
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicencePolyoxometalate-encapsulated metal–organic frameworks for photocatalytic uranium isolation†
Zhimin
Dong‡
a,
Dongling
Zeng‡
a,
Zifan
Li
a,
Junjie
Chen
a,
Youqun
Wang
a,
Xiaohong
Cao
 a,
Guoping
Yang
*a,
Zhibin
Zhang
*a,
Yunhai
Liu
a and
Feng
Yang
*b
a,
Guoping
Yang
*a,
Zhibin
Zhang
*a,
Yunhai
Liu
a and
Feng
Yang
*b
aState Key Laboratory of Nuclear Resources and Environment, East China University of Technology, Nanchang 330013, China. E-mail: erick@ecut.edu.cn; zhbzhang@ecut.edu.cn
bDepartment of Chemistry, Southern University of Science and Technology, Shenzhen 518055, China. E-mail: yangf3@sustech.edu.cn
First published on 22nd October 2024
Abstract
Recycling uranium (U) via adsorption and controlled conversion is crucial for the sustainable development of nuclear energy, in which photocatalytic reduction of U(VI) from aqueous solutions is considered one of the most effective strategies. The primary challenge in the photocatalytic elimination of U(VI) resides in the demand for photocatalysts with exceptional properties for effective U(VI) adsorption and charge separation. Herein, we developed the hybrids of polyoxometalate@Cu-metal–organic frameworks (POM@Cu-MOFs) through a self-assembly strategy and demonstrated the efficient removal of U(VI) via synergistic adsorption and photocatalysis. The abundant oxygen-rich groups in POM served as the adsorption sites, endowing POM@Cu-MOFs with a remarkable removal capacity (1987.4 mg g−1 under light irradiation) to remove 99.4% of UO22+. The attraction of electrons from Cu atoms within Cu-MOFs effectively accelerated the carrier dynamics due to their pronounced electronegativity. A mechanism associated with the synergetic effects of adsorption and photocatalytic reduction of U(VI) was proposed. This work provides a feasible approach for efficiently eliminating U(VI) from aqueous solutions in environmental pollution cleanup using the POM@Cu-MOF photocatalyst.
Introduction
Uranium, as the most important resource for nuclear power, has an irreplaceable role but is also known as a global environmental contaminant due to its inherent radio and chemo-toxicities.1 Motivating the development of uranium enrichment strategies is vital for both the sustainability of nuclear energy and environmental governance. Among various strategies, adsorption is considered the most promising one due to its high removal efficiency, economic productivity, and easy operation.2,3 However, the adsorption capacity will be limited once the active sites of the adsorbent reach saturation. Currently, photocatalytic reduction has attracted increasing interest because of its high efficiency, reusability of photocatalysts, environmental friendliness, and low energy input.4 What is even more noteworthy is its capacity to harness solar light for facile conversion of soluble and mobile U(VI) into insoluble and immobile uranium precipitates, which is paramount in the immobilization of uranium.5 Various photocatalysts, including TiO2 particles,6 graphitic carbon nitride,7,8 and composite materials,9 have been utilized for U(VI) removal. By far, it remains a significant challenge to explore an environmentally friendly photocatalyst that effectively combines adsorption and photocatalytic reduction mechanisms to enhance the removal ability for U(VI).The surface reaction plays a crucial role in the photocatalytic process. This involves the target substance adhering to the material's surface, and subsequently interacting with electrons or holes generated from photoexcitation, leading to the oxidation or reduction process. To improve both catalytic activity and selectivity, various surface engineering strategies have been employed, including the construction of an active surface with high adsorption and activation capacity, the control of surface properties to ensure that more carriers can reach the surface-active site to participate in the reaction, and the improvement of the electron or hole reduction or oxidation capacity of the catalyst on its surface.10–12
Polyoxometalate (POM), a metal oxygen cluster with an oxygen-enriched surface, exhibits rapid and reversible multielectron-transfer reactions while maintaining a stable structure.13 These remarkable attributes endow POMs with excellent photocatalytic reduction and adsorption capacities, facilitating the segregation and immobilization of radioactive waste.14 However, the performance and stability of pure POM may be limited by agglomeration, disordered arrangements, and a restricted surface area.15 Therefore, selecting an appropriate solid matrix for dispersing POM would be a promising alternative strategy. Recently, MOFs have emerged as excellent platforms for various applications due to their unique porous structures and chemical stability, thus showing great promise for applications in lithium-ion batteries, gas separation, luminescent sensors, supercapacitors, photocatalysis, and ion capture.16–21 For instance, Wang et al. reported POM-based MOFs with enhanced U(VI) removal efficiency by three distinct sorption mechanisms (complexation, chemical reduction, and photocatalytic reduction).22 The study on the super-sodalite cage constructed from POM reported by Xu et al. also showed effective U(VI) adsorption capacity.23 Therefore, incorporating POM and MOFs with diverse sorption mechanisms would be a feasible approach for constructing effective sorbents that are still rarely explored.
Here, we developed a feasible self-assembly strategy to construct POM@Cu-MOFs, in which redox POM units are uniformly confined in the pores of MOFs that are constructed through the in situ growth of Cu2(CO2)4 paddle wheel-based structures. Serving as both the photocatalyst and adsorbent for efficient U(VI) removal, POM@Cu-MOFs exhibit synergetic advantages by combining the photocatalysis activity of POM with the strong pre-enrichment capability of MOFs, thereby enhancing the uranium removal efficiency.
