
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceUnlocking regioselectivity: steric effects and conformational constraints of Lewis bases in alkyllithium-initiated butadiene polymerization†
Jian
Tang
 a,
Yuan
Fu
a,
Jing
Hua
*a,
Jiahao
Zhang
a,
Shuoli
Peng
a and
Zhibo
Li
b
a,
Yuan
Fu
a,
Jing
Hua
*a,
Jiahao
Zhang
a,
Shuoli
Peng
a and
Zhibo
Li
b
aKey Laboratory of Rubber-Plastics, Ministry of Education / Shandong Provincial Key Laboratory of Rubber-plastics, Qingdao University of Science and Technology, Qingdao 266042, P.R. China. E-mail: huajing72@qust.edu.cn
bCollege of Polymer Science and Engineering, Qingdao University of Science and Technology, 53 Zhengzhou Road, 266042, Qingdao, China
First published on 19th November 2024
Abstract
In nonpolar solvents, alkyllithium-initiated 1,3-butadiene polymerization exhibits high 1,4-selectivity, which shifts towards 1,2-selectivity upon the addition of Lewis bases. For the past 50 years, the prevailing hypothesis has suggested that Lewis bases primarily influence regioselectivity through electronic effects. However, our study reveals that steric hindrance also plays a crucial role. Using X-ray single-crystal diffraction, we analyzed the structure of the active species and proposed a new model for the chain-growth transition state. Techniques such as in situ NMR spectroscopy, isotope labeling studies, and density functional theory (DFT) calculations were employed to compare the impact of electronic and steric effects of various Lewis bases on regioselectivity. Our findings demonstrate that during 1,4-addition, the butadiene monomer is forced into close proximity with the Lewis base ligand, leading to significant steric interference and thus favoring 1,2-addition. Furthermore, we applied the concepts of “conformational restriction” to explain the enhanced 1,2-selectivity observed with ring-containing Lewis bases such as 1,2-dipiperidylethane. Building on this understanding, we have designed several highly efficient and cost-effective Lewis bases which achieves close to 100% 1,2-selectivity under mild conditions and significantly outperforms the best previously reported Lewis base, 1,2-dipiperidylethane, across a broad temperature range.
Introduction
Anionic polymerization, a fundamental method in polymer synthesis, is notable for its “living” and “controlled” characteristics.1–6 It's extensively utilized for the synthesis of monodisperse polymers,7–9 multiblock copolymers,10–14 hyperbranched polymers,15–17 and telechelic polymers,18,19 playing a crucial role in polymerizing various polar and nonpolar monomers, particularly conjugated dienes.20–23 This versatility and precision in polymer synthesis highlight the importance of deeply understanding the initiator systems that drive these reactions.Among various initiators, alkyllithium is the most common and widely used for diene anionic polymerization.24,25 The regioselectivity of anionic polymerization initiated by alkyllithium is strongly influenced by Lewis bases, known as polar modifiers or polar additives.20,24,26,27 For instance, butadiene polymerization typically exhibits a high 1,4-selectivity in the absence of Lewis bases, whereas the addition of Lewis bases increases the 1,2-unit content. Diethyl ether, tetrahydrofuran (THF), tetramethylethylenediamine (TMEDA), and 1,2-dipiperidylethane (DiPip) are among the most employed Lewis bases, showing significant differences in their ability to influence polymerization selectivity.6,20,28,29
Deciphering the active centers and mechanisms of polymerization remains a significant challenge. The selectivity mechanisms of stereoregular polymerization catalyzed by transition metals and rare earth elements are well-explored,30–36 but those of butadiene anionic polymerization are less understood. In the context of polybutadiene, stereoselectivity narrowly refers to the ability of the catalyst (or initiator) to control the chiral arrangement of neighboring 1,2-units (i.e., isotactic, syndiotactic, or atactic). Meanwhile, regioselectivity indicates the system's capacity to control the ratio of 1,2- to cis/trans-1,4-structural units. Given that stereoselective control is nearly unachievable in the anionic polymerization of dienes, the focus typically centers on regioselectivity. Research has long concentrated on understanding how Lewis bases influence the regioselectivity of anionic polymerization, as well as the correlation between the structure of these bases and the resulting polymer microstructure. The initial theory posits that higher solvent polarity leads to an increased 1,2-unit content in polymers.37,38 This theory partially explains the prevalence of polymers with high 1,2-structure in polar solvents like THF and ether. Nevertheless, it does not adequately account for certain experimental observations, particularly the marked increase in 1,2-unit content observed when minute quantities of TMEDA (less than 0.1 wt%) are added to nonpolar solvents. Consequently, THF and similar “so-called” solvents have increasingly been considered as acting primarily as Lewis bases, influencing the polymerization mechanism through this role.
