b and
Changhai
Xu
*a
b,
Jinmei
Du
a,
Yang
Jiang
a,
Dagang
Miao
a,
Dongpeng
Yan
*b and
Changhai
Xu
*a
aCollege of Textiles & Clothing, Qingdao University, Qingdao, Shandong 266071, China. E-mail: changhai_xu@qdu.edu.cn
bBeijing Key Laboratory of Energy Conversion and Storage Materials, Key Laboratory of Radiopharmaceuticals, Ministry of Education, College of Chemistry, Beijing Normal University, Beijing 100875, China. E-mail: yandp@bnu.edu.cn
First published on 27th September 2024
Abstract
Developing color-tunable ultralong room temperature phosphorescence (RTP) materials with variable afterglow is essential for applications in displays, sensors, information encryption, and optoelectronic devices. However, designing full-color ultralong RTP for persistent luminescence remains a significant challenge. Here, we propose a straightforward strategy to achieve predictable full-color afterglow using readily available disperse dyes in polymeric systems, via the phosphorescence resonance energy transfer (PRET) process. We incorporated the unconventional luminophore tetraacetylethylenediamine (TAED) into polyurethane (PU) to create a polymer host with green afterglow. By adding three typical disperse dyes as guests, we achieved a modulated afterglow covering the full visible light spectrum. Leveraging PRET processes between TAED and the disperse dyes, we achieved a prediction accuracy of 88.89% for afterglow color, surpassing well-developed coloration dye systems. This work thus introduces a novel method to obtain easily predictable ultralong RTP emission and establishes an on-demand design strategy for constructing disperse dye-based full-color afterglow, effectively linking fundamental color science to practical customization.
Introduction
Ultralong room temperature phosphorescence (RTP) with long-lived luminescence, large Stokes shift, and high triplet exciton utilization has significant potential in data encryption,1–3 environmental sensing,4,5 information storage,6–9 bioimaging,10,11 color displays,12–15 and organic light-emitting diodes (OLEDs).16,17 However, triplet excitons are easily quenched by intermolecular collisions or oxygen, complicating the achievement of efficient RTP. To optimize molecular RTP systems, several strategies have been proposed: (i) enhancing spin–orbit coupling (SOC) by introducing carbonyl groups, heteroatoms, and halogens to populate triplet excitons;18,19 (ii) employing techniques like crystallization engineering,20–22 H-aggregation,21–24 supramolecular assembly,25 and embedding organic phosphors into rigid substrates26–28 to suppress nonradiative deactivation pathways. To date, most of one phosphor-based persistent luminescence materials produce monochromatic afterglow colors, making the attainment of multi-color or full-color afterglow difficult. Recently, new methods such as molecular doping and external stimuli have been developed to achieve colorful persistent luminescence,29–32 corresponding to the static and dynamic adjustment of RTP, respectively. Molecular doping, especially dye doping based on Förster or phosphorescence resonance energy transfer (FRET or PRET),33–37 is straightforward, with the selection of suitable energy donors being crucial.Unconventional luminescent molecules are gaining attention due to their ease of synthesis, environmental friendliness, and excellent biocompatibility.38 Unlike classical luminophores, which consist of significantly conjugated fragments, nonconventional systems feature non-conjugated or short-conjugated structures based on electron-rich units. Effective through-space conjugation (TSC) and conformational rigidity between these units give nonconventional luminophores a long lifetime and a broad emission band, making them promising candidates for donor species in energy transfer.39 While the construction of donor–acceptor pairs for color-tunable RTP is well-documented, predicting the final emissive color remains challenging.
Disperse dyes, a key segment of the dye industry, provide a wide variety and a comprehensive color range, making up about half of all dyes. Unlike fluorescence color matching, the colors of disperse dyes can be predicted through color science, allowing calculation of permissible colors within tolerance ranges.40 Applying this predictive theory to RTP could significantly streamline the selection of doping phosphors and determination of target afterglow colors, thus advancing multi-color persistent luminescence research. However, multi-color afterglow based on disperse dyes has not been explored until now.
Leveraging the excellent processability, machinability, and scalability of host–guest doping polymers, we designed efficient and predictable afterglow systems using industrial disperse dyes. We selected polyurethane (PU) as the polymer matrix and incorporated the unconventional luminophore tetraacetylethylenediamine (TAED) to create the energy donor PU@TAED. This system has a long RTP lifetime of 1.18 s and a broad emission range from blue to red. We then introduced three classical disperse dyes-disperse blue 183:1 (Dye-B), disperse yellow 114 (Dye-Y), and disperse red 145 (Dye-R)-into PU@TAED as energy acceptors, resulting in blue, yellow, and red persistent luminescence with lifetimes of 1.13, 1.1, and 0.9 s, respectively, via the PRET process. Applying subtractive color mixing theory, we developed a series of predictable full-color afterglow systems in disperse dye-doped PU@TAED, achieving 88.89% accuracy in predicted afterglow colors within the tolerance range of MacAdam ellipses. To the best of our knowledge, this marks the first use of subtractive color mixing theory to achieve full-color luminescence. Therefore, this work not only provides an effective strategy for predicting and constructing full-color ultralong RTP but also establishes a new method for on-demand color manipulation in polychromatic and machinable materials.