Results and discussion
Synthesis and structural characterization of POM@Cu-MOFs
The POM@Cu-MOF was constructed through an in situ self-assembly approach, by encapsulating Keggin-type POMs into the porous structure of HKUST-1 (Fig. 1a). The scanning electron microscopy (SEM) image shows that the as-synthesized POM@Cu-MOFs feature a massive structure formed by the aggregation of nanoparticles with rough surfaces (Fig. S1†). Besides, the SEM-energy dispersive X-ray spectroscopy (EDS) mapping shows Cu, Mo, O, P, and C elements uniformly distributed on POM@Cu-MOFs, indicating the uniform dispersion of POM in channels of Cu-MOFs (Fig. S1†). Among them, the Cu, Mo, and P contents determined by EDS are found to be 28.4, 15.2, and 1.9 wt%, respectively. The X-ray diffraction (XRD) patterns of Cu-MOFs and POM@Cu-MOFs depict good crystalline structures of heterojunctions (Fig. 1b). From the FT-IR spectrum of Cu-MOFs, the band at 728 cm−1 is attributed to the Cu–O bond,24 and the bands at 1508, 1384, and 1405 cm−1 are attributed to the asymmetric and symmetrical stretching of the carboxylic acid group, respectively (Fig. 1c).25 The intensive bands at 1370 and 1643 cm−1 correspond to the stretching of C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C and the symmetrical stretching of the CO bond.26 All the above characteristic bands of Cu-MOFs are also observed in the FT-IR spectrum of POM@Cu-MOFs, indicating that the framework of Cu-MOFs is preserved. Besides, the emerging bands at 600–1100 cm−1 in the FT-IR spectrum of POM@Cu-MOFs correspond to P–O, MoO, and Mo–O–Mo bonds of POM, demonstrating the confined assembly of POM into the Cu-MOF (Fig. 1c and S2†).27
C and the symmetrical stretching of the CO bond.26 All the above characteristic bands of Cu-MOFs are also observed in the FT-IR spectrum of POM@Cu-MOFs, indicating that the framework of Cu-MOFs is preserved. Besides, the emerging bands at 600–1100 cm−1 in the FT-IR spectrum of POM@Cu-MOFs correspond to P–O, MoO, and Mo–O–Mo bonds of POM, demonstrating the confined assembly of POM into the Cu-MOF (Fig. 1c and S2†).27
 | ||
| Fig. 1 Characterization of POM@Cu-MOFs and Cu-MOFs. (a) Synthetic procedure of POM@Cu-MOFs. (b) XRD patterns. (c) FT-IR spectra. (d) N2 adsorption–desorption isotherms. (e) BJH model derived mesopore size distribution. | ||
The N2 adsorption–desorption isotherms and pore size distribution were obtained to demonstrate the encapsulation of POM into Cu-MOFs (Fig. 1d and e). The Cu-MOFs exhibits the typical type I isotherm,28,29 indicating the microporous adsorption behaviour. However, the POM@Cu-MOFs exhibit a certain adsorption capacity in the low-pressure region and hysteresis loop in the medium–high pressure region, revealing the appearance of increased microporosity in the POM@Cu-MOFs attributed to the encapsulation of POM.30,31 The substantially decreased specific surface area of POM@Cu-MOFs (872.5 m2 g−1) compared with Cu-MOFs (1365.7 m2 g−1), calculated by using the Brunauer–Emmett–Teller (BET) method, also suggests the encapsulation of POMs into the Cu-MOF framework. The average pore size of Cu-MOFs calculated by the Barrett–Joyner–Halenda (BJH) method was found to be approximately 3.5 nm, which decreased to 3.1 nm after the formation of POM@Cu-MOFs.
The XPS spectra of POM@Cu-MOFs further demonstrate the surface compositions and valence states (Fig. S3†). The signals of C 1s for Cu-MOFs and POM@Cu-MOFs both exhibit three peaks at 288.2, 285.8, and 284.3 eV, which could be assigned to O–CO, C–O, and C–C (Fig. 2a), respectively.32 The O 1s spectra of Cu-MOFs and POM@Cu-MOFs both show four peaks related to Cu–O, C–O, O–CO, and O–H (Fig. 2b).33 As shown in Fig. 2c, the Cu element in Cu-MOFs with strong spectral peaks at 954.8 eV and 935.0 eV correspond to Cu 2p1/2 and Cu 2p3/2, respectively.34,35 For POM@Cu-MOFs, the peaks at 133.8 and 134.7 eV, and 235.6 and 232.5 eV are attributed to P 2p3/2 and 2p1/2, and Mo 3d5/2 and 3d3/2, respectively (Fig. 2d and e).36 Compared with the spectra of the Cu-MOFs, the POM@Cu-MOFs showed increased binding energies of Cu 2p. It is worth noting that Cu 2p in POM@Cu-MOFs shows an upshift toward high binding energy compared with Cu-MOFs, which indicates the electron transfer from the Cu-MOF to POM (Fig. 2f).
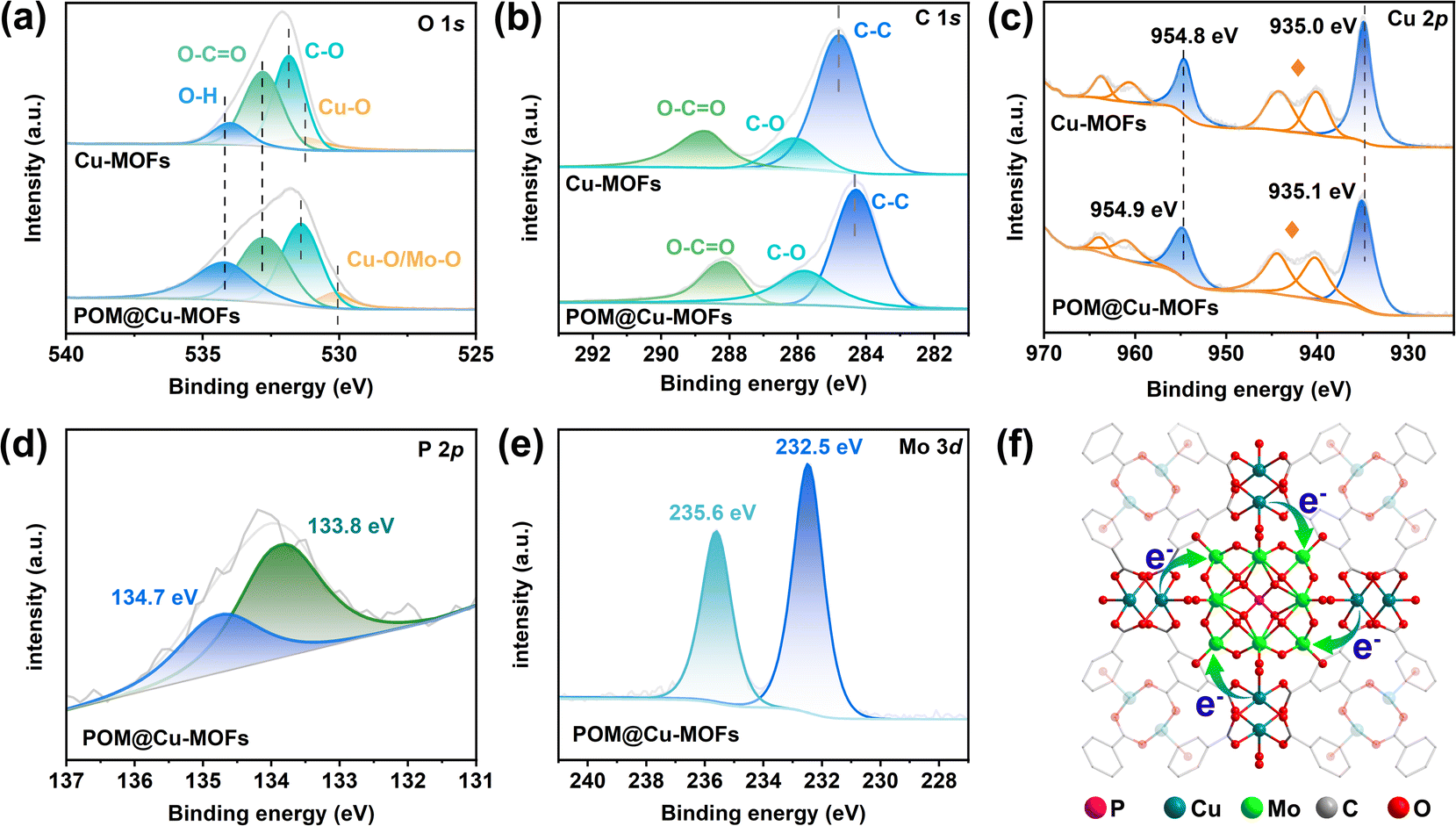 | ||
| Fig. 2 XPS spectrum of Cu-MOFs and POM@Cu-MOFs. (a–e) High-resolution spectra of C 1s (a), O 1s (b), Cu 2p (c), P 2p (d), and Mo 3d (e). Cu satellite peaks are marked by stars. (f) Interfacial electron transfer from Cu-MOFs to POM. | ||
Adsorption and photocatalytic reduction of uranium
To evaluate the uranium removal performance of Cu-MOFs and POM@Cu-MOFs, a series of adsorption and photocatalytic experiments were carried out. There is no removal efficiency without a photocatalyst (Fig. S4†), indicating that a catalyst is essential for uranium reduction. The removal capacity of U(VI) by POM@Cu-MOFs is higher than that of Cu-MOFs in a broad range of uranium concentrations (20–200 mg L−1) (Fig. 3a and S5†), implying that the encapsulation of POM into Cu-MOFs enhances the stability and improves the efficiency of multi-electron transfer. For solutions with initial UO22+ concentrations at 20 and 50 mg L−1, POM@Cu-MOFs thoroughly eliminate UO22+ from the solution in a short period under light, outperforming Cu-MOFs under the same conditions (Fig. S5a and b†). Uranium was removed through adsorption in the dark. With increasing UO22+ concentration, the uranium in the solution was not completely removed due to the limited active sites. On increasing the initial concentration, the ability of POM@Cu-MOFs to remove U(VI) under light conditions does not significantly decrease, while the effect of adsorption alone is significantly inferior to that of the synergy of adsorption and reduction. | ||