On the other hand, the more recent and widely accepted theory argues that Lewis bases contribute to regioselectivity by modifying the charge distribution of the chain-end allylic carbanion.24,26,39,40 This modification leads to a concentration of electron density on the γ-carbon, thereby promoting 1,2-propagation. This electron effect-based theory partially elucidates the role of Lewis bases in anionic polymerization regioselectivity. However, some intriguing phenomena still contradict these existing theories, casting a shadow of uncertainty over the mechanism of anionic polymerization.
In this study, we meticulously revisited and examined existing theories through extensive experimental work, identifying both the strengths and shortcomings of previous mechanisms. Following these findings, we proposed a new model for the active center, derived from X-ray single-crystal diffraction studies of active species. Building on this new model, we introduced a mechanism mainly dictated by steric hindrance to elucidate the influence of Lewis bases on the regioselectivity of butadiene polymerization. This proposed active center model and regioselective mechanism have been strongly supported through in situ NMR experiments, isotope labeling studies, 2H-NMR, and DFT studies. With these new insights into the mechanism, we have successfully designed highly effective Lewis bases capable of facilitating the synthesis of polybutadiene with an unprecedented near-100% 1,2-unit content, effective across a broad temperature range.
Results and discussion
Initially, we reviewed the reported potential reaction mechanisms of lithium-initiated living anionic polymerization of butadiene. It is widely acknowledged that the structure of the final structural unit before the next monomer addition into the C–Li bond is uncertain due to possible isomerization of the terminal allylic group.40,41 As shown in Scheme 1, the mainstream view26,39,40 that proposed by Morton and his co-worker holds that in nonpolar solvents, the majority of electron density resides on the α-carbon of the chain-end allylic group, leading to the nucleophilic attack on butadiene at this site and resulting in 1,4-addition. However, in the presence of Lewis bases, the chain-end allyl anion become highly delocalized, transitioning into π-allyllithium. Moreover, the electron density of the chain-end allylic group shifts from the α-carbon to the γ-carbon, causing the nucleophilic attack on butadiene to occur at the γ-carbon, thereby facilitating 1,2-addition. This theory is primarily supported by the experiments of Young et al.39 and Fraenkel et al.,42 their findings revealed that 1,2-addition in dienes is attributed to ionic allylic species instead of covalent secondary allyllithium, and the addition of Lewis bases to hydrocarbon solvents leads to a high-field shift in the γ-H chemical shifts, a clear sign of elevated electron density at the γ-carbon. | ||
| Scheme 1 Existing mainstream mechanisms for Lewis base-induced 1,2-addition in anionic polymerization of butadiene. | ||
Another viewpoint,43 as depicted in Scheme 1b, suggests that Lewis bases induce a transition of the active species from the anti-isomer to the syn-isomer, thereby favoring the production of 1,2-polybutadiene. The main basis for this mechanism is that Lewis bases such as 6 DiPip promote the formation of syn-active species, which in turn enhance the 1,2-selectivity.
Although widely accepted, particularly Morton's theory,26 we still encountered certain experimental phenomena that this mechanism fails to explain. Particularly, Lewis bases with strikingly similar structures exhibited vastly different effects on the 1,2-addition ratio (e.g., 6 DiPip and 7 DiAze), despite their expected similar electronic effects. To assess the reliability of this mechanism, we selected a series of Lewis bases and tested their impact on anionic polymerization (Table 1). Furthermore, we used in situ13C-NMR to analyze the electron distribution of the chain-end allyl anions (Fig. 1).42,44
| Entry | Lewis base | Conv. |
![[M with combining macron]](https://www.rsc.org/images/entities/i_char_004d_0304.gif) n
(103)
n
(103) |
PDI | Microstructured (%) | |||
|---|---|---|---|---|---|---|---|---|
| 1,2 | Trans-1,4 | Cis-1,4 | Trans/cis | |||||
a Polymerization conditions: in n-hexane at 20 °C for 10 h, designed molecular weight = 30![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000, [Bd] = 0.1 g mL−1, [Lewis base]/[Li] = 1.5 (molar ratio).