Results and discussion
Photophysical properties and structural analysis of TAED and DAED
Amide compounds, key nonconventional luminophores, efficiently achieve RTP through n–π* transitions facilitated by nitrogen and oxygen heteroatoms, which readily promote SOC and intersystem crossing (ISC). Recent studies highlight that efficient RTP in nonconventional luminophores relies on the clustering of functional units, proper molecular packing, and intra/intermolecular interactions.41,42 Key contributors to this phenomenon include the clustering-triggered emission/phosphorescence (CTE/CTP) mechanism and TSC of electron-rich substructures.38,39,43In this work, we firstly focuses on N,N′-diacetylethylenediamine (DAED) and TAED, acylamide-based non-conjugated systems, to delve deeper into their RTP mechanisms. As shown in Fig. 1a, both DAED and TAED crystals emit blue light under 365 nm UV irradiation; however, only TAED displays a cyan afterglow lasting over 4 s after the UV light is removed. We examined their prompt and delayed photoluminescence (PL) spectra. DAED's prompt PL spectra exhibit a single emission band with peaks at 416 nm and 438 nm, consistent across excitation wavelengths from 300 nm to 380 nm (Fig. S1†). Conversely, TAED shows a bathochromic shift in emission, ranging from 412 nm to 485 nm as the excitation wavelength varies from 340 nm to 440 nm, likely due to the balance of singlet and triplet excitons (Fig. S2a†). The delayed PL spectra (1 ms delay) indicate that TAED emits at 473 nm with a lifetime of 283 ms when excited at 410 nm, characteristic of RTP due to the long lifetime and large Stokes shift (Fig. S2b†). When the excitation wavelength shifts to 340 nm, the RTP emission moves to 497 nm, extending the lifetime to 643 ms (Fig. S3 and Table S3†). This excitation-dependent RTP suggests the involvement of multiple triplet excitons from various clustered chromophores in TAED.
 | ||
Fig. 1 (a) Molecular structures, packing modes, and afterglow photographs of DAED and TAED. (b and d) Hirshfeld surfaces and fingerprint plots showing H⋯O![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C interactions for DAED and TAED, respectively. (c and e) Calculated excitation energies, SOC coefficients (ξ), and orbitals of DAED and TAED. C interactions for DAED and TAED, respectively. (c and e) Calculated excitation energies, SOC coefficients (ξ), and orbitals of DAED and TAED. | ||
To understand the differing RTP properties between TAED and DAED, we thoroughly analyzed their crystal structures (Fig. 1a). Both compounds exhibit highly distorted conformations with effective intermolecular interactions such as C–H⋯O and N–H⋯O contacts. Notably, TAED has a shorter CO bond length (1.211 Å) compared to DAED (1.239 Å), and a higher CO bond density (98 Å3 per unit cell) versus DAED's 70.9 Å3 per unit cell, indicating more π electrons and lone-pair (n) electrons in TAED (Table S1†). The powder X-ray diffraction (PXRD, Fig. S4†) data align with the SCXRD results, confirming their pure phase. Additionally, five atoms are coplanar in DAED, while eight atoms are coplanar in TAED, suggesting greater electron delocalization and increased conformational rigidity in TAED (Fig. S5†). These factors indicate that TAED's RTP is originated from CTE mechanism.
Hirshfeld surface (HS) analysis,44 π-electron distribution diagrams, and SOC constants (ξ) were investigated for TAED and DAED (Fig. 1b–e and S6†). The 2D fingerprint plots from HS (Fig. 1b and d) quantify intermolecular interactions, revealing more H⋯CO interactions in TAED (36.7%) than in DAED (27.5%), which are beneficial to the ultralong RTP. The π-electronic distribution diagrams (insets of Fig. 1c and e) show that, unlike in DAED, the vertical orbital of nitrogen in TAED connects two carbonyl groups, expanding electron delocalization and enhancing TSC.