| Fig. 3 Photocatalytic removal of U(VI). (a) Uranium removal by Cu-MOFs and POM@Cu-MOFs using the photocatalytic reduction method compared to the adsorption method in the dark with an initial uranium concentration of ∼200 mg L−1 (m/V = 0.1 g L−1, pH = 4.0, and CCa2+ = 0.001 mol L−1). (b) Plots of −ln(Ce/C0) as a function of reaction time (t) over POM@Cu-MOF and Cu-MOF catalysts fitted by using the pseudo-first-order kinetics model. The R2 factors for K1–K4 are found to be 0.96, 0.95, 0.99, and 0.99, respectively. (c) Removal rate of photocatalysis and adsorption at different pH values (CU(VI) = 50 mg L−1, m/V = 0.1 g L−1, and CCa2+ = 0.001 mol L−1). (d) Reusability of POM@Cu-MOF catalysts (CU(VI) = 50 mg L−1, m/V = 0.1 g L−1, pH = 4.0, and CCa2+ = 0.001 mol L−1). | ||
When the initial concentration of UO22+ is 100 and 200 mg L−1 and the solid–liquid ratio is 0.1 g L−1, the saturated adsorption capacity under dark conditions is measured to be 709.6 and 1637.4 mg g−1, respectively (Fig. 3a and S5c†). In contrast, POM@Cu-MOFs still show high removal capacity even at high initial concentrations under light conditions, achieving 985.7 and 1987.4 mg g−1, respectively. POM@Cu-MOFs offer notable advantages over Cu-MOFs owing to the synergistic effect of light-induced adsorption and photocatalysis. Compared with the photocatalysts reported elsewhere, POM@Cu-MOFs display superior removal capacity (Table S1†),14,22,37–40 highlighting their promising prospects as an effective photocatalyst for uranium-bearing wastewater treatment. We fitted the data by using the pseudo-first-order and the pseudo-second-order models,41,42 respectively. As shown in Fig. 3b and S6,† the R2 factor for the quasi-first-order dynamics (R2 = 0.96) is superior to that of the quasi-second-order dynamics (R2 = 0.95), indicating that the pseudo-first-order model provides a more precise depiction of the reaction kinetics. These results affirm that the rate-limiting step for the removal of U(VI) is attributed to the photocatalytic reduction reaction. Therefore, the enhanced adsorption performance of the POM@Cu-MOFs is more conducive to enhancing photocatalytic reduction performance for U(VI). Then the reaction rate constant (k) was calculated by using the equation −ln(Ce/C0) = kt, where C0 and Ce represent the initial and final concentrations of U(VI), respectively. Under light irradiation, the k values for U(VI) photoreduction by Cu-MOFs and POM@Cu-MOFs are found to be 0.006 and 0.061 min−1, respectively. Under light irradiation, the highest k value for POM@Cu-MOFs is 2.9 times larger than that under dark conditions (Fig. 3b).
The pseudo-first-order kinetics derived from the linear correlation between −ln(Ce/C0) and reaction time (t) (Fig. S7†) indicates that adsorption of UO22+ on the catalyst surface is the rate-determining step in terms of the Langmuir–Hinshelwood model.28 At different initial concentrations of UO22+, POM@Cu-MOFs remove 99.4% of UO22+, which is higher than that removed by Cu-MOFs. The photocatalytic reduction rate constant of POM@Cu-MOFs is 11–20 times larger than that of Cu-MOFs at a UO22+ concentration of 20–200 mg L−1, indicating the higher photocatalytic efficiency of POM@Cu-MOFs.
Additionally, the existence of uranium-containing wastewater containing numerous coexisting ions may potentially affect photocatalytic reduction. With particular emphasis on Ca2+ as a major competing ion, we conducted experiments using various concentrations of CaCl2 in the range of 0.001–1 mol L−1 to evaluate its influence on the photocatalysis efficiency. The removal efficiency of uranium by photocatalytic reduction exceeds 80% across all concentration conditions (Fig. S8†). Therefore, the presence of CaCl2 does not significantly impact the photocatalytic reduction efficiency.
Fig. 3c illustrates the impact of pH on the adsorption and photocatalytic reactions of U(VI) solution. The removal efficiency decreases from 86.3% to 31.8% on increasing the pH value from 3.0 to 5.5 without light irradiation, which increases first and then decreases under light irradiation. Therefore, the optimal pH value for photocatalytic reduction of U(VI) is 4.0. The surface potential of the material becomes more negative with increasing pH value (Fig. S9†). This surface charge, being negative in nature, is more conducive to the adsorption of positively charged UO22+ ions due to the electrostatic interactions.43 Therefore, the surface of the material carries negative charges, and the electrostatic attraction between the negatively charged surface and positively charged UO22+ ions plays a significant role in the photocatalytic removal of U(VI). The evaluation of a photocatalyst’s in treatment efficiency must consider not just its U(VI) removal kinetics in a complex environment but also its recyclability and stability during catalyst development.44 Essentially undiminished photocatalytic reduction efficiency of U(VI) (90%) is achieved during 5 reaction cycles using one-batch POM@Cu-MOFs recovered after each run of the evaluation (Fig. 3d), indicating that POM@Cu-MOFs have excellent structural stability and good cyclic reusability.