b Determined by GPC in THF at 35 °C against a polystyrene standard, and universally calibrated with the Mark–Houwink equation.
c Considering THF is a monodentate ligand with a relatively low complex stability constant, it was employed as the solvent.
d Determined by 1H-NMR and 13C-NMR. 000, [Bd] = 0.1 g mL−1, [Lewis base]/[Li] = 1.5 (molar ratio).
b Determined by GPC in THF at 35 °C against a polystyrene standard, and universally calibrated with the Mark–Houwink equation.
c Considering THF is a monodentate ligand with a relatively low complex stability constant, it was employed as the solvent.
d Determined by 1H-NMR and 13C-NMR.
|
||||||||
| 1 | 1 Glyme | >99% | 32.7 | 1.09 | 78.1 | 11.3 | 10.6 | 1.1 |
| 2 | 2 THFc | >99% | 39.0 | 1.10 | 91.3 | 3.7 | 4.0 | 0.9 |
| 3 | 3 TMEDA | >99% | 34.0 | 1.11 | 82.7 | 10.6 | 6.7 | 1.6 |
| 4 | 4 TEEDA | >99% | 35.2 | 1.09 | 86.4 | 8.0 | 5.6 | 1.4 |
| 5 | 5 DiPyr | >99% | 31.4 | 1.09 | 92.2 | 5.4 | 2.4 | 2.3 |
| 6 | 6 DiPip | >99% | 33.5 | 1.10 | 97.7 | 1.9 | 0.4 | 4.8 |
| 7 | 7 DiAze | >99% | 33.4 | 1.18 | 82.7 | 10.9 | 6.4 | 1.7 |
 | ||
| Fig. 1 13C-NMR study of Lewis base effects on γ-C electron density in “living” polybutadiene. (a) 13C-NMR study process. (b) 13C-NMR spectra of active species (γ-C region, C6D6). (c) Relative charge distribution at the “living” chain ends with various Lewis bases as determined by 13C-NMR spectra, and the corresponding 1,2-unit content of the polymers measured by 1H-NMR. | ||
Fig. 1b illustrates that the addition of various Lewis bases to “living” polybutadienyllithium leads to higher field shifts in the 13C-NMR chemical shifts of γ-C, though the degree of shift varies among the bases. The efficacy of Lewis bases in decreasing γ-C chemical shifts (correlating with the electron density at γ-C) was ranked as 3 TMEDA > 5 DiPyr > 7 DiAze > 4 TEEDA > 6 DiPip.29 However, their ability to promote 1,2-addition was in the order of 6 DiPip> 5 DiPyr > 4 TEEDA > 3 TMEDA ≈ 7 DiAze (Fig. 1b and Table 1). Contrary to expectations, no clear correlation was observed between the ability of Lewis bases to enhance γ-C electron density and their effectiveness in promoting 1,2-addition. Particularly, while 6 DiPip demonstrated a substantially higher capability to increase the 1,2-unit content in the polymer than other Lewis bases,28 its effect on elevating γ-C electron density was relatively low, challenging the previous mechanism.26
With greater precision, we measured the relative distribution of negative charges on the chain-end allylic group at the α and γ positions in the presence of various Lewis bases, using the technique developed by Bywater and Worsfold (Fig. 1c).44 Without any Lewis base, the charge ratio between the γ and α positions is approximately 2:8; however, with the introduction of various Lewis bases, this ratio shifts to around 4:6. This differs significantly from the 1,2- to 1,4-unit ratio in the corresponding polymer (ranging from 4:1 to 50:1). This discrepancy is unexpected, as previous theories suggested that the charge ratio at these positions should closely reflect the proportion of 1,2-units in the polymer. Moreover, despite the use of different Lewis bases, the distribution of negative charges at the γ and α positions showed minimal changes, indicating that the electronic effects of these amines in this system are almost consistent. Overall, charge distribution analyses estimate that electronic effects contribute merely about 40% to the enhancement of 1,2-addition, with this contribution being relatively consistent across different Lewis base types. Given the significant variation in polymerization selectivity with different Lewis bases, we can conclude that electronic effects are not the sole factor determining regioselectivity in this system.