Key factors for achieving ultralong RTP include stronger TSC, higher electron density, and shorter CO bond lengths. The singlet-triplet energy gap (ΔEst) and SOC constants (ξ) between the singlet and triplet excited states evaluate the probability of ISC. Fig. 1c and e show that the ΔEst for DAED between S1 and T4 is 0.349 eV, while for TAED, it is 0.098 eV between S1 and T5/T6. The ξ(S0–T5/T6) for DAED is 15.74 cm−1, compared to 26.93 cm−1 in TAED. TAED's smaller ΔEst and larger ξ indicate a lower energy barrier for excited electrons migrating from S1 to the T energy level through ISC, enhancing RTP. Additionally, TAED's broad emission range (400–700 nm) and ultralong lifetime (643 ms) make it a promising candidate for energy donor applications in molecular doped systems.
Synthesis and photophysical properties of TAED matrix (PU@TAED)
PU forms rigid polymer matrix rich in amidogen groups and dissolves well in acetone (Scheme S1†). Given TAED's high solubility in acetone (0.8 g ml−1), PU@TAED can be synthesized by incorporating TAED into PU using acetone as a solvent (Fig. 2a). A PU@TAED film with 0.1 wt% TAED displays blue fluorescence and a green afterglow that lasts about 7 s after UV light is turned off under ambient conditions (Fig. 2c). The film shows strong phosphorescence emission peaks at 503 and 540 nm, with lifetimes of 1.17 and 1.18 s, respectively, which are significantly longer than those of TAED alone (Fig. 3b and Table S3†). This enhancement is due to the interaction between the amidogen groups in PU and the carbonyl groups in TAED, forming a dense hydrogen-bond network that creates a rigid microenvironment and restricts molecular motion, thus facilitating long-lived and efficient RTP emission.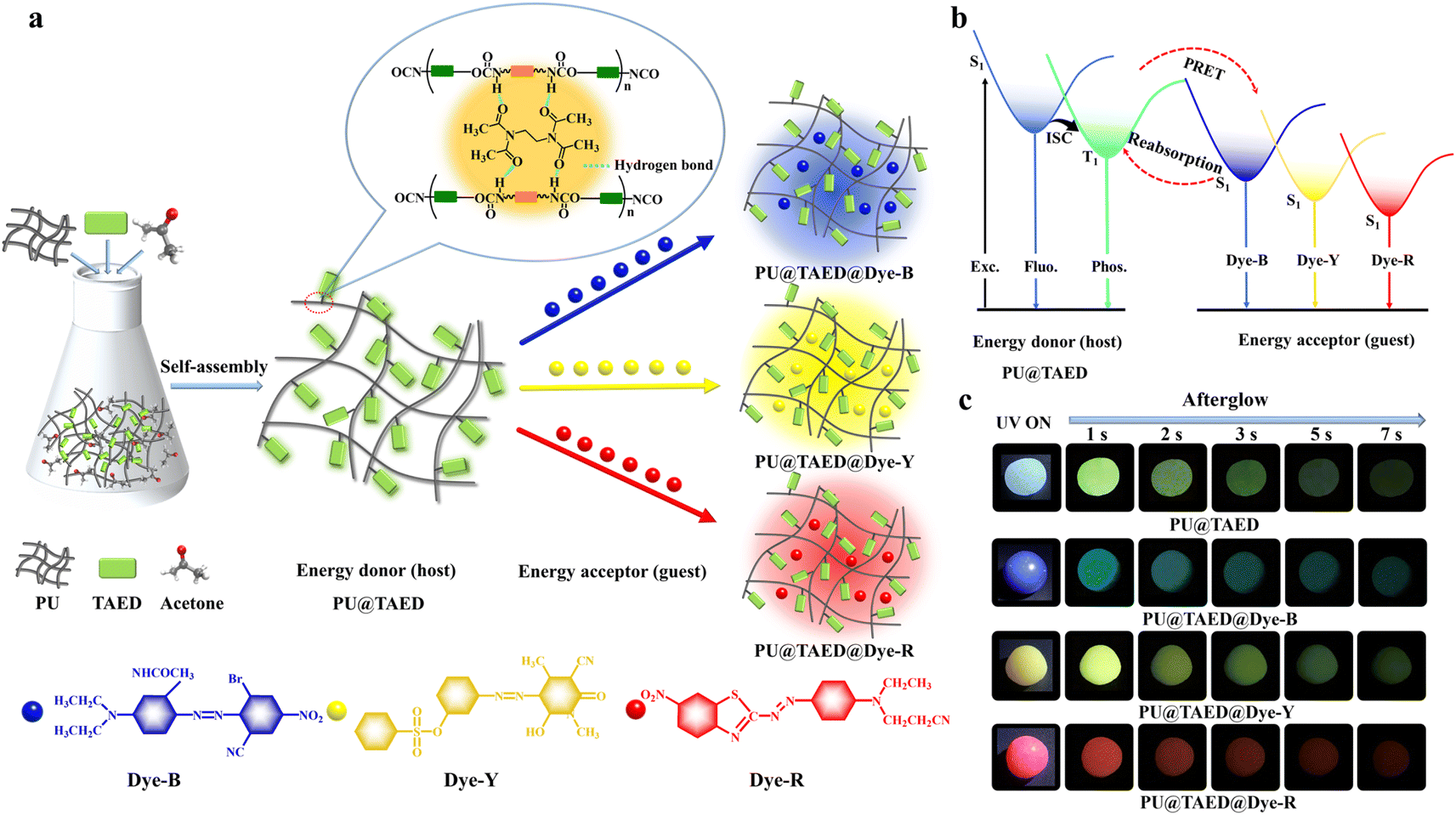 | ||
| Fig. 2 (a) Schematic diagram for preparing ultralong RTP polymeric systems with full-color afterglow, and molecular structures of the polymeric host of PU and disperse dyes: disperse blue 183:1 (Dye-B), disperse yellow 114 (Dye-Y), and disperse red 145 (Dye-R). (b) Simplified Jablonski diagram for the PRET process. (c) Photographs of PU@TAED, PU@TAED@Dye-B, PU@TAED@Dye-Y, and PU@TAED@Dye-R after removing the 365 nm excitation source. | ||
 | ||
| Fig. 3 (a) Steady-state prompt and delayed spectra of PU@TAED, PU@TAED@Dye-B, PU@TAED@Dye-Y, and PU@TAED@Dye-R. Inset: corresponding photographs under and after removing the 365 nm UV lamp. (b–d) Phosphorescence lifetimes and CIE coordinate diagrams of PU@TAED, PU@TAED@Dye-B, PU@TAED@Dye-Y, and PU@TAED@Dye-R. | ||
Unlike TAED, PU@TAED exhibits excitation-independent RTP with consistent peaks at 503 and 540 nm (Fig. S7†). This behavior is attributed to the tighter cluster conformations of electron-rich carbonyl units in TAED when combined with polymeric chains, resulting in lower triplet energy levels and reduced vibrational dissipation.45 Consequently, the delayed spectrum of PU@TAED shows a bathochromic shift compared to TAED. The broad green afterglow emission (380–700 nm) and ultralong lifetime indicate that PU@TAED is an ideal host for various luminescent guests, enabling the construction of full-color, highly efficient afterglow polymeric systems via the PRET process (Fig. 2b).
Mechanism and feasibility verification of the PRET for full-color afterglow
