
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Magnetic circularly polarized luminescence from spin–flip transitions in a molecular ruby†
Alessio
Gabbani
 ab,
Maxime
Poncet
c,
Gennaro
Pescitelli
a,
Laura
Carbonaro
a,
J.
Krzystek
d,
Enrique
Colacio
e,
Claude
Piguet
c,
Francesco
Pineider
ab,
Lorenzo
Di Bari
a,
Juan-Ramón
Jiménez
*e and
Francesco
Zinna
*a
ab,
Maxime
Poncet
c,
Gennaro
Pescitelli
a,
Laura
Carbonaro
a,
J.
Krzystek
d,
Enrique
Colacio
e,
Claude
Piguet
c,
Francesco
Pineider
ab,
Lorenzo
Di Bari
a,
Juan-Ramón
Jiménez
*e and
Francesco
Zinna
*a
aDipartimento di Chimica e Chimica Industriale, University of Pisa, Via Moruzzi 13, 56124, Pisa, Italy. E-mail: francesco.zinna@unipi.it
bDepartment of Physics and Astronomy, University of Florence, Via Sansone 1, 50019, Sesto Fiorentino, Italy
cDepartment of Inorganic and Analytical Chemistry, University of Geneva, 30 Quai E. Ansermet, CH-1211 Geneva 4, Switzerland
dNational High Magnetic Field Laboratory, Florida State University, Tallahassee, Florida 32310, USA
eDepartamento de Química Inorgánica, Facultad de Ciencias, University of Granada, Unidad de Excelencia en Química (UEQ), Avda. Fuente Nueva s/n, 18071, Granada, Spain. E-mail: jrjimenez@ugr.es
First published on 1st October 2024
Abstract
Magnetic circularly polarized luminescence (MCPL), i.e. the possibility of generating circularly polarized luminescence in the presence of a magnetic field in achiral or racemic compounds, is a technique of rising interest. Here we show that the far-red spin–flip (SF) transitions of a molecular Cr(III) complex give intense MCD (magnetic circular dichroism) and in particular MCPL (gMCPL up to 6.3 × 10−3 T−1) even at magnetic fields as low as 0.4 T. Cr(III) doublet states and SF emission are nowadays the object of many investigations, as they may open the way to several applications. Due to their nature, such transitions can be conveniently addressed by MCPL, which strongly depends on the zero field splitting and Zeeman splitting of the involved states. Despite the complexity of the nature of such states and the related photophysics, the obtained MCPL data can be rationalized consistently with the information recovered with more established techniques, such as HFEPR (high-frequency and -field electron paramagnetic resonance). We anticipate that emissive molecular Cr(III) species may be useful in magneto-optical devices, such as magnetic CP-OLEDs.
Introduction
Molecular complexes based on d-metals offer a diverse and intriguing photophysics,1 with applications ranging from photocatalysis,2 optoelectronics,3,4 imaging5,6 and photodynamic therapy.7–9 To understand the often non-trivial photophysics at play, the use of less common spectroscopic techniques may be beneficial. In turn, this is necessary to exploit the full potential of those systems and to give indications for a rational design of the ligands and complexes.A particularly interesting case is observed when metal-centred excited states differ only by spin configuration with respect to the ground state.10 Such configurations are called spin–flip (SF) states and they may display sharp phosphorescent transitions (SF-transitions), forbidden by electric transition moment, with lifetimes up to a millisecond. SF luminescence was observed in the case of V(II)/V(III),11–13 Mn(IV),14,15 Mo(III),16,17 Re(IV),17 and in particular, remarkable results in terms of emission efficiency were obtained in the case of (pseudo)octahedral Cr(III) complexes.18–20 Such complexes show luminescence associated with the doublet states 2T1/2E (Fig. 1) with quantum yields up to 30% with narrow bands in the far red or near infrared region.18 These features, reminiscent of those of the ruby gemstone, can be obtained in a molecular compound with octahedral-like geometry (i) to avoid excited state distortions and (ii) to induce strong ligand field splitting, needed to shift the 4T2 states toward higher energy thus preventing deactivation of the SF states due to back intersystem crossing (BISC).19 Complexes featuring SF states may be exploited as optical probes for oxygen,20 pressure21 and temperature,22 photocatalysis23–25 and photocathodic solar cells.19,26
 | ||
| Fig. 1 (a) Structure of the complex. (b) Electronic states of [Cr(dqp)2]3+ in an ideal octahedral (Oh) and D2 geometry; note the 2T1/2E level inversion from Oh to D2 geometry. | ||
Concerning the luminescence activity associated with the SF transitions, the most promising results have been achieved so far by employing two families of Cr(III) complexes: [Cr(ddpd)2]3+ (ddpd = N,N′-dimethyl-N,N′′-dipyridin-2-ylpyridine-2,6-diamine)18,27,28 and [Cr(dqp)2]3+ (dqp = 2,6-di(quinolin-8-yl)pyridine).29,30 In those cases, the first coordination sphere is roughly octahedral, but the arrangement of the tridentate organic ligand around the Cr-center defines a λ/δ chirality in an overall D2 geometry.31 Thanks to the electric dipole forbidden nature of the SF transition, in enantiopure form, such compounds display highly circularly polarized luminescence (CPL), with dissymmetry factors (glum) on the order of 10−1.30–34 Such values are comparable with those obtained for the f–f transitions of chiral lanthanide(III) complexes,35–39 but Cr offers the advantage of being cheaper, kinetically inert and more abundant than lanthanides.40