Additionally, deuterium labeling technology was employed to study electron distribution in the “living” chain-end. Terminating polymerization in the presence of different Lewis bases with methanol-d4 led to deuterium replacing lithium at the polybutadiene chain-end, resulting in deuterium-labeled polybutadiene (Fig. 2a).45 This reaction essentially involves the chain-end allylic carbanions abstracting D+ from deuterated methanol, thereby reflecting the charge distribution in these chain-end carbanions. Fig. 2b demonstrate that, despite the addition of Lewis bases, deuterium predominantly resides at the terminal α-position (indicative of 1,4-addition), with only about 25% at the γ-position (indicative of 1,2-addition), which is consistent with previous observations.24 This indicates that even with the addition of Lewis bases, the electron density in the chain-end allylic group remains concentrated at the α-carbon—this would logically lead to a higher 1,4-addition selectivity. However, in practice, the polymerization reaction predominantly results in 1,2-addition (Table 1). It is worth noting that while both 2H NMR and 13C NMR analyses provide consistent conclusions regarding charge distribution, there are quantitative differences between the two methods. A more detailed discussion of these differences is provided in the ESI.†
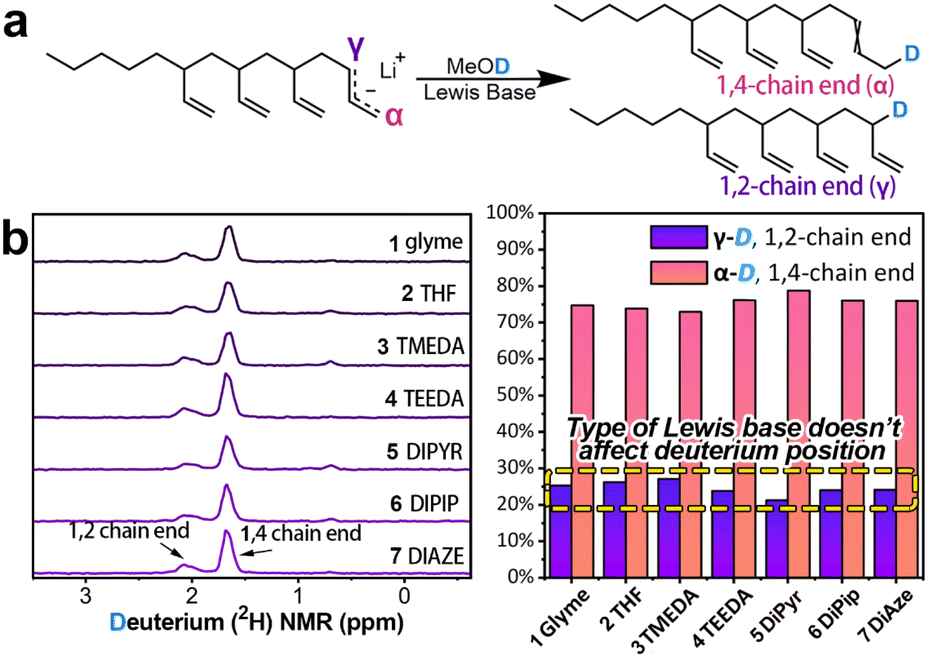 | ||
| Fig. 2 Study of chain-end charge distribution using isotopic labeling. (a) Quenching the polymerization reaction with methanol-d4 to label the chain-end charge distribution. (b) 2H NMR spectrum of the labeled polymer, showing that the type of Lewis base has minimal influence on the chain-end charge distribution. | ||
These results suggest that the negative charge on the “living” chain-end predominantly localizes on the α-carbon, rather than the γ-carbon, regardless of the Lewis base added. Furthermore, the electron density on the γ-carbon does not correlate directly with the 1,2-selectivity, suggesting that the electronic effects of these various Lewis bases on the active species are nearly identical. However, the structure of the Lewis base has a significant impact on the selectivity of the polymerization reaction, indicating that factors other than electronic effects play an important role in this process.