Efficient PRET relies on high degree of spectral overlap between the emission spectrum of the energy donor and the absorption spectrum of the energy acceptor. Additionally, close proximity and appropriate dipole–dipole orientation between donor and acceptor are crucial.36,46 We used three easily available disperse dyes (Dye-B, Dye-Y, Dye-R) as sensitized guests and PU@TAED as the host at various doping percentages to identify optimal ratios for full-color afterglow emission. We analyzed the absorption spectra of the three dyes (Fig. 4a–c and S8†) to investigate the PRET processes. The phosphorescence spectrum of PU@TAED (400–700 nm) overlaps significantly with the absorption spectra of Dye-B (400–700 nm), Dye-Y (335–475 nm), and Dye-R (350–700 nm), indicating potential for efficient PRET and color-tunable persistent luminescence. We systematically examined the photophysical properties of films doped with these dyes at concentrations ranging from 0.1 wt% to 1.0 wt%. For PU@TAED@Dye-B, the optimal doping concentration was determined to be 0.5 wt%. The delayed PL spectra showed long-lived afterglow emission peaks at 435 and 503 nm with lifetimes exceeding 1 s, confirming effective PRET (Fig. S9–S12†). For PU@TAED@Dye-Y and PU@TAED@Dye-R, the intensity of delayed PL spectra at 540 nm and 600 nm, respectively, increased with doping concentrations from 0.1 wt% to 0.5 wt%, and then plateaued, indicating 0.5 wt% as optimal. Higher concentrations resulted in shorter afterglow lifetimes (Fig. S13–S20†). Thus, the optimal doping ratio for all three dyes is 0.5 wt%, producing afterglow materials with blue, yellow, and red emissions, corresponding to CIE coordinates of (0.174, 0.252), (0.318, 0.561), and (0.530, 0.372), respectively (Fig. 2c, 3a and c). | ||
| Fig. 4 (a–c) Normalized UV absorption spectra of Dye-B, Dye-Y, and Dye-R and the phosphorescence spectrum of PU@TAED. (d–f) XPS of PU, PU@TAED, and PU@TAED@Dye-B films. (g–i) Schematic illustration of hydrogen bonds between the phosphors and polymer matrix in PU, PU@TAED, and PU@TAED@Dye-B films. | ||
Luminescent materials using the PRET strategy often involve a decline in donor lifetime due to energy transfer to the acceptor. Fig. 3b and d show that PU@TAED@Dye-B, PU@TAED@Dye-Y, and PU@TAED@Dye-R have shorter lifetimes than pristine PU@TAED, confirming energy transfer. Interestingly, unlike prior systems,47,48 the lifetimes of the donors in these systems have not decreased significantly. This could be due to the broad absorption spectrum of the PU@TAED@Dye composite materials (Fig. S21†), enabling the modulation of donor lifetimes through radiative energy transfer via reabsorption.49–51 X-ray photoelectron spectroscopy (XPS) profiles for PU, PU@TAED, and PU@TAED@Dye-B (Fig. 4d–f) show similar C 1s peaks for C–C/C–N (284.6 eV) and C–O (286.0 eV) in all three polymers. However, the CO peak positions differ, centered at 287.8 eV for PU, 288.2 eV for PU@TAED, and 288.4 eV for PU@TAED@Dye-B. The higher binding energy indicates decreased electron cloud density, suggesting that TAED and dyes increase the electron delocalization area of PU@TAED, enhancing TSC and preserving triplet excitons for efficient RTP emission (Fig. 4g–i).
Direct doping of dye molecules into PU yielded PU@Dye-B, PU@Dye-Y, and PU@Dye-R, all exhibiting weak phosphorescence signals with microsecond-scale lifetimes (Fig. S22–S26†). This control experiment indicates that the long-lived triplet excitons of the PU@TAED host are the sole source of the ultralong afterglow characteristic in the disperse dyes guest via the PRET process. Hydrogen bonding interactions between TAED and the PU matrix play a crucial role in achieving multicolor ultralong RTP emission. Further investigation into the internal structural composition of these films was conducted using Fourier transform infrared spectra (FTIR). As shown in Fig. S27,† a stretching vibration peak of C–H in the methyl group appeared at 1375 cm−1 in PU@TAED, PU@TAED@Dye-B, PU@TAED@Dye-Y, and PU@TAED@Dye-R, indicating successful introduction of TAED. Additionally, the FTIR spectra of PU@TAED exhibited a longer wavelength at 3419 cm−1 compared to PU at 3318 cm−1 (Fig. S28†), suggesting that the carbonyl groups of TAED strengthened the hydrogen bond association between TAED and PU, resulting in a more rigid network and increased RTP emissions.52,53