A different technique to study the circular polarization of the emitted light is magnetic CPL (MCPL), where the physical origin of the CP emission is not the chirality of the material, but the effect is triggered by the application of an external magnetic field. This technique belongs to the family of magneto-optical spectroscopies, along with the more common Faraday rotation and magnetic circular dichroism (MCD).41–43 In a MCPL experiment, the circular polarization of the luminescence is studied, when the sample is placed under a magnetic field collinear with the emission direction, and excited with unpolarized light.44,45 Unlike CPL, MCPL may be displayed by both chiral and achiral luminescent systems and it does not depend on the enantiomer chirality. Indeed, CPL and MCPL follow very different selection rules. CPL is gauged by the scalar product mng·μgn, where m and μ are the magnetic and electric transition moments between the ground state g and excited state(s) n.46 On the other hand, several mechanisms can lead to MCPL. Relatively strong signals are predicted in the case of orbital or spin degenerate ground or excited states, where the degeneracy is removed by the magnetic field due to the Zeeman effect. In these cases, the MCPL signal depends on mggμngμgn and mnnμngμgn products (mgg and mnn are the static magnetic dipole moments of the ground and excited state), for a degenerate ground or excited state respectively.47,48 Those expressions are associated to the so-called Faraday A- and C-terms.44,47,49,50 Emitting compounds characterized by a strong spin–orbit coupling, which allows for a significant mixing of states, are good candidates for magneto-optical spectroscopies, including MCPL.
MCPL has been thus studied in the case of f–f transitions(III) of lanthanide complexes,51–54 d-metals (such as Ru(II), Ir(III) and Pt(II) complexes),55–58 organic and metallo-organic compounds,59–69 as well as other metal-based materials.70–72 MCPL and MCD were also reported in early studies of Cr(III) inorganic structures.73,74 MCD was also used to study far red/near infrared (NIR) transitions in the case of Ir(III) or Pt(II) complexes,75,76 where their observation through emission, and thus MCPL, is challenging. On the other hand, emissive Cr(III) compounds are potentially a more suitable platform to address metal transitions through MCPL. On a fundamental level, an analysis of the MCPL spectrum can elucidate the nature of the excited and ground states, Zeeman effects, etc., and along with other techniques can help to understand the full picture of the photophysics of a complex system.44 Moreover, MCPL-active compounds have recently found applications in OLEDs able to emit circularly polarized electroluminescence in a magnetic field (MCP-OLEDs),77–81 therefore there is also a practical interest in unveiling different types of emitters endowed with significant MCPL.
In the following, we investigate the racemic [Cr(dqp)2](PF6)3 material by MCPL and MCD (Fig. 1b). As introduced above, [Cr(dqp)2]3+ is one of the archetypes of a molecular ruby and the same concept shown here may be applied to similar systems.
Results and discussion
Optical and magneto-optical studies reported here were performed on deaerated acetonitrile solutions of the racemic compound ([Cr(dqp)2](PF6)3). The photoluminescence (PL) spectrum of [Cr(dqp)2]3+ shows two main emission bands centred at 750 and 729 nm (Fig. 2), associated to the SF transitions (2E/2T1 → 4A2) from the sublevels of the doublet excited states (see Fig. 1a) to the ground state. The lower energy band appears more intense than the higher energy one by a factor of 1.53 at 300 K, due to a higher Boltzmann population with an energy gap of ≈420 cm−1 between the two bands.27 | ||
| Fig. 2 PL and MCPL at 0.4 T, normalized for the PL maximum, of the Cr3+ complex dissolved in deareated acetonitrile, along with the corresponding fittings according to the rigid-shift model (see the text). | ||
We measured the MCPL emission at 300 K under a magnetic field of ±0.4 T generated by a permanent magnet, exciting the sample at 365 nm (see the ESI and Fig. S1† for the details of the measurement set-up). In these conditions, a relatively strong and slightly asymmetric bisignate MCPL band was observed corresponding to the higher energy doublet transition (Fig. 2). Such band has a cross-over point at 725 nm, matching the maximum of the corresponding photoluminescence band. This derivative-like shape is consistent with a signal originating from Zeeman-split states (see below). The MCPL strength can be quantified by a magnetic field (H) normalized dissymmetry factor (gMCPL), defined as:
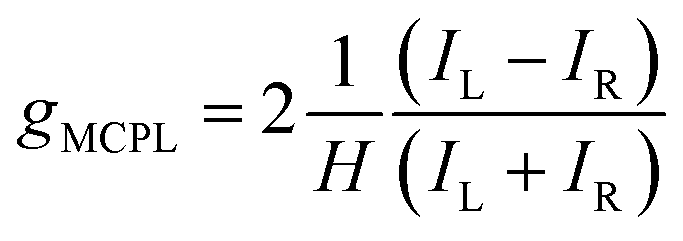 | (1) |
 | (2) |
Positive and negative fields are here defined as parallel and anti-parallel to the k-vector, i.e. the propagating direction of light. Baseline effects and possible artefact signals due to photoselection cause some deviation from the mirror image relationship expected for spectra obtained under opposite magnetic field, especially in the case of small signals (Fig. S3a†). The data treated according to eqn (2) ensures that such artefacts, which are magnetic field-independent, are eliminated and only the true MCPL is recovered (Fig. S3b†).