Photoelectric properties
To delve into the optoelectronic attributes of the materials, the optical performance of POM, Cu-MOFs, and POM@Cu-MOFs was characterized. The UV-vis diffuse reflectance spectrum of Cu-MOFs alone shows strong adsorption below 400 nm, while the light absorption of POM is limited to below 550 nm (Fig. 4a). In contrast, POM@Cu-MOFs display a red shift and show significant adsorption in the range of 200–800 nm, indicating that POM@Cu-MOFs broaden the light absorption range and modulate the band structure. By further analysing the transformed Kubelka Munk function versus absorbed light energy, the band gap of semiconductor photocatalysts can be calculated according to the formula:45| (αhν)1/n = A(hν − Eg) | (1) |
 | ||
| Fig. 4 Charge transport dynamics of Cu-MOFs and POM@Cu-MOFs. (a) UV/vis spectra of POM, Cu-MOFs, and POM@Cu-MOFs. (b) Electrochemical Mott–Schottky plots of Cu-MOFs. (c) EPR spectra of Cu-MOFs and POM@Cu-MOFs under visible light (400–800 nm) irradiation. (d and e) Temperature-dependent PL spectra of POM@Cu-MOFs and Cu-MOFs. (f) Evolution of PL intensity of Cu-MOFs and POM@Cu-MOFs as a function of the reciprocal of temperature from 80 to 300 K. | ||
The bandgaps of Cu-MOFs, POM, and POM@Cu-MOFs are calculated to be 3.02, 2.25, and 2.31 eV, respectively (Fig. S10a†). From the Mott–Schottky curves, the positive slopes of the plots suggest that Cu-MOFs are a n-type semiconductor (Fig. 4b).46 From the intercept on the abscissa, the obtained flat band potential (Efb) is −1.27 eV (vs. Ag/AgCl), with the conduction band nearly overlapping the flat band potential, and the position of Efb relative to the normal hydrogen electrode (NHE) is 0.2 higher than the flat band potential;47 thus, the conduction potential (ECB) of Cu-MOFs is calculated to be −1.01 V vs. NHE. Moreover, the flat band potential of POM is around −0.28 V vs. NHE.15 These data, combined with the band gap energies, allow us to calculate the valence band (EVB) edges of Cu-MOFs and POM at 2.01 eV and 1.97 eV (vs. NHE), respectively. The band structure of the POM@Cu-MOFs, along with the ECB value of Cu-MOFs and POM is more negative than the U(VI) reduction potential (UO22+/U4+, +0.267 V) and (UO22+/UO2, +0.411 V). Therefore, the conduction band of POM@Cu-MOFs can provide sufficient driving force for photocatalytic reduction from U(VI) to U(IV).
Additionally, electrochemical impedance spectroscopy (EIS) was performed to further probe the properties of charge transport of POM@Cu-MOF heterostructures. POM@Cu-MOFs exhibited a smaller radius in its semicircle compared to Cu-MOFs, indicating a lower charge transfer resistance and higher interfacial charge separation efficiency (Fig. S10b†). Photoluminescence spectroscopy is widely used to evaluate the charge recombination probability, in which the low spectral intensity implies a lower recombination rate of photogenerated electrons and holes, while higher spectral intensity indicates higher recombination efficiency of photogenerated electrons and holes.48 The PL spectra show that the emission intensity of POM@Cu-MOFs is reduced compared to that of Cu-MOFs (Fig. S10c†). Therefore, the incorporation of POM and MOFs significantly inhibits the recombination rate of electron–hole pairs, highlighting the advantages of POM in improving carrier transfer for enhanced visible photocatalytic activity.
The primary active species involved in the photocatalytic reduction mechanism were identified through a series of trapping experiments. The results of the quenching experiments were obtained by adding 0.01 mol L−1 radical scavenger (Fig. S11†). Interestingly, the presence of methanol enhanced the photocatalytic performance when the electronic scavenger (AgNO3), hole scavenger (methanol), ˙O2− scavenger (p-benzoquinone, BQ), and hydroxyl radical scavenger (t-butylalcohol, TBA) were added to the reaction solution, respectively.49,50 Addition of AgNO3 could inhibit photocatalytic reduction, indicating that electrons are indeed active species involved in the reaction process. In contrast, the presence of methanol can greatly enhance the photocatalytic performance, enabling complete degradation of U(VI) within 40 min. Furthermore, both TBA and BQ exhibit distinct effects on the removal rate of U(VI) under visible light irradiation, achieving 97% and 89% removal rates for U(VI), respectively. During the photocatalytic process, h+, ˙OH, and ˙O2− species are generally employed as oxidizing agents. Thus, scavenging these oxidative radicals and holes can efficiently prevent re-oxidation of U(VI) and reduce recombination between photogenerated holes and electrons to improve the recovery ability, indicating that e− are the main active species. To further verify the details of the photoelectrons in the as-prepared POM@Cu-MOFs, electron paramagnetic resonance (EPR) was performed. As shown in Fig. 4c, the Cu-MOFs exhibit a weak EPR signal with a g factor of 2.003, suggesting a relatively low photoelectron density.51 Conversely, POM@Cu-MOFs exhibit a much stronger EPR signal due to the increased concentration of photogenerated electrons in the system. This is attributed to the formation of a heterojunction between POM and Cu-MOFs, where photogenerated electrons can transfer freely between POM and Cu-MOFs. We performed the EPR experiment by using 5,5-dimethyl-1-pyrroline N-oxide (DMPO) as a free radical trapping agent to further confirm the presence of ˙O2− and ˙OH radicals. Under dark conditions, no EPR signal was observed in an aqueous suspension containing POM@Cu-MOFs. By contrast, the characteristic EPR signals of DMPO–˙O2− and DMPO–˙OH species were observed after light irradiation, and the intensity of these signals increased with the duration of illumination (Fig. S12†). This observation suggests that the photocatalytic removal of U(VI) by POM@Cu-MOFs is facilitated by the generation of more photogenerated electrons and the dissociation of more O2 and H2O on the active sites of the catalyst under illumination.
In addition, temperature-dependent PL spectroscopy was employed to reveal exciton dissociation kinetics in Cu-MOFs and POM@Cu-MOFs (Fig. 4d and e). As the temperature decreases, the fluorescence intensity gradually increases along with enhanced charge carrier recombination and emission excitons. Then, the dependence of the material's fluorescence intensity on temperature was calculated by using the Arrhenius equation,52 resulting in values of 53.18 and 27.25 meV for Cu-MOFs and POM@Cu-MOFs, respectively (Fig. 4f). Accordingly, Eb of POM@Cu-MOFs is smaller than that of Cu-MOFs, indicating that exciton dissociation in POM@Cu-MOFs is more likely to occur in Cu-MOFs, which can more effectively dissociate into free carriers and improve charge transfer.