Predictable panchromatic RTP system
By leveraging the feasibility of color mixing calculations in disperse dyes and the high controllability and modulation flexibility of host–guest polymers, it is theoretically feasible to create high-performance RTP materials with customizable afterglow colors. As a proof of concept, we have developed predictable full-color afterglow systems by blending three primary disperse dyes in various proportions. To enhance the efficiency and quality of afterglow color matching, the development of an afterglow color prediction model based on the concentration ratios of the three primary disperse dyes is crucial.Following the principle of subtractive color mixing, the final color coordinates represent the weighted average of the monochromatic coordinates, where the weight is the stimulus m of the monochromatic coordinates. We established the relationship between monochromatic stimulus m and monochromatic concentration by coloring PU films with different concentration gradients of monochromatic dyes, as illustrated in Fig. 5a. The mixed color coordinates (xmix, ymix) can be determined based on the color coordinates (x, y) of the single color and the stimulus m values at different concentrations, as demonstrated in eqn (1-1).
 | (1-1) |
We synthesized 27 luminescent PU films by blending three primary dyes in varying concentration ratios. The CIE coordinate diagram in Fig. 5b compares the 27 sets of afterglow spectral test results with the model prediction results. Remarkably, 24 sets of actual and predicted results fall within the tolerance range of MacAdam ellipses, indicating an impressive prediction accuracy of 88.89% for this color prediction model. This accuracy surpasses that of well-developed doping dye systems (Table S2†), underscoring the effectiveness of this color prediction model. The full-color afterglow pagoda diagram and phosphorescence spectra, derived from 15 typical host–guest afterglow films, are shown in Fig. 5c and S29.†
 | ||
| Fig. 5 (a) Curve showing the relationship between monochromatic stimulus (m) and monochromatic concentration. (b) CIE coordinate diagram comparing real and predicted values of PU@TAED with different dyes using the MacAdam ellipses model. (c) Afterglow photographs of PU@TAED with different dyes films in daylight, under, and after turning off the 365 nm UV lamp. (d) The predictive process of a four-leaf clover anti-counterfeiting label. (e) Schematic diagram depicting the evolution of the decryption process. (f) Time-resolved anti-counterfeiting label of the Qingdao University logo designed on clothes. | ||
The color prediction formula generates a full-color phosphorescence library by substituting different concentration ratios of the three primary disperse dyes. From this library, the desired color and its corresponding concentration ratio of the three primary dyes can be selected. To validate the practicality of this color-matching theory, we developed a new four-leaf clover anti-counterfeiting label as a demonstration. By substituting different concentration ratios of the three primary dyes into the formula, the pre-designed color can be achieved. This process results in the creation of a four-leaf clover anti-counterfeiting label (Fig. 5d and S31†). The results showed that the required and actual phosphorescence colors were within the tolerance range of MacAdam ellipses, significantly enhancing the efficiency and quality of phosphorescence color matching.