The same SF transitions were also studied in absorption through MCD, on a concentrated solution of the complex under ±1.4 T magnetic field (Fig. 3b), generated by an electromagnet. A derivative-like signal was observed associated to the higher energy doublet transition, similar to the corresponding MCPL band. No significant MCD corresponding to the lower energy SF transition was detected. Similarly to the MCPL, MCD dissymmetry factor (gMCD) can be defined as:
 | (3) |
 | ||
| Fig. 3 MCD and extinction spectra of an acetonitrile solution of [Cr(dqp)2]3+, for the higher energy region (a) and for the SF transition region (b). The data are normalized for concentration, optical path and applied magnetic field. | ||
To rationalize these results, it is worth analysing the states involved in the SF transitions giving origin to the MCD and MCPL spectra. In the complex, the presence of the helically twisted tridentate dqp suppresses any symmetry plane and lowers the overall symmetry to D2. In such symmetry, the orbital degeneracy of the excited states is removed, and 2T1 and 2E states are split into 3 and 2 components respectively (Fig. 1a), the main MCPL signal at 729 nm is associated with the lower component of the doubly degenerate 2E state. A small contribution to the PL, giving non-significant MCPL, is observed at approximately 710 nm, possibly stemming from the higher energy component of the 2E state (Fig. S2†). Moreover, even in the absence of an external field, the orbitally-nondegenerate quartet ground state (4A2) is split by the zero-field splitting (ZFS) into two Kramers doublets (KD) |±1/2〉 and |±3/2〉 (Fig. 4).
 | ||
| Fig. 4 ZFS- and Zeeman-split levels at 0.4 T corresponding to the main MCPL bands. The MCPL transitions are represented by the dotted arrows along with the expected sign. The + and − symbols indicate left and right circularly polarized emissive transitions respectively. The energies of ground state sublevels (for H0∥z) are obtained through HFEPR analysis (see Fig. 5 and Table S3†). As considerable mixing occurs (see Table S3†), the predominant character of the Ms state is indicated by a ∼ symbol. Purple colour indicates strongly mixed states. | ||
To extract the ZFS and the corresponding spin Hamiltonian D and E parameters, we followed the method proposed by van Slageren et al. (see ESI†).83 As the first step, the energies corresponding to the 3 4A2 → 4T2 term-to-term transitions were determined by a phenomenological deconvolution of the 360–500 nm region of the MCD spectrum (Fig. S5†). With this procedure, we identified the following energies, centred approximately at 24![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 220, 24922 and 25630 cm−1 (Table S1†). From these values (see ESI† for the formulae in a D2 geometry), we calculated D and E parameters as approximately 0.51 and 0.16 cm−1, respectively, with a rhombicity factor E/D = 0.32, close to the maximum (1/3). The corresponding ZFS was calculated to be ≈1.1 cm−1 as
220, 24922 and 25630 cm−1 (Table S1†). From these values (see ESI† for the formulae in a D2 geometry), we calculated D and E parameters as approximately 0.51 and 0.16 cm−1, respectively, with a rhombicity factor E/D = 0.32, close to the maximum (1/3). The corresponding ZFS was calculated to be ≈1.1 cm−1 as  (see Fig. 4). Despite the large error of the method (see for instance the standard errors on the fitting coefficient in Table S1†), the values are comparable with those found for the analogue complex [Cr(ddpd)2]3+,83 and the values found by HFEPR (high-frequency and -field electron paramagnetic resonance) on our complex (see below).
(see Fig. 4). Despite the large error of the method (see for instance the standard errors on the fitting coefficient in Table S1†), the values are comparable with those found for the analogue complex [Cr(ddpd)2]3+,83 and the values found by HFEPR (high-frequency and -field electron paramagnetic resonance) on our complex (see below).
We now focus our analysis on the MCPL and PL spectra at the NIR SF transitions. In the presence of an external magnetic field, the degeneracy of the KDs of the quartet ground states is removed by Zeeman effect giving 4 spin states: ideally |+1/2〉, |−1/2〉, |+3/2〉 and |−3/2〉. Similarly, the doublet excited states split into |+1/2〉 and |−1/2〉 states.
In the following, we focus only on the higher energy SF band around 729 nm, producing the main signal. In a reasonably simplified scheme, we consider only the transitions among Zeeman levels with ΔMs = ±1.84 A ΔMs > 0 corresponds to a positive MCPL transition and vice versa, therefore a total of two closely spaced positive and two negative MCPL contributions are expected (Fig. 4).