The morphology of POM@Cu-MOFs remains essentially unchanged after the photocatalytic reduction of U(VI) under visible light irradiation (Fig. S13a†). Besides this, amorphous sheets are also observed, which was ascribed to the products of uranium-bearing precipitates, as SEM-EDS mapping demonstrates that U is uniformly distributed in POM@Cu-MOFs after photoreduction (Fig. S13b†). The electrons generated within photoexcited POM@Cu-MOFs reduce the soluble U(VI) adsorbed on the surface of POM@Cu-MOFs to form the insoluble uranium-bearing precipitate, thus achieving the reduction and isolation of U(VI). The XRD peak of the POM@Cu-MOF catalyst has minimal alterations after adsorption and photocatalysis compared to that of the original sample (Fig. 5a). The new IR bands appearing at 918.4 and 1534.9 cm−1 for the catalyst after adsorption and photocatalytic reduction are ascribed to the vibration of UO22+ and uranyl hydroxy species, respectively (Fig. 5b).53 These results confirm the complexing ability of POM@Cu-MOFs for UO22+. XPS was performed to evaluate the stability of the POM@Cu-MOF catalyst after adsorption and irradiation. The valence state of uranium after adsorption and the photocatalytic reduction reaction is inconsistent (Fig. S14a†). U 4f7/2 and 4f5/2 can be divided into two peaks: 380.6 eV and 391.5 eV for U(IV), and 383.1 eV and 394.7 eV for U(VI), indicating the coexistence of U(VI) and U(IV) species after the photocatalytic reduction of U(VI) by POM@Cu-MOFs.54 For the uranium complexation produced after POM@Cu-MOF absorption of U(VI) under dark conditions, only the peak of U(VI) is present at 382.2 and 393.2 eV (Fig. 5c). After the photocatalytic reduction, the XPS peaks of Mo and P in POM shift to a lower binding energy (Fig. 5d and e), indicating spontaneous electron transfer at the interface. Meanwhile, compared with the spectra of POM@Cu-MOFs after absorption of U(VI), the POM@Cu-MOFs show increased binding energies of Cu 2p, C 1s, and lattice oxygen in O 1s (Fig. 5f and S14b, c†). More importantly, the XPS peak intensity of oxygen-containing species adsorbed on the POM@Cu-MOF surface (Oads) in O 1s increases after the reaction because unsaturated UO groups are formed during the reduction process (Fig. S12d†). These results demonstrate that the electrons transfer from Cu-MOFs to POM and finally to UO22+.
 | ||
| Fig. 5 Mechanism of photocatalytic removal of U(VI). (a) XRD patterns and (b) FT-IR spectra of POM@Cu-MOFs in photocatalytic reduction and absorption reaction alone. Comparison of the XPS spectra of POM@Cu-MOFs after photocatalytic reduction and absorption: (c) U 4f, (d) Mo 3d, (e) P 2p, and (f) Cu 2p. | ||
Based on the above results, a possible mechanism for reducing U(VI) is proposed (Fig. 6). Due to the porous architecture, the Cu-MOF exhibits commendable adsorption capacity and POM displays reversible multi-electron transfer reactions, endowing POM@Cu-MOFs with notable adsorption and photocatalytic processes. The reaction process as follows: (1) the oxo-metal cluster and P–O bonds within POM@Cu-MOFs were the active coordination sites for UO22+ adsorption, allowing UO22+ in solution to be effectively bound to the catalyst surface; (2) when exposed to light, the complexation of POM and Cu-MOFs could efficiently inhibit the recombination of photogenerated electrons and holes to accelerate the migration of electrons. Ultimately, the adsorbed UO22+ is reduced to an insoluble uranium-bearing precipitate via e− and ˙O2− generated in the photocatalytic process. (3) Photoexcited electrons from Cu-MOFs can easily transfer to POM, because the CB levels of Cu-MOFs are lower than those of POM, thus improving UO22+ reduction and facilitating activation of H2O into active ˙OH radicals. (4) The holes in the VB were transferred from POM to Cu-MOFs, and H2O molecules were consumed at the same time to generate ˙OH. POM, with excellent electrical conductivity, facilitates the separation of photogenerated electrons, thereby improving photocatalytic performance. Free radicals and the reaction process can be described by using the following equation:
| POM@Cu-MOFs + hν → e− + h+ | (2) |
| H2O + h+ → OH + H+ | (3) |
| UO22+ + e− → UO2+x (s, amorphism) | (4) |
| O2 + e− → ˙O2− | (5) |
| UO22+ + 2O2− → UO2+x (s, amorphism) + O2 | (6) |
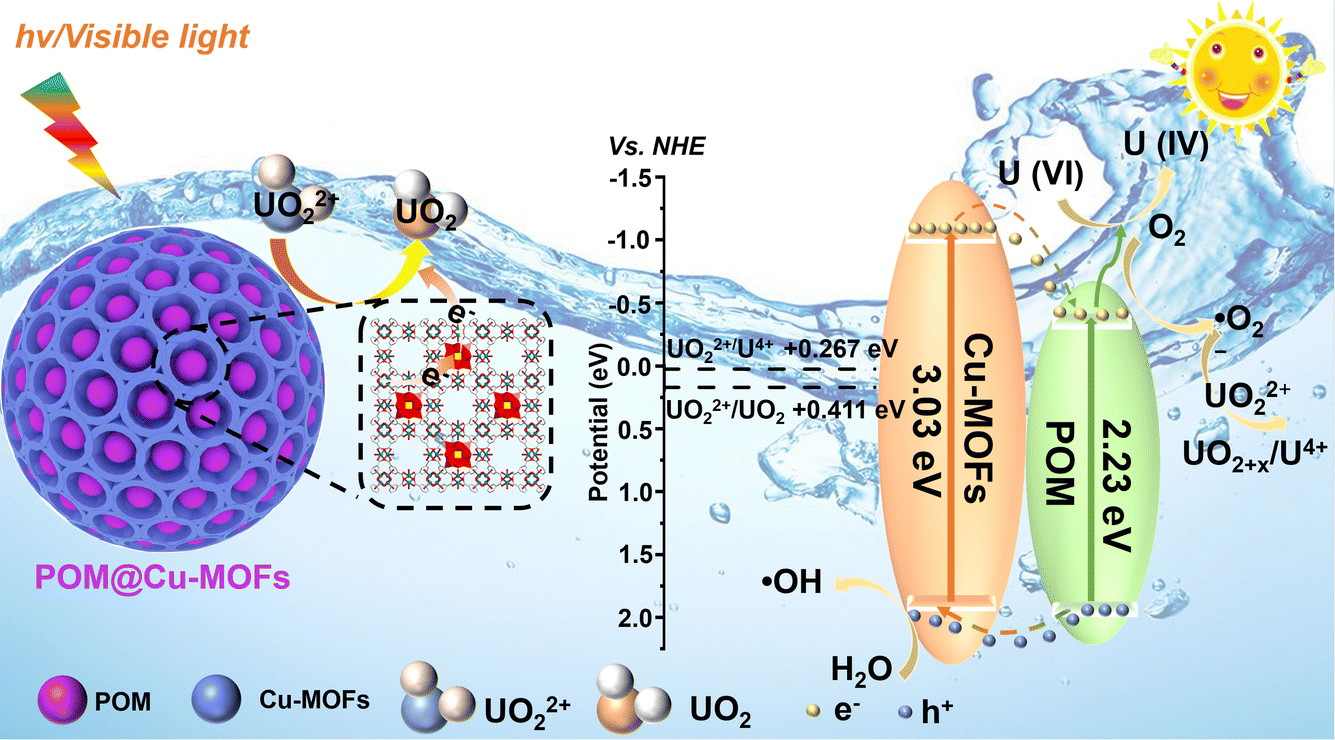 | ||
| Fig. 6 A plausible photoreduction mechanism of U(VI) over the POM@Cu-MOF heterojunction under light irradiation. | ||
Conclusion
The encapsulated POM@Cu-MOFs catalyst has been constructed and achieved excellent removal performance for U(VI) through synergetic adsorption and photocatalysis. The photogenerated electrons efficiently transferred from Cu-MOFs to POM to reduce soluble U(VI) to insoluble U(IV). Under light irradiation, the maximum removal capacity of U(VI) on POM@Cu-MOFs reached 1987.4 mg g−1, which is 1.2 times larger than that under dark conditions. Meanwhile, the photocatalytic reaction rate (0.061 min−1) is 10.2 times greater than that of Cu-MOFs (0.006 min−1). In the presence of a high concentration of competing metal cations (Ca2+), POM@Cu-MOFs still maintained high selectivity for U(VI), and 99% of U(VI) was removed from aqueous solutions with the concentration ranging from 10 to 200 mg L−1, affirming the promising potential of this material in practical applications.Experimental section
Chemicals
Copper(II) acetate monohydrate (Cu(OAc)2·H2O, 99%), phosphomolybdic acid (H3PMo12O40·xH2O, AR), ethanol, trimesic acid (H3BTC, AR), ethylene glycol (AR), silver nitrate (AgNO3, 98%), t-butylalcohol (TBA, AR), p-benzoquinone (BQ, AR), and methanol were obtained from Adamas-beta®. All chemicals were used as received without any further purification.Synthesis of Cu-MOFs
The synthesis process of Cu-MOFs was adapted from the literature.42 0.875 g of Cu(NO3)2·3H2O was dissolved in 12 mL of deionized water to obtain solution A. 0.42 g H3BTC was dissolved in 12 mL ethanol to obtain solution B. Solution B was added dropwise to solution A, stirred for 30 min at room temperature, and then transferred to an autoclave, and reacted at 110 °C for 18 h. After cooling to room temperature, Cu-MOFs were collected by centrifugation and cleaning with deionized water and ethanol, respectively.Synthesis of POM@Cu-MOFs