To unravel the real form of the active species, we synthesized allyllithium-Lewis base complexes, which serve as models for the active center, and tried to grow their single crystals. Specifically, we first prepared tetraallyltin, then synthesized solid allyllithium through a Sn–Li exchange reaction.46 Subsequently, we attempted to prepare allyllithium complexes by mixing allyllithium with various Lewis base ligands (Fig. 3). Due to the high self-ignition propensity and poor stability of these complexes, only the complex 6a with 6 DiPip as the ligand yielded crystals suitable for X-ray diffraction. We tried initiating butadiene polymerization with 6a. The resulting polymer showed a structure nearly identical to that obtained from the BuLi/6 DiPip system, with a very narrow molecular weight distribution. MALDI-TOF-MS analysis of the polymer confirmed the presence of allyl end groups (Fig. S4 and S5†), demonstrating that complex 6a successfully initiated butadiene polymerization. X-ray diffraction analysis revealed that the complex 6a is a mononuclear compound, adopting a distorted tetrahedral geometry. The 6 DiPip chelates to the Li+ ion in a κ2-N,N bidentate mode, while the allyl anion coordinates in an η3-coordination mode at the Li+ ion. The torsion angle (C1–C3–N1–N2) formed by the line connecting C1 and C2 in the allyl anion and the line connecting N1 and N2 in the DiPip ligand is 81.705°, suggesting that the axes of the two ligands are almost orthogonal. The chelation angle C1–Li1–C3 [68.00(11)] in the complex is relatively small due to the small bite size of the allyl ligand.47 The C1–C2 and C2–C3 bond lengths in the allyl group are very close, indicating a high degree of delocalization of the allyl electrons.
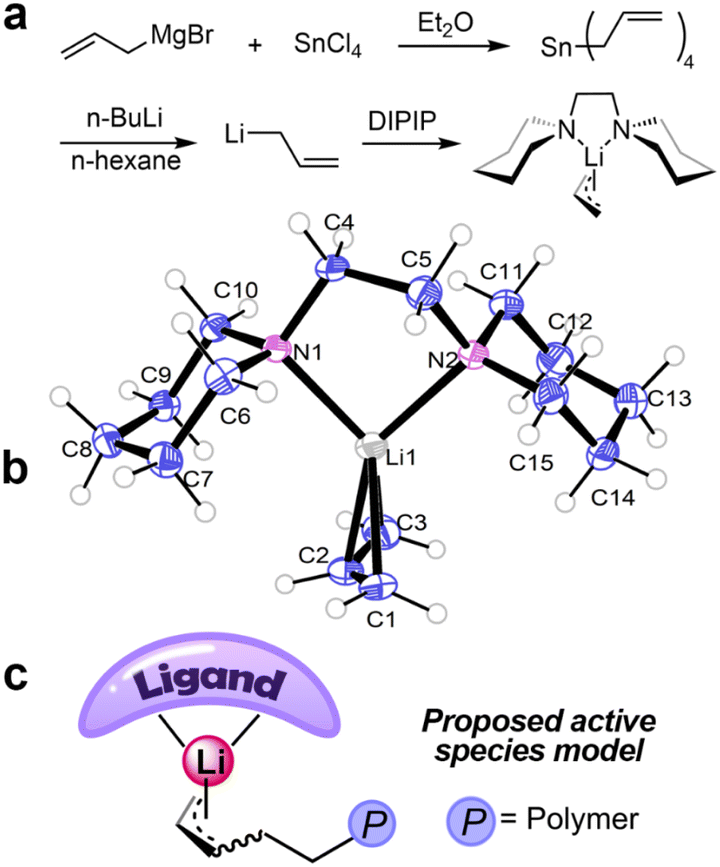 | ||
| Fig. 3 (a) Synthetic route of complex 6a. (b) ORTEP drawings of complex 6a. Ellipsoids at the 45% probability level. The unit cell contains two highly similar asymmetric units. The figure only depicts one for clarity. Selected distances (Å) and angles (degrees). Li1–N1 2.170(3), Li1–N2 2.161(3), Li1–C1 2.237(3), Li1–C2 2.093(3), Li1–C3 2.208(3), C1–C2 1.356(4), C2–C3 1.388(4), C1–Li1–C3 68.00(11). (c) The proposed active center model. | ||
Drawing insights from the X-ray single-crystal diffraction analysis, we have developed a model for the active species, as depicted in Fig. 3c. This model suggests that the active species is a Lewis base complex of polybutadienyllithium, similar to coordination polymerization active species,33,48,49 but without monomer coordination.