When the bands are separated by an energy much smaller than the bandwidth, the spectral features can be conveniently modelled by using the so-called rigid-shift approximation. This method is usually applied to model A- and C-Faraday terms in MCD.43,85,86 Within this model, PL and MCPL spectra are fitted simultaneously using a home-built MATLAB code, employing fitting functions that hold shared parameters. The emission is fitted with a bell-shaped function centred on the energy barycentre (unsplit levels), while the MCPL is fitted with equal but opposite functions, displaced by the small field-induced splitting (see ESI and Fig. S6†). All the functions, used for both the PL and MCPL, share the same shape and bandwidth, and are therefore determined simultaneously in the fitting. To carry out this procedure, we used the four energy levels of the 4A2 state determined by HFEPR at 0.4 T (see below), as fixed parameters. The fitting obtained through this model, by using pseudo-Voigt functions, closely retraces the experimental PL and MCPL data (Fig. 2 and ESI†). Similar results were obtained using purely Gaussian or Lorentzian functions, but pseudo-Voigt functions retrace better the PL and MCPL line shapes (Fig. S7 and Table S2†). The overestimation of the model with respect to the experimental MCPL data may be due to the fact that, in the case of ZFS with E ≠ 0, the four states associated with 4A2 cannot be described by a pure Ms quantum number as they are significantly mixed (see below). This would therefore impact the underlying assumption that each transition is completely circularly polarized. Notice that the lifetime of the excited state being sufficiently long (τobs = 1.2 ms),33 the population of its Zeeman levels follows Boltzmann distribution. The Zeeman splitting being much smaller than room temperature thermal energy (207 cm−1 at 298 K), the Zeeman levels of the doublet excited states are almost equally populated, as 1 − (ΔE/kbT) ≈ 0.998. Fig. 4 quantitatively summarizes the fine structure of the electronic level involved in the MCPL emission of the main band.
To corroborate the analysis and demonstrate the consistency of our approach, we performed magnetometry and EPR characterization. DC magnetometry, studied in the 2–300 K temperature range, confirms a quartet ground state (4A2, see ESI and Fig. S8†). Saturation magnetization at 2 K is consistent with what expected for isolated Cr(III) cations with g = 2 and S = 3/2. Upon cooling, the χMT product remains almost constant until about 10 K and then sharply decreases to reach a value of 1.78 cm3 mol−1 K at 2 K. This decrease is due to the ZFS and Zeeman interactions. The simultaneous fitting of the susceptibility and magnetization data with the ZFS Hamiltonian using the PHI software87 leads to a |D| value of 0.72 cm−1, which is rather consistent with the values extracted from MCD and HFEPR spectroscopies.
To confirm the determined D and E values from MCD measurements, the Cr(III) compound was studied by HFEPR spectroscopy.88 The shape and amplitude of the low-temperature (30 K) spectra of the powder sample “as is” (i.e. unconstrained) strongly suggest field-induced alignment (i.e. torquing) of the crystallites.89 Indeed, the resulting spectra could be very well simulated as originating from a single crystal oriented with the z-axis of the ZFS tensor parallel to the magnetic field H0 (Fig. 5). At 270 GHz, the three dominating peaks between 8 and 12 T represent the allowed ΔMs = ±1 transitions between the spin sublevels of the S = 3/2 spin state of Cr(III). The weak peaks in the 3–6 T range are the nominally forbidden ΔMs = ±2 and ±3 transitions. The structure visible on the peaks is an artefact that can be attributed to imperfect field alignment and is not simulated.
 | ||
| Fig. 5 Top: EPR spectrum of a polycrystalline sample containing the Cr(III) compound at 30 K and 270 GHz (black trace) accompanied by its simulation (red trace) using the following spin Hamiltonian parameters: S = 3/2, D = 0.42 cm−1, E = 0.14 cm−1 (E/D = 0.33, maximum rhombicity limit), giso = 1.99. The simulation assumed a single crystal oriented with the z-axis of the ZFS tensor parallel to the magnetic field H0. Bottom: representation of the energy levels for a S = 3/2 spin state at 270 GHz. The HFEPR transitions between the spin sublevels are marked with red arrows. | ||
In order to extract the full set of frequency-independent spin Hamiltonian parameters, the sample was constrained using n-eicosane and pressed into a pellet (Fig. S9†). The resulting spectrum is accompanied by a simulation, this time assuming a powder distribution of the crystallites in space. The simulation parameters are modified relative to those used above to account for the slight asymmetry of the central line: D = 0.43 cm−1, E = 0.14 cm−1 (E/D = 0.325), gx = 1.99, gy,z = 1.98.
To finalize the values of frequency-independent spin Hamiltonian parameters, we built a two-dimensional map of turning points in pellet spectra and applied the tunable-frequency methodology90 by fitting the parameters simultaneously to that map. This resulted in the following values: D = −0.436(7) cm−1, E = −0.144(7) cm−1 (E/D = 0.31), giso = 1.980(4) (Fig. S10†). The negative sign of D reproduced single-frequency spectra better than a positive value.