0.2 g of Cu(OAc)2·H2O and 0.1 g of H3PMo12O40·xH2O were dissolved in 10 mL of deionized water (denoted as solution A). 0.14 g of H3BTC was dissolved in 10 mL of ethanol (denoted as solution B). Then solution A and solution B were mixed and reacted for 30 min. The final indigo blue powder was obtained after washing with ethanol three times and drying.General characterization
SEM and EDS were performed on a Hitachi S-4800 FESEM. XRD characterization was conducted on a Bruker D8 Advance diffractometer (2θ range: 5° to 80°, Cu Kα radiation: λ = 0.15418 nm; a scan rate of 0.05° s−1). The specific surface area and pore size distribution were calculated by using the BET method and BJH desorption branch, respectively, on a Micromeritics ASAP 2460 apparatus. Fourier-transform infrared spectroscopy was conducted on a Nicolet 6700 (Thermo, USA) in the wavelength range of 4000–500 cm−1. The valence state and surface energy state distribution of photocatalysts were obtained using XPS (Escalab 250Xi, Thermo Fisher Scientific, USA). UV-vis diffuse reflectance spectroscopy (UV-vis DRS) was conducted on a Hitachi U-300 with the scanning range from 200 nm to 800 nm and a white standard of BaSO4 was used as the reference. The detection of hydroxyl radicals by spin trapping EPR was carried out using a Bruker model A300 spectrometer. The photoluminescence (PL) emission spectra were collected on a FluoroMAX-4 spectrometer with an excitation wavelength of 350 nm.Temperature-dependent photoluminescence spectroscopy
The temperature-dependent PL spectroscopy instrument is composed of a sample table, a fluorescence emitter, a liquid nitrogen constant temperature controller, and a fluorescence receiver. The sample tank was first filled with the sample, pressed with a slide, and then the sample table was placed in the instrument. Therefore, the fluorescence emission port was aligned with the sample tank. The liquid nitrogen constant temperature controller was then turned on, and liquid nitrogen was added to the sample table. The fluorescence data of the samples at different low temperatures were measured successively.Photoelectrochemical test
Electrochemical tests such as electrochemical impedance spectroscopy (EIS) and transient photocurrent response (TPCR) were conducted, and Mott–Schottky (M–S) curves were measured on a conventional three-electrode configuration using an electrochemical analyzer CHI 660D electrochemical workstation (ChenHua, Shanghai, China). More specifically, a three-electrode system consisting of 0.1 mol per L K3[Fe(CN)6] and 0.1 mol per L KCl as an electrolyte, a platinum wire electrode as the counter electrode, an Ag/AgCl electrode as the reference electrode, and a glassy carbon electrode as the working electrode were used for acquiring EIS and M–S curves. The working electrode was prepared as follows: 1 mg of the sample was ultrasonically dispersed in 1 mL of deionized water to obtain a suspension. 200 μL of the suspension was coated on the surface of the glassy carbon electrode and then the glassy carbon electrode covered with the sample was dried in an oven, and this was repeated several times. The transient photocurrent test was performed with 0.5 mol per L Na2SO4 aqueous solution as the electrolyte and a 300 W xenon lamp as the light source. The light/light avoidance treatment was performed at certain time intervals during the test.Evaluation of photocatalytic activity
All photocatalytic experiments were conducted in a photocatalytic reactor at RT and ambient pressure. Visible light was obtained with a 420 nm cut-off filter on a 300 W xenon lamp, and the photocatalytic reduction of U(VI) was performed. The thermal influence triggered by light irradiation cannot be disregarded, which negatively affects the efficiency of photocatalysis, particularly when the Curie temperature closely approaches room temperature. To mitigate this effect, we meticulously controlled the water temperature of our photo reactor, keeping the average temperature at a steady level of 25 ± 0.5 °C throughout the entire catalytical process via the constant circulation of water. 10 mg of the sample was added to 100 mL of uranium solution, adjusting the pH by using a small amount of NaOH or Na2CO3 solution. The reaction solution was extracted at certain time intervals under light, filtered with a filter membrane (0.45 μm), and then the U(VI) concentration was measured by arsine azo III spectrophotometry at a wavelength of 650 nm via UV-vis spectroscopy. The amount of U uptake by Cu-MOFs and POM@Cu-MOFs is calculated using the following equation:| qt = (C0 − Ct)V/m | (7) |
| U(VI) removal rate = (C0 − Ce)/C0 × 100% | (8) |
Reusability test
After each photocatalytic reaction, the resulting materials and insoluble uranyl compounds formed on the surface were separated from the used solution by filtration. The filter membrane was then soaked in 50 mL 0.2 mol per L of Na2CO3 solution for 5 min to disperse the solid material in the solution, and then stirred for 6 h. Na2CO3 was selected as the eluent because the insoluble uranyl compounds formed during photocatalysis can be dissolved in Na2CO3 solution. The CO32− competed for coordination to form soluble uranyl carbonate with higher coordination strength. Subsequently, the photocatalyst was separated from the uranium solution by filtration, and washed and dried for reuse. The filtrate was recycled for the next elution, and through multiple cycles, a high concentration of uranyl carbonate solution can be obtained to achieve the recovery and enrichment of uranyl ions.Data availability
All data have been included in the main text and ESI.†Author contributions
Z. D., G. Y., Z. Z., and F. Y. conceived and designed the experiments. Z. D. and D. Z. prepared the POM@Cu-MOFs and measured the adsorption and photocatalysis performance. Z. L., Y. W., X. C., and Y. L. helped analyse the data. Z. D., D. Z., G. Y., Z. Z., and F. Y. drafted and revised the manuscript. All authors discussed the results and reviewed the manuscript.Conflicts of interest
The authors declare no competing financial interests.Acknowledgements
This work was financially supported by the National Natural Science Foundation of China (22276030, U2167223, 22376025, 22222504, and 92161124), the National Key Research and Development Program of China (2021YFA0717400), the Shenzhen Basic Research Project (JCYJ20210324104808022), and the Guangdong Pearl River Talent Plan (2021QN02C104).Notes and references
- C. Tsouris, Nat. Energy, 2017, 2, 17022 CrossRef.