The versatility of multi-color phosphorescence expands the application prospects of host–guest polymers, especially in multi-step encryption and anti-counterfeiting. Using a predictable multi-color phosphorescence system, we designed a multi-step encrypted anti-counterfeiting system with different colors for “QDU COLOR SCIENCE” using PU@Dye-B, PU@TAED@Dye-Y, PU@Dye-G, and PU@TAED@Dye-R (Fig. 5e). The brightly colorful label “QDU COLOR SCIENCE” is visible under daylight or UV light. When the 365 nm UV light is turned off, the label displays the letters “DU OR CIE” for up to 2 s, masking the actual information “DOI”. After 5 s, the true information “DOI” is revealed. Thus, host–guest polymers with predictable phosphorescence and an efficient anti-counterfeiting system show great potential in high-security data storage applications. Additionally, leveraging the good abrasion resistance and stability of the PU matrix, we created the logo of Qingdao University using PU@TAED and PU@TAED@Dye-B/Dye-Y (2:1) on clothes through hot stamping, establishing a simple and easily distinguishable anti-counterfeiting system and enhancing the added value of fabrics (Fig. 5f).
Conclusions
In summary, we have developed an effective method for creating customizable full-color afterglow materials in host–guest polymer systems. This approach relies on a significant overlap between the host's phosphorescence and the guest's absorption, activating the PRET process. The controllability and flexibility of our polymers allow for easy generation of a full-color afterglow library by adjusting the guest dyes and their concentrations, resulting in colors ranging from blue to green to red. Furthermore, by using subtractive color mixing calculations on disperse dyes, we have designed a predictive polymer afterglow system based on PU@TAED@disperse dyes. The predicted afterglow colors are highly accurate, with 88.89% falling within the MacAdam ellipses' tolerance range. This work not only enables the development of full-color host–guest afterglow materials through the PRET process but also provides valuable insights for designing afterglow colors for multi-color RTP materials on demand.Data availability
Experimental procedures, details of the calculations, and additional data can be found in the ESI.†Author contributions
G. Xiao and X. Wang contributed equally to this work. C. Xu, D. Yan, G. Xiao and X. Wang designed the project. G. Xiao and X. Wang synthesized the samples and carried out the measurements. All authors analyzed the experimental data. G. Xiao, X. Wang, D. Yan and C. Xu co-wrote the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This study was supported by the National Natural Science Foundation of China (Grant No. 21978117, 22275021, 22405148), the Major Scientific and Technological Innovation Project of Shandong Province (No. 2023CXGC010610) and the Qingdao Postdoctoral Science Foundation (No. QDBSH20230102082).Notes and references
- R. Gao, M. S. Kodaimati and D. Yan, Chem. Soc. Rev., 2021, 50, 5564–5589 RSC.
- Y. Su, S. Z. F. Phua, Y. Li, X. Zhou, D. Jana, G. Liu, W. Q. Lim, W. K. Ong, C. Yang and Y. Zhao, Sci. Adv., 2018, 4, eaas9732 CrossRef PubMed.
- Z. Chen, X. Chen, D. Ma, Z. Mao, J. Zhao and Z. Chi, J. Am. Chem. Soc., 2023, 145, 16748–16759 CrossRef CAS PubMed.
- H. Lee, J. Kim, H. Kim, J. Kim and S. Kwon, Nat. Mater., 2010, 9, 745–749 CrossRef CAS PubMed.
- L. Pan, S. Sun, A. Zhang, K. Jiang, L. Zhang, C. Dong, Q. Huang, A. Wu and H. Lin, Adv. Mater., 2015, 27, 7782–7787 CrossRef CAS PubMed.
- Y.-J. Ma, X. Fang, G. Xiao and D. Yan, Angew. Chem., Int. Ed., 2022, 61, e202114100 CrossRef CAS PubMed.