Altogether, these results are in good agreement with the ones that we found by MCPL and MCD experiments. Finally, the nature of the Ms states associated with 4A2 was evaluated by calculating the mixing coefficients at 0.4 T (Tables S3 and S4, see also Fig. S11† for an expansion of HFEPR levels at low field). The coefficients show (in the case of H0∥z, see Table S3†) a strong mixing between two inner |+3/2〉 and |−1/2〉 levels, responsible for the avoided level crossing near 1 T visible in Fig. 5 (bottom), while the outer levels retain mostly their |+1/2〉 and |−3/2〉 character.
Conclusions
Beyond the many areas of interest in Cr(III) SF transitions of the molecular ruby [Cr(dqp)2]3+, we show here that they also display a strong MCPL activity. MCD and MCPL techniques are here applied to elucidate the fine structure of the levels involved in the SF transitions. In particular, the analysis of SF transition through MCPL was consistent with the energies found with the more established EPR spectroscopy. Such possibilities offered by magneto-optical techniques can be exploited to gather more insight into the photophysics of SF transitions in related cases, e.g. in view of optical read-out of molecular qubits. Moreover, it opens up new opportunities such as applications in magneto-optical and magnetoelectronic devices.Data availability
The data supporting this article have been included as part of the ESI.†Author contributions
Conceptualization: F. Z.; data curation: A. G., G. P., F. P., J. R. J., F. Z.; formal analysis: A. G., F. Z.; resources: M. P., C. P., J. R. J.; funding acquisition: L. D. B., C. P., F. P., J. R. J.; investigation: A. G., L. C., E. C., J. K., F. Z. methodology: F. Z., G. P. writing – original draft: A. G., J. R. J., F. Z.; writing – review & editing: all authors.Conflicts of interest
There are no conflicts to declare.Acknowledgements
F. P. acknowledges MUR through PRIN-PNRR project (P20229723Z) J. R. J. thanks Ministerio de Ciencia Innovación y Universidades for a Ramón y Cajal contract (grant RYC2022-037255-I) funded by MCIN/AEI/10.13039/501100011033 and FSE+. E. C. and J. R. J. acknowledge Ministerio de Ciencia e Innovación for financial support (project PID2022-138090NB-C21 funded by MCIN/AEI/10.13039/501100011033/FEDER,UE). Part of this work was done at the National High Magnetic Field Laboratory which is funded by the US National Science Foundation (Cooperative Agreement DMR-212856) and the State of Florida. Dr A. Ozarowski (NHMFL) is acknowledged for his EPR fit and simulation software SPIN and help with solving the level mixing problem. Prof. L. Sorace (University of Florence) is acknowledged for fruitful discussions. We would like to thank Mr D. Michelotti (University of Pisa) for technical support with the MCPL set-up.Notes and references
- V. Balzani, A. Credi and M. Venturi, Coord. Chem. Rev., 1998, 171, 3–16 CrossRef CAS.
- J. Twilton, C. (Chip) Le, P. Zhang, M. H. Shaw, R. W. Evans and D. W. C. MacMillan, Nat. Rev. Chem, 2017, 1, 1–19 CrossRef.
- H. Xu, R. Chen, Q. Sun, W. Lai, Q. Su, W. Huang and X. Liu, Chem. Soc. Rev., 2014, 43, 3259–3302 RSC.
- W. C. H. Choy, W. K. Chan and Y. Yuan, Adv. Mater., 2014, 26, 5368–5399 CrossRef CAS PubMed.
- M. P. Coogan and V. Fernández-Moreira, Chem. Commun., 2013, 50, 384–399 RSC.
- J. Berrones Reyes, M. K. Kuimova and R. Vilar, Curr. Opin. Chem. Biol., 2021, 61, 179–190 CrossRef CAS PubMed.
- Y. Wu, S. Li, Y. Chen, W. He and Z. Guo, Chem. Sci., 2022, 13, 5085–5106 RSC.
- T. W. Rees, P.-Y. Ho and J. Hess, ChemBioChem, 2023, 24, e202200796 CrossRef CAS PubMed.
- C. B. Smith, L. C. Days, D. R. Alajroush, K. Faye, Y. Khodour, S. J. Beebe and A. A. Holder, Photochem. Photobiol., 2022, 98, 17–41 CrossRef CAS PubMed.
- W. R. Kitzmann, J. Moll and K. Heinze, Photochem. Photobiol. Sci., 2022, 21, 1309–1331 CrossRef CAS PubMed.
- M. Dorn, J. Kalmbach, P. Boden, A. Kruse, C. Dab, C. Reber, G. Niedner-Schatteburg, S. Lochbrunner, M. Gerhards, M. Seitz and K. Heinze, Chem. Sci., 2021, 12, 10780–10790 RSC.
- M. Dorn, D. Hunger, C. Förster, R. Naumann, J. van Slageren and K. Heinze, Chem.–Eur. J., 2023, 29, e202202898 CrossRef CAS PubMed.