- F. Wu, N. Pu, G. Ye, T. Sun, Z. Wang, Y. Song, W. Wang, X. Huo, Y. Lu and J. Chen, Sci. Technol., 2017, 51, 4606–4614 CrossRef CAS PubMed.
- Y. Xie, Z. Y. Liu, Y. Y. Geng, H. Li, N. Wang, Y. P. Song, X. L. Wang, J. Chen, J. C. Wang, S. Q. Ma and G. Ye, Chem. Soc. Rev., 2023, 52, 97–162 RSC.
- Y. Zhang, J. Xu, J. Zhou and L. Wang, Chin. J. Catal., 2022, 43, 971–1000 CrossRef CAS.
- Z. Y. Wang, Q. H. Meng, R. C. Ma, Z. K. Wang, Y. J. Yang, H. Y. Sha, X. J. Ma, X. H. Ruan, X. Q. Zou, Y. Yuan and G. S. Zhu, Chem, 2020, 6, 1683–1691 CAS.
- Z. Dong, S. Hu, Z. Li, J. Xu, D. Gao, F. Yu, X. Li, X. Cao, Y. Wang, Z. Zhang, Y. Liu and X. Wang, Small, 2023, 2300003 CrossRef CAS PubMed.
- S. Li, X. Yang, Z. Cui, Y. Xu, Z. Niu, P. Li, D. Pan and W. Wu, Applied Catalysis B: Environment and Energy, 2021, 298, 120625 CrossRef CAS.
- X. Wu, C. Lu, J. Liu, S. Song and C. Sun, Applied Catalysis B: Environment and Energy, 2017, 217, 232–240 CrossRef CAS.
- S. Liu, Z. Wang, Y. Lu, H. Li, X. Chen, G. Wei, T. Wu, D. J. Maguire, G. Ye and J. Chen, Applied Catalysis B: Environment and Energy, 2021, 282, 119523 CrossRef CAS.
- K. Maeda, K. Teramura, D. Lu, N. Saito, Y. Inoue and K. Domen, Angew. Chem., Int. Ed., 2006, 118, 7970–7973 CrossRef.
- L. Mao, B. Zhai, J. Shi, X. Kang, B. Lu, Y. Liu, C. Cheng, H. Jin, E. Lichtfouse and L. Guo, ACS Nano, 2024, 18, 13939–13949 CrossRef CAS PubMed.
- T. Chen, K. Yu, C. Dong, X. Yuan, X. Gong, J. Lian, X. Cao, M. Li, L. Zhou, B. Hu, R. He, W. Zhu and X. Wang, Coord. Chem. Rev., 2022, 467, 214615 CrossRef CAS.
- C. Xing, G. Yu, J. Zhou, Q. Liu, T. Chen, H. Liu and X. Li, Applied Catalysis B: Environment and Energy, 2022, 315, 121496 CrossRef CAS.
- Z. Zhang, Z. Li, Z. Dong, F. Yu, Y. Wang, Y. Wang, X. Cao, Y. Liu and Y. Liu, Chin. Chem. Lett., 2022, 33, 3577–3580 CrossRef CAS.
- D. Y. Du, J. S. Qin, S. L. Li, Z. M. Su and Y. Q. Lan, Chem. Soc. Rev., 2014, 43, 4615–4632 RSC.
- C. J. Yin, L. F. Xu, Y. S. Pan and C. L. Pan, ACS Appl. Energy Mater., 2020, 3, 10776–10786 CrossRef CAS.
- T. Ponmuthuselvi, S. Saravanakumar and S. Viswanathan, Nanoscale Adv., 2020, 2, 2073 RSC.
- L. Yan, G. Ashna, K. Saima, H. Patricia, M. H. Lan, W. J. Zhang and X. F. Chen, Chem. Eng. J., 2022, 435, 134975 CrossRef CAS.
- P. Cheng, X. H. Wang, J. Markus, M. A. Wahab, S. Chowdhury, R. J. Xin, S. M. Alshehri, Y. Bando, Y. Yamauchi and Y. V. Kaneti, J. Colloid Interface Sci., 2023, 638, 220–230 CrossRef CAS PubMed.
- Z. S. Fan, Y. V. Kaneti, S. Chowdhury, Y. Yamauchi and F. B. Zhang, Chem. Eng. J., 2023, 462, 142094 CrossRef CAS.
- S. Chowdhury, N. L. Torad, M. Godara, A. A. M. El-Amir, G. Gumilar, A. Ashok, M. R. Karim, I. A. Alnaser, W. Chaikittisilp, N. Ray, Y. Yamauchi and Y. V. Kaneti, Chem. Eng. J., 2024, 480, 147990 CrossRef CAS.
- H. Zhang, W. Liu, A. Li, D. Zhang, X. Li, F. Zhai, L. Chen, L. Chen, Y. Wang and S. Wang, Angew. Chem., Int. Ed., 2019, 58, 16110–16114 CrossRef CAS PubMed.
- Y. Dong, Z. Dong, Z. Zhang, Y. Liu, W. Cheng, H. Miao, X. He and Y. Xu, ACS Appl. Mater. Interfaces, 2017, 9, 22088–22092 CrossRef CAS PubMed.