Based on this model, we conducted a DFT study of the chain propagation process. Utilizing the Gaussian 09 (ref. 50) and OCRA 5.0.3 (ref. 51) packages, we examined the transition states and their barriers in chain propagation step in presence of Lewis bases 1–6. It's well-known that the “living” chain-end can be either syn-π-allyllithium or anti-π-allyllithium, depending on the type of ligand and reaction conditions.43 DFT results indicate a preference for anti-π-allyllithium at the chain-end with 1 glyme, 2 THF, and 3 TMEDA as ligands, while 4 TEEDA, 5 DiPyr, 6 DiPip favor syn-π-allyllithium. This is corroborated by our in situ13C-NMR and deuterium labeling experiments, as shown in Fig. 2b and c.
The DFT calculations provide a clear and compelling explanation for the regioselectivity in the chain-growth process of butadiene polymerization. Considering the existence of cis- and trans-configurations of butadiene monomers, syn- and anti-configurations of allyllithium, and the possibilities of 1,2- or 1,4-addition, a total of 2 × 2 × 2 = 8 possible chain propagation scenarios were computationally simulated. We performed DFT calculations for each of these scenarios with various Lewis bases as ligands. For example, using 3 TMEDA as a ligand (Fig. 4a), polybutadienyllithium species A dissociates upon addition of 3 TMEDA to form the mononuclear Li complex B. The anti-π-allyl configuration exhibits lower free energy compared to the syn-π-allyl configuration. Notably, not all Lewis bases lead to a predominant anti-π configuration; this is thought to be related to the steric hindrance of the Lewis base, as discussed in detail later. During chain propagation, the monomer approaches the active species through van der Waals forces and undergoes addition via transition states TS1-4 or TS5-8, leading to either 1,2- or 1,4-polybutadiene. Both cis and trans butadiene, as well as anti and syn forms of the active species, can lead to 1,2- or 1,4-PB, though the barriers differ. It is important to note that, as an irreversible process, anionic polymerization is governed by kinetic control.52 According to transition state theory (TST), the reaction rate is determined by the pathway with the lowest energy barrier. Thus, the barrier difference between 1,4- and 1,2-propagation directly influences the selectivity of the polymerization. Thus, even though TS2 and TS4 have the highest barriers for 1,2-growth, the actual 1,2-addition rate depends on TS1, which has the lowest energy. Therefore, the 1,2-chain propagation barrier (TS1: 18.6 kcal mol−1) is lower than that for 1,4-chain propagation (TS6: 20.0 kcal mol−1), resulting in a 1,2-unit dominance in the product PB.
 | ||
| Fig. 4 (a) Energy profile of butadiene anionic polymerization with TMEDA as ligand and potential transition states. (Energies were calculated at ωB97X -2-D3(BJ)53/def2-QZVPP level) (b) and (c) Visualizing how steric hindrance influences energy barriers of 1,2 and 1,4-additions. | ||
Crucially, during the transition state of 1,4-addition, the monomer is closer to the ligand than in 1,2-addition, facing greater steric hindrance, thus leading to a higher barrier for 1,4-addition. This results in a higher energy barrier for 1,4-addition compared to 1,2-addition (Fig. 4b and c). This steric hindrance-driven regioselectivity mechanism is vividly demonstrated in ESI Movie S1.†
In addition to 3 TMEDA, we have calculated and summarized the energy barriers for chain propagation with other Lewis bases, detailed in Fig. 5, S148 to S153† and Table 2. Regardless of the Lewis base used, the energy barrier for 1,2-propagation is consistently lower than that for 1,4-propagation by approximately 1–3 kcal mol−1. According to TST theory, this suggests that the rate of 1,2-propagation is about 10 to 100 times faster than that of 1,4-propagation, which is consistent with the high 1,2-unit content observed in the resulting polybutadiene.20,24 When ethers serve as ligands, the activation energy for chain propagation is considerably lower than with amines, approximately 17.4 kcal mol−1, corresponding with the experimental findings that polymerizations with ethers usually proceed much faster than those with amines.41,53,54 When employing amines as ligands, the energy barriers for 1,2-addition are closely similar, approximately 18.5 kcal mol−1, while the 1,4-addition barriers vary significantly with the ligand. We conducted a quantitative analysis of the steric hindrance of these Lewis bases using the SambVca 2 program55 (Fig. S6†). The results show a significant correlation between the parameter percent buried volumes and the 1,2-selectivity of the system.