- M. Dorn, J. Kalmbach, P. Boden, A. Päpcke, S. Gómez, C. Förster, F. Kuczelinis, L. M. Carrella, L. A. Büldt, N. H. Bings, E. Rentschler, S. Lochbrunner, L. González, M. Gerhards, M. Seitz and K. Heinze, J. Am. Chem. Soc., 2020, 142, 7947–7955 CrossRef CAS PubMed.
- J. P. Harris, C. Reber, H. E. Colmer, T. A. Jackson, A. P. Forshaw, J. M. Smith, R. A. Kinney and J. Telser, Can. J. Chem., 2017, 95, 547–552 CrossRef.
- N. R. East, R. Naumann, C. Förster, C. Ramanan, G. Diezemann and K. Heinze, Nat. Chem., 2024, 1–8 Search PubMed.
- W. R. Kitzmann, D. Hunger, A.-P. M. Reponen, C. Förster, R. Schoch, M. Bauer, S. Feldmann, J. van Slageren and K. Heinze, Inorg. Chem., 2023, 62, 15797–15808 CrossRef CAS PubMed.
- Q. Yao and A. W. Maverick, Inorg. Chem., 1988, 27, 1669–1670 CrossRef CAS.
- C. Wang, S. Otto, M. Dorn, E. Kreidt, J. Lebon, L. Sršan, P. Di Martino-Fumo, M. Gerhards, U. Resch-Genger, M. Seitz and K. Heinze, Angew. Chem., Int. Ed., 2018, 57, 1112–1116 CrossRef CAS PubMed.
- S. Treiling, C. Wang, C. Förster, F. Reichenauer, J. Kalmbach, P. Boden, J. P. Harris, L. M. Carrella, E. Rentschler, U. Resch-Genger, C. Reber, M. Seitz, M. Gerhards and K. Heinze, Angew. Chem., Int. Ed., 2019, 58, 18075–18085 CrossRef CAS PubMed.
- C. Wang, S. Otto, M. Dorn, K. Heinze and U. Resch-Genger, Anal. Chem., 2019, 91, 2337–2344 CrossRef CAS PubMed.
- S. Otto, J. P. Harris, K. Heinze and C. Reber, Angew. Chem., Int. Ed., 2018, 57, 11069–11073 CrossRef CAS PubMed.
- S. Otto, N. Scholz, T. Behnke, U. Resch-Genger and K. Heinze, Chem.–Eur. J., 2017, 23, 12131–12135 CrossRef CAS PubMed.
- W. R. Kitzmann and K. Heinze, Angew. Chem., Int. Ed., 2023, 62, e202213207 CrossRef CAS PubMed.
- T. H. Bürgin, F. Glaser and O. S. Wenger, J. Am. Chem. Soc., 2022, 144, 14181–14194 CrossRef PubMed.
- C. Wang, H. Li, T. H. Bürgin and O. S. Wenger, Nat. Chem., 2024, 1–9 Search PubMed.
- B. Doistau, G. Collet, E. A. Bolomey, V. Sadat-Noorbakhsh, C. Besnard and C. Piguet, Inorg. Chem., 2018, 57, 14362–14373 CrossRef CAS PubMed.
- S. Otto, M. Grabolle, C. Förster, C. Kreitner, U. Resch-Genger and K. Heinze, Angew. Chem., Int. Ed., 2015, 54, 11572–11576 CrossRef CAS PubMed.
- S. Otto, C. Förster, C. Wang, U. Resch-Genger and K. Heinze, Chem.–Eur. J., 2018, 24, 12555–12563 CrossRef CAS PubMed.
- J.-R. Jiménez, M. Poncet, B. Doistau, C. Besnard and C. Piguet, Dalton Trans., 2020, 49, 13528–13532 RSC.
- J.-R. Jiménez, B. Doistau, C. M. Cruz, C. Besnard, J. M. Cuerva, A. G. Campaña and C. Piguet, J. Am. Chem. Soc., 2019, 141, 13244–13252 CrossRef PubMed.
- M. Poncet, A. Benchohra, J.-R. Jiménez and C. Piguet, ChemPhotoChem, 2021, 5, 880–892 CrossRef CAS.
- C. Dee, F. Zinna, W. R. Kitzmann, G. Pescitelli, K. Heinze, L. D. Bari and M. Seitz, Chem. Commun., 2019, 55, 13078–13081 RSC.
- J.-R. Jiménez, M. Poncet, S. Míguez-Lago, S. Grass, J. Lacour, C. Besnard, J. M. Cuerva, A. G. Campaña and C. Piguet, Angew. Chem., Int. Ed., 2021, 60, 10095–10102 CrossRef PubMed.
- J.-R. Jiménez, S. Míguez-Lago, M. Poncet, Y. Ye, C. L. Ruiz, C. M. Cruz, A. G. Campaña, E. Colacio, C. Piguet and J. M. Herrera, J. Mater. Chem. C, 2023, 11, 2582–2590 RSC.