- K. Jiang, L. Zhang, J. Lu, C. Xu, C. Cai and H. Lin, Angew. Chem., Int. Ed., 2016, 55, 7231–7235 CrossRef CAS PubMed.
- G. Xiao, X. Fang, Y.-J. Ma and D. Yan, Adv. Sci., 2022, 9, 2200992 CrossRef CAS PubMed.
- B. Xu, Z. Song, M. Zhang, Q. Zhang, L. Jiang, C. Xu, L. Zhong, C. Su, Q. Ban, C. Liu, F. Sun, Y. Zhang, Z. Chi, Z. Zhao and G. Shi, Chem. Sci., 2021, 12, 15556–15562 RSC.
- G. Zhang, G. M. Palmer, M. Dewhirst and C. L. Fraser, Nat. Mater., 2009, 8, 747–751 CrossRef CAS PubMed.
- J. Yang, Y. Zhang, X. Wu, W. Dai, D. Chen, J. Shi, B. Tong, Q. Peng, H. Xie, Z. Cai, Y. Dong and X. Zhang, Nat. Commun., 2021, 12, 4883 CrossRef CAS PubMed.
- M. Li and Z. Xia, Chem. Soc. Rev., 2021, 50, 2626–2662 RSC.
- K. Wan, B. Tian, Y. Zhai, Y. Liu, H. Wang, S. Liu, S. Li, W. Ye, Z. An, C. Li, J. Li, T. D. James and Z. Chen, Nat. Commun., 2022, 13, 5508 CrossRef CAS PubMed.
- F. Nie and D. Yan, Angew. Chem., Int. Ed., 2023, 62, e202302751 CrossRef CAS PubMed.
- Y. Liu, X. Huang, Z. Niu, D. Wang, H. Gou, Q. Liao, K. Xi, Z. An and X. Jia, Chem. Sci., 2021, 12, 8199–8206 RSC.
- T. Wang, X. Su, X. Zhang, X. Nie, L. Huang, X. Zhang, X. Sun, Y. Luo and G. Zhang, Adv. Mater., 2019, 31, 1904273 CrossRef CAS PubMed.
- R. Kabe, N. Notsuka, K. Yoshida and C. Adachi, Adv. Mater., 2016, 28, 655–660 CrossRef CAS PubMed.
- W. Zhao, Z. He, J. W. Y. Lam, Q. Peng, H. Ma, Z. Shuai, G. Bai, J. Hao and B. Z. Tang, Chem, 2016, 1, 592–602 CAS.
- J. Yang, X. Zhen, B. Wang, X. Gao, Z. Ren, J. Wang, Y. Xie, J. Li, Q. Peng, K. Pu and Z. Li, Nat. Commun., 2018, 9, 840 CrossRef PubMed.
- C. Xing, Z. Qi, B. Zhou, D. Yan and W.-H. Fang, Angew. Chem., Int. Ed., 2024, 63, e202402634 CrossRef CAS PubMed.
- K. M. Steed and J. W. Steed, Chem. Rev., 2015, 115, 2895–2933 CrossRef CAS PubMed.
- G. Xiao, Y.-J. Ma, Z. Qi, X. Fang, T. Chen and D. Yan, Chem. Sci., 2024, 15, 3625–3632 RSC.
- Z. An, C. Zheng, Y. Tao, R. Chen, H. Shi, T. Chen, Z. Wang, H. Li, R. Deng, X. Liu and W. Huang, Nat. Mater., 2015, 14, 685–690 CrossRef CAS PubMed.
- E. Lucenti, A. Forni, C. Botta, L. Carlucci, C. Giannini, D. Marinotto, A. Previtali, S. Righetto and E. Cariati, J. Phys. Chem. Lett., 2017, 8, 1894–1898 CrossRef CAS PubMed.
- J. Wang, Z. Huang, X. Ma and H. Tian, Angew. Chem., Int. Ed., 2020, 59, 9928–9933 CrossRef CAS PubMed.
- M. S. Kwon, D. Lee, S. Seo, J. Jung and J. Kim, Angew. Chem., Int. Ed., 2014, 53, 11177–11181 CrossRef CAS PubMed.
- B. Zhou and D. Yan, Matter, 2024, 7, 1950–1976 CrossRef.
- Y. Zhang, Y. Su, H. Wu, Z. Wang, C. Wang, Y. Zheng, X. Zheng, L. Gao, Q. Zhou, Y. Yang, X. Chen, C. Yang and Y. Zhao, J. Am. Chem. Soc., 2021, 143, 13675–13685 CrossRef CAS PubMed.
- G. Xiao, B. Zhou, X. Fang and D. Yan, Research, 2021, 2021, 9862327 CAS.