 | ||
| Fig. 5 Comparison of minimal energy barriers (calculated at wB97X-2-D3(BJ)/def2-QZVPP level) for 1,2, cis-1,4 and trans-1,4 butadiene additions with various Lewis bases, including corresponding 1,2-unit content data from polymerization experiments. | ||
| Addition mode | Configuration (allyl-BD) | 3 TMEDA | 4 TEEDA | 5 DiPyr | 6 DiPip | 1 Glyme | 2 THF |
|---|---|---|---|---|---|---|---|
| a Energies were calculated at ωB97X-2-D3(BJ)56/def2-QZVPP level. | |||||||
| 1,2 | Syn-trans | 18.6 | 18.8 | 18.3 | 18.8 | 17.2 | 18.2 |
| Syn-cis | 22.9 | 23.1 | 23.7 | 24.7 | 20.5 | 21.1 | |
| Anti-trans | 19.2 | 19.4 | 18.5 | 18.9 | 17.5 | 17.6 | |
| Anti-cis | 23.8 | 24.2 | 23.9 | 23.2 | 21.2 | 23.1 | |
| Trans-1,4 | Syn-trans | 21.4 | 21.2 | 21.2 | 21.9 | 20.7 | 21.2 |
| Syn-cis | 20.0 | 23.0 | 23.1 | 23.5 | 19.3 | 20.4 | |
| Cis-1,4 | Anti-trans | 22.9 | 21.1 | 22.0 | 22.5 | 21.5 | 21.6 |
| Anti-cis | 20.5 | 23.0 | 22.8 | 24.0 | 19.8 | 20.3 | |
Based on the above discussion, larger Lewis bases should lead to higher 1,2-selectivity in polymerization reactions. However, some Lewis bases, such as 7 DiAze, deviate from this rule despite their larger size compared to 6 DiPip, showing poorer 1,2-selectivity. To explain these exceptions, we utilized the concept of “conformational constraint”. As illustrated in Fig. 6b, ligands like 4 TEEDA and 7 DiAze facilitate monomer addition by allowing internal rotation of the C–C bonds in their ethyl or azepanyl groups. This conformational flexibility, however, reduces the 1,2-selectivity during polymerization. In contrast, for 6 DiPip, the high conformational change barrier of its cyclohexyl groups makes it difficult to adjust to a conformation favorable for 1,4-monomer addition. The calculated conformational change barrier of 4 TEEDA, 7 DiAze, and 6 DiPip are 2.7, 1.8, and 11.2 kcal mol−1, respectively, supporting our estimates of their flexibility. Furthermore, the conformational change barrier of all other Lewis bases correlates well with the 1,2-unit content in the polymers.
 | ||
| Fig. 6 (a) Correlation between conformational change barriers of Lewis bases and 1,2-unit content in polybutadiene. (b) Conformational flexibility in 4 TEEDA and 7 DiAze leading to reduced regioselectivity, while enhanced regioselectivity in 6 DiPip due to conformational constraint effects. | ||
To further validate the “conformational constraint” theory, we synthesized a series of Lewis bases 8–11 (Fig. 6a and 7). Compared to 6 DiPip, 8 MeDiPip has methyl groups at the 3,5-positions of its piperidinyl rings, slightly increasing its volume. However, the conformational change barrier for 8 shows no significant difference from that of 6. Consistent with expectations, when used in butadiene polymerization, the 1,2-unit content of the polymer produced with 8 closely mirrors that of 6. Compared to 6 DiPip, Lewis Bases 9–11 feature substitutions at the 4-position of the cyclohexyl rings with O, N–Me, and N–iPr, respectively. It is well-known that the flexibility decreases progressively from C–O–C to C–C–C to C–N(Me)–C to C–N(iPr)–C structures. This is evidenced by the conformational change barriers of 9, 10, 11, and 6, which are 7.7, 12.2, 13.4 and, 11.2 kcal mol−1, respectively. Correspondingly, the 1,2-unit contents in the polybutadienes produced with these Lewis bases are 92.5%, 98.2%, 99.8%, and 97.7%, respectively, showing a strong correlation.