- O. G. Willis, F. Zinna and L. Di Bari, Angew. Chem., Int. Ed., 2023, 62, e202302358 CrossRef CAS PubMed.
- L. Llanos, P. Cancino, P. Mella, P. Fuentealba and D. Aravena, Coord. Chem. Rev., 2024, 505, 215675 CrossRef CAS.
- H.-Y. Wong, W.-S. Lo, K.-H. Yim and G.-L. Law, Chem, 2019, 5, 3058–3095 CAS.
- F. Zinna and L. Di Bari, Chirality, 2015, 27, 1–13 CrossRef CAS PubMed.
- B. Doistau, J.-R. Jiménez and C. Piguet, Front. Chem., 2020, 8, 555 CrossRef CAS PubMed.
- C. Förster and K. Heinze, Chem. Soc. Rev., 2020, 49, 1057–1070 RSC.
- P. N. Schatz, A. J. McCaffery, W. Suetaka, G. N. Henning, A. B. Ritchie and P. J. Stephens, J. Chem. Phys., 1966, 45, 722–734 CrossRef CAS.
- P. J. Stephens, W. Suëtaak and P. N. Schatz, J. Chem. Phys., 1966, 44, 4592–4602 CrossRef CAS PubMed.
- W. R. Mason, A practical guide to magnetic circular dichroism spectroscopy, Wiley-Interscience, Hoboken, NJ, 2007 Search PubMed.
- F. Zinna and G. Pescitelli, Eur. J. Org Chem., 2023, 26, e202300509 CrossRef CAS.
- M. Fusè, G. Mazzeo, S. Ghidinelli, A. Evidente, S. Abbate and G. Longhi, Spectrochim. Acta, Part A, 2024, 319, 124583 CrossRef PubMed.
- J. P. Riehl and F. S. Richardson, Chem. Rev., 1986, 86, 1–16 CrossRef CAS.
- J. P. Riehl and F. S. Richardson, J. Chem. Phys., 1977, 66, 1988–1998 CrossRef CAS.
- L. D. Barron, Molecular Light Scattering and Optical Activity, Cambridge University Press, 2009 Search PubMed.
- P. J. Stephens, J. Chem. Phys., 1970, 52, 3489–3516 CrossRef CAS.
- Z. Nelson, L. Delage-Laurin and T. M. Swager, J. Am. Chem. Soc., 2022, 144, 11912–11926 CrossRef CAS PubMed.
- F. S. Richardson and H. G. Brittain, J. Am. Chem. Soc., 1981, 103, 18–24 CrossRef CAS.
- T. Wu, J. Kapitán, V. Andrushchenko and P. Bouř, Anal. Chem., 2017, 89, 5043–5049 CrossRef CAS PubMed.
- H. Yoshikawa, G. Nakajima, Y. Mimura, T. Kimoto, Y. Kondo, S. Suzuki, M. Fujiki and Y. Imai, Dalton Trans., 2020, 49, 9588–9594 RSC.
- D. A. Gálico and M. Murugesu, Adv. Opt. Mater., 2024, 2401064, DOI:10.1002/adom.202401064.
- M. Kitahara, S. Suzuki, K. Matsudaira, S. Yagi, M. Fujiki and Y. Imai, ChemistrySelect, 2021, 6, 11182–11187 CrossRef CAS.
- E. Krausz, G. Moran and H. Riesen, Chem. Phys. Lett., 1990, 165, 401–406 CrossRef CAS.
- K. Matsudaira, A. Izumoto, Y. Mimura, Y. Kondo, S. Suzuki, S. Yagi, M. Fujiki and Y. Imai, Phys. Chem. Chem. Phys., 2021, 23, 5074–5078 RSC.
- K. Matsudaira, Y. Mimura, J. Hotei, S. Yagi, K. Yamashita, M. Fujiki and Y. Imai, Chem.–Asian J., 2021, 16, 926–930 CrossRef CAS PubMed.
- S. Jena, S. K. Behera, J. Eyyathiyil, M. Kitahara, Y. Imai and P. Thilagar, Adv. Opt. Mater., 2023, 11, 2300923 CrossRef CAS.
- S. Ghidinelli, S. Abbate, G. Mazzeo, L. Paoloni, E. Viola, C. Ercolani, M. P. Donzello and G. Longhi, Chirality, 2020, 32, 808–816 CrossRef CAS PubMed.
- S. Ghidinelli, S. Abbate, G. Mazzeo, R. Paolesse, G. Pomarico and G. Longhi, ACS Omega, 2021, 6, 26659–26671 CrossRef CAS PubMed.
- Q. Jin, S. Chen, Y. Sang, H. Guo, S. Dong, J. Han, W. Chen, X. Yang, F. Li and P. Duan, Chem. Commun., 2019, 55, 6583–6586 RSC.
- H. Toda, N. Hara, M. Fujiki and Y. Imai, RSC Adv., 2021, 11, 1581–1585 RSC.
- H. Toda, S. Otake, A. Ito, M. Miyasaka, M. Fujiki and Y. Imai, ChemPhysChem, 2021, 22, 2058–2062 CrossRef CAS PubMed.