- Y. Lei, W. Dai, J. Guan, S. Guo, F. Ren, Y. Zhou, J. Shi, B. Tong, Z. Cai, J. Zheng and Y. Dong, Angew. Chem., Int. Ed., 2020, 59, 16054–16060 CrossRef CAS PubMed.
- J.-A. Li, L. Zhang, C. Wu, Z. Huang, S. Li, H. Zhang, Q. Yang, Z. Mao, S. Luo, C. Liu, G. Shi and B. Xu, Angew. Chem., Int. Ed., 2023, 62, e202217284 CrossRef CAS PubMed.
- X.-Y. Lou and Y.-W. Yang, J. Am. Chem. Soc., 2021, 143, 11976–11981 CrossRef CAS PubMed.
- F. Lin, H. Wang, Y. Cao, R. Yu, G. Liang, H. Huang, Y. Mu, Z. Yang and Z. Chi, Adv. Mater., 2022, 34, 2108333 CrossRef CAS PubMed.
- X.-Y. Dai, M. Huo and Y. Liu, Nat. Rev. Chem, 2023, 7, 854–874 CrossRef CAS PubMed.
- D. Wang, J. Gong, Y. Xiong, H. Wu, Z. Zhao, D. Wang and B. Z. Tang, Adv. Funct. Mater., 2023, 33, 2208895 CrossRef CAS.
- C. Xing, B. Zhou, D. Yan and W.-H. Fang, CCS Chem., 2023, 5, 2866–2876 CrossRef.
- B. Zhou and D. Yan, Adv. Funct. Mater., 2019, 29, 1807599 CrossRef.
- S. Tang, T. Yang, Z. Zhao, T. Zhu, Q. Zhang, W. Hou and W. Z. Yuan, Chem. Soc. Rev., 2021, 50, 12616–12655 RSC.
- T. Yang, Y. Li, Z. Zhao and W. Z. Yuan, Sci. China Chem., 2023, 66, 367–387 CrossRef CAS.
- J. A. C. R. Yule, J. Opt. Soc. Am., 1938, 28, 419–430 CrossRef.
- T. Zhu, T. Yang, Q. Zhang and W. Z. Yuan, Nat. Commun., 2022, 13, 2658 CrossRef CAS PubMed.
- N. Jiang, G.-F. Li, B.-H. Zhang, D.-X. Zhu, Z.-M. Su and M. R. Bryce, Macromolecules, 2018, 51, 4178–4184 CrossRef CAS.
- H. Sun, R. Ding, S. Lv, S. Zhou, S. Guo, Z. Qian and H. Feng, J. Phys. Chem. Lett., 2020, 11, 4962–4969 CrossRef CAS PubMed.
- P. R. Spackman, M. J. Turner, J. J. McKinnon, S. K. Wolff, D. J. Grimwood, D. Jayatilaka and M. A. Spackman, J. Appl. Crystallogr., 2021, 54, 1006–1011 CrossRef CAS PubMed.
- X. Chen, W. Luo, H. Ma, Q. Peng, W. Z. Yuan and Y. Zhang, Sci. China Chem., 2018, 61, 351–359 CrossRef CAS.
- X. Zou, N. Gan, M. Dong, W. Huo, A. Lv, X. Yao, C. Yin, Z. Wang, Y. Zhang, H. Chen, H. Ma, L. Gu, Z. An and W. Huang, Adv. Mater., 2023, 35, 2210489 CrossRef CAS PubMed.
- F. Nie, K.-Z. Wang and D. Yan, Nat. Commun., 2023, 14, 1654 CrossRef CAS PubMed.
- R. Gao and D. Yan, Chem. Sci., 2017, 8, 590–599 RSC.
- T. Kuroda, K. Fujii and K. Sakoda, J. Phys. Chem. C, 2010, 114, 983–989 CrossRef CAS.
- D. Li, Z. Liu, M. Fang, J. Yang, B. Z. Tang and Z. Li, ACS Nano, 2023, 17, 12895–12902 CrossRef CAS PubMed.
- H. Diab, C. Arnold, F. Ledee, G. Trippe-Allard, G. Delport, C. Vilar, F. Bretenaker, J. Barjon, J.-S. Lauret, E. Deleporte and D. Garrot, J. Phys. Chem. Lett., 2017, 8, 2977–2983 CrossRef CAS PubMed.
- X. Zhou, X. Zhao, X. Bai, Q. Cheng and Y. Liu, Adv. Funct. Mater., 2024, 34, 2400898 CrossRef CAS.
- F. Nie and D. Yan, Nat. Commun., 2024, 15, 5519 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4sc05741d |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2024 |