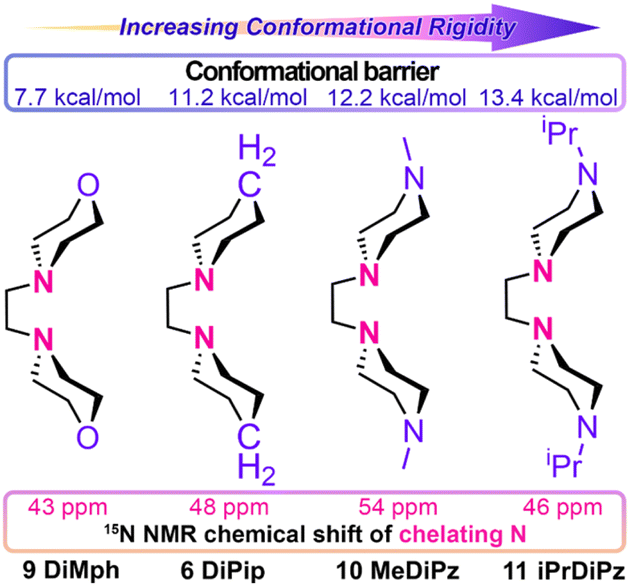 | ||
| Fig. 7 Influence of CH2, O, N–Me, and N–iPr substitutions on the conformational rigidity of Lewis bases and the electronic properties of chelating N atoms. | ||
These findings provide strong support for the “conformational constraint theory”. On one hand, it is obvious that the flexibility of C–O–C structures is much higher than that of C–C–C structures, which enables 9 to easily adjust its conformation to accommodate the higher steric hindrance during 1,4-addition, thereby reducing its 1,2-selectivity. Conversely, the much lower flexibility of C–N(iPr)–C increases the rigidity of 11, resulting in the highest 1,2-selectivity among the Lewis bases tested, at 99.8%, significantly exceeding the levels previously reported for 6 DiPip. On the other hand, the introduction of O and N atoms markedly changes the electronic effect of the ligands. As illustrated in Fig. 7, the 15N-NMR chemical shifts of the coordinating N atoms in ligands 6, 9, 10, and 11 are 48, 43, 54, and 46 ppm, respectively, indicating substantial variations in electron density of N atoms. However, similar to the results shown in Fig. 1b and c, there appears to be no clear correlation between the electronic effects of the Lewis bases and their 1,2-selectivity.
Remarkably, the enhanced conformational constraint effect allows 11iPrDiPz, when used in butadiene polymerization, to not only significantly exceed all known Lewis bases in 1,2-unit content (>99.8%) at room temperature but also to maintain excellent performance at higher temperatures. At 40 °C and 60 °C, the 1,2-selectivity remains above 98% and 93%, respectively. In contrast, under the same conditions, 6 DiPip achieves only 95% and 86% selectivity, while 7 DiAze, lacking these conformational constraints, reaches only 74% and 60% (Fig. S1†).
Building on the discussion of Lewis bases' effects on 1,2-selectivity, we must also consider their influence on the distribution of cis-1,4 and trans-1,4 units, which are influenced by similar, yet distinctly impactful, steric interactions. This consideration is crucial for ruling out the second mechanism proposed in Scheme 1.
We have observed an interesting phenomenon: ligands that exhibit high 1,2-selectivity also tend to produce polymers with a higher ratio of trans-1,4 to cis-1,4 units (see entries 5 and 6 in Table 1). We believe that this intriguing coincidence arises because both the cis-1,4, trans-1,4 and the 1,2-selectivity are governed by steric effects. It is crucial to understand that the changes in the syn/anti ratio of active species and the 1,2/1,4-unit proportion are parallel phenomena, both resulting from the steric effects, and not causally related.
The DFT calculations indicate that when Lewis bases with significant steric hindrance are involved in polymerization, the energy barrier for trans-1,4 addition is lower than that for cis-1,4 addition, matching the cis–trans ratio in product polymer. Fig. 8 clearly shows that, compared to the “C-shaped” anti-allyl species (leading to cis-1,4-units), the “zig-zag-shaped” syn-allyl active species (leading to trans-1,4-units) encounters less steric hindrance when bulkier Lewis bases