- J. Li, J. Li, Y. Chen, P. Tai, P. Fu, Z. Chen, P. Yap, Z. Nie, K. Lu and B. He, ACS Nano, 2024, 18, 16184–16198 CrossRef CAS PubMed.
- L. Li, X. L. Liu, H. Y. Geng, B. Hu, G. W. Song and Z. S. Xu, J. Mater. Chem. A, 2013, 1, 10292 RSC.
- F. A. Sofi, K. Majid and O. Mehraj, J. Alloys Compd., 2018, 737, 798–808 CrossRef CAS.
- W. Sun, Y. Si, H. Jing, Z. Dong, C. Wang, Y. Zhang, L. Zhao, W. Feng and Y. Yan, Chem. Res. Chin. Univ., 2018, 34, 464–469 CrossRef CAS.
- J. Sun, M. Shang, M. Zhang, S. Yu, Z. Yuan, X. Yi, S. Filatov and J. Zhang, Carbohydr. Polym., 2022, 293, 119720 CrossRef CAS PubMed.
- Y. Wang, Y. Wang, L. Zhang, C. S. Liu and H. Pang, Inorg. Chem. Front., 2019, 6, 2514–2520 RSC.
- A. M. Varghese, K. S. K. Reddy, N. Bhoria, S. Singh, J. Pokhrel and G. N. Karanikolos, Chem. Eng. J., 2021, 420, 129677 CrossRef CAS.
- M. Jin, X. Qian, J. Gao, J. Chen, D. K. Hensley, H. C. Ho, R. J. Percoco, C. M. Ritzi and Y. Yue, Inorg. Chem., 2019, 58, 8332–8338 CrossRef CAS PubMed.
- J. Xie, Y. Zhang, Y. Han and C. Li, ACS Nano, 2016, 10, 5304–5313 CrossRef CAS PubMed.
- J. Jie, P. Jeong, S. Wan, H. Kwang, Y. Choi, P. Li, J. Kang, Z. Ma, H. Jung, K. Ohchan and D. Kim, Applied Catalysis B: Environment and Energy, 2022, 317, 1211751 Search PubMed.
- K. Zhang, Z. Yang, X. Mao, X. L. Chen, H.-H. Li and Y. Y. Wang, ACS Appl. Mater. Interfaces, 2020, 12, 55316–55323 CrossRef CAS PubMed.
- W. Zhao, Y. Long, Y. He, J. Cai and M. Liu, Microporous Mesoporous Mater., 2022, 344, 112207 CrossRef CAS.
- Q. Li, Y. Wu, X. Ye, Y. Zeng and M. Ding, Appl. Catal., A, 2022, 633, 118533 CrossRef CAS.
- H. Li, F. Zhai, D. Gui, X. Wang, C. Wu, D. Zhang, X. Dai, H. Deng, X. Su, J. Diwu, Z. Lin, Z. Chai and S. Wang, Applied Catalysis B: Environment and Energy, 2019, 254, 47–54 CrossRef CAS.
- X. Zhong, Y. Liu, T. Hou, Y. Zhu and B. Hu, J. Environ. Chem. Eng., 2022, 10, 107170 CrossRef CAS.
- X. Han, Y. Wang, X. Cao, Y. Dai, Y. Liu, Z. Dong, Z. Zhang and Y. Liu, Appl. Surf. Sci., 2019, 484, 1035–1040 CrossRef CAS.
- Q. Zhang, D. Zhao, S. Feng, Y. Wang, J. Jin, A. Alsaedi, T. Hayat and C. Chen, J. Colloid Interface Sci., 2019, 552, 735–743 CrossRef CAS PubMed.
- K. Vanish, S. Vinamrita, K. Ki-Hyun, E. Eilhann and Y. Sherif A, Coord. Chem. Rev., 2021, 447, 214148 CrossRef.
- Z. M. Dong, Z. F. Li, D. L. Zeng, Z. P. Cheng, Y. C. Wang, Y. Dai, X. H. Cao, Y. Q. Wang, Z. B. Zhang and Y. H. Liu, Sep. Purif. Technol., 2023, 304, 122255 CrossRef CAS.
- Z. Dai, Y. Zhen, Y. Sun, L. Li and D. Ding, Chem. Eng. J., 2021, 415, 129002 CrossRef CAS.
- M. J. Comarmond, T. E. Payne, J. J. Harrison, S. Thiruvoth, H. K. Wong, R. D. Aughterson, G. R. Lumpkin, K. Müller and H. Foerstendorf, Environ. Sci. Technol., 2011, 45, 5536–5542 CrossRef CAS PubMed.
- Z. Zhang, C. Liu, Z. Dong, Y. Dai, G. Xiong, Y. Liu, Y. Wang, Y. Wang and Y. Liu, Appl. Surf. Sci., 2020, 520, 146352 CrossRef CAS.
- H. Yang, M. Hao, Y. Xie, X. Liu, Y. Liu, Z. Chen, X. Wang, N. Geoffrey and S. Ma, Angew. Chem., Int. Ed., 2023, 30, e202303129 Search PubMed.
- L. Wu, X. Yang, T. Chen, Y. Li, Q. Meng, L. Zhu, W. Zhu, R. He and T. Duan, Chem. Eng. J., 2022, 427, 131773 CrossRef CAS.
- Y. Dong, J. Zhang, Y. Yang, L. Qiu, D. Xia, K. Lin, J. Wang, X. Fan and R. Fan, Angew. Chem., Int. Ed., 2019, 58, 17610–17615 CrossRef CAS PubMed.
- P. Liang, L. Yuan, K. Du, L. Wang, Z. Li, H. Deng, X. Wang, S. Z. Luo and W. Shi, Chem. Eng. J., 2021, 420, 129831 CrossRef CAS.
- W. Zhang, Z. Deng, J. Deng, C. T. Au, Y. Liao, H. Yang and Q. Liu, J. Mater. Chem. A, 2022, 10, 22419–22427 RSC.
- E. Hu, Q. Chen, Q. Gao, X. Fan, X. Luo, Y. Wei, G. Wu, H. Deng, S. Xu, P. Wang, L. Liu, R. He, X. Chen, W. Zhu and Y. Zhu, Funct. Mater., 2024, 19, 2312235 Search PubMed.
- J. Wang, Y. Wang, W. Wang, Z. Ding, R. Geng, P. Li, D. Pan, J. Liang, H. Qin and Q. Fan, Chem. Eng. J., 2020, 383, 123193 CrossRef CAS.
- S. Zhang, L. Chen, Z. Qu, F. Zhai, X. Yin, D. Zhang, Y. Shen, H. Li, W. Liu, S. Mei, G. Ji, C. Zhang, X. Dai, Z. Chai and S. Wang, Chem, 2017, 9, 3172–3184 Search PubMed.
- W. Wang, Q. Luo, L. Li, Y. Wang, X. Huo, S. Chen, X. Du and N. Wang, Adv. Funct. Mater., 2023, 33, 2302913 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4sc05349d |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2024 |