- N. Hara, M. Kitahara, T. Sugimura, H. Toda, M. Shizuma, A. Ito, M. Miyasaka, M. Fujiki and Y. Imai, Phys. Chem. Chem. Phys., 2021, 23, 8236–8240 RSC.
- R. Amasaki, M. Kitahara, T. Kimoto, M. Fujiki and Y. Imai, Eur. J. Inorg. Chem., 2022, 2022, e202101066 CrossRef CAS.
- T. Tomikawa, Y. Kitagawa, K. Yoshioka, K. Murata, T. Miyatake, Y. Hasegawa and K. Ishii, J. Mater. Chem. C, 2023, 11, 2831–2835 RSC.
- D. H. Metcalf, T. C. VanCott, S. W. Snyder, P. N. Schatz and B. E. Williamson, J. Phys. Chem., 1990, 94, 2828–2832 CrossRef CAS.
- Z. Gasyna, D. H. Metcalf, P. N. Schatz, C. L. McConnell and B. E. Williamson, J. Phys. Chem., 1995, 99, 5865–5872 CrossRef CAS.
- T. Kimoto, Y. Mimura, M. Fujiki and Y. Imai, Chem. Lett., 2021, 50, 916–919 CrossRef CAS.
- Y. Mimura, M. Fujiki and Y. Imai, Chem. Phys. Lett., 2021, 767, 138353 CrossRef CAS.
- R. Amasaki, M. Kitahara, S. Tanaka and Y. Imai, Eur. J. Inorg. Chem., 2024, 27, e202300621 CrossRef CAS.
- R. A. Shatwell and A. J. McCaffery, Mol. Phys., 1975, 30, 1489–1504 CrossRef CAS.
- A. J. McCaffery, P. Brint, R. Gale and R. A. Shatwell, Chem. Phys. Lett., 1973, 22, 600–602 CrossRef CAS.
- K. Murata and K. Ishii, Eur. J. Inorg. Chem., 2017, 2017, 5103–5107 CrossRef CAS.
- K. Ishii, J. Wada and K. Murata, J. Phys. Chem. Lett., 2020, 11, 9828–9833 CrossRef CAS PubMed.
- K. Hara, A. Morimoto, K. Matsudaira, S. Suzuki, S. Yagi, M. Fujiki and Y. Imai, ChemPhotoChem, 2022, 6, e202100253 CrossRef CAS.
- T. Kuroda, M. Kitahara, S. Yagi and Y. Imai, Front. Chem., 2023, 11, 1281168 CrossRef CAS PubMed.
- M. Kitahara, K. Hara, S. Suzuki, H. Iwasaki, S. Yagi and Y. Imai, Org. Electron., 2023, 119, 106814 CrossRef CAS.
- S. Suzuki, Y. Yamamoto, M. Kitahara, R. Shikura, S. Yagi and Y. Imai, J. Mater. Chem. C, 2024, 12, 3430–3436 RSC.
- Y. Imai, R. Amasaki, Y. Yanagibashi, S. Suzuki, R. Shikura and S. Yagi, Magnetochemistry, 2024, 10, 39 CrossRef CAS.
- Y. Imai, ChemPhotoChem, 2021, 5, 969–973 CrossRef CAS.
- S. Lenz, H. Bamberger, P. P. Hallmen, Y. Thiebes, S. Otto, K. Heinze and J. van Slageren, Phys. Chem. Chem. Phys., 2019, 21, 6976–6983 RSC.
- E. I. Solomon, M. L. Neidig and G. Schenk, in Comprehensive Coordination Chemistry II, Elsevier, 2003, pp. 339–349 Search PubMed.
- A. Gabbani, G. Petrucci and F. Pineider, J. Appl. Phys., 2021, 129, 211101 CrossRef CAS.
- A. Gabbani, G. Campo, V. Bonanni, P. van Rhee, G. Bottaro, C. de Julián Fernández, V. Bello, E. Fantechi, F. Biccari, M. Gurioli, L. Armelao, C. Sangregorio, G. Mattei, P. Christianen and F. Pineider, J. Phys. Chem. C, 2022, 126, 1939–1945 CrossRef CAS.
- N. F. Chilton, R. P. Anderson, L. D. Turner, A. Soncini and K. S. Murray, J. Comput. Chem., 2013, 34, 1164–1175 CrossRef CAS PubMed.
- J. Krzystek, A. Ozarowski and J. Telser, Coord. Chem. Rev., 2006, 250, 2308–2324 CrossRef CAS.
- T. Dubroca, A. Ozarowski, Y. Sunatsuki, J. Telser, S. Hill and J. Krzystek, Appl. Magn. Reson., 2024, 55 DOI:10.1007/s00723-024-01706-3.
- J. Krzystek, S. A. Zvyagin, A. Ozarowski, S. Trofimenko and J. Telser, J. Magn. Reson., 2006, 178, 174–183 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4sc04718d |
| This journal is © The Royal Society of Chemistry 2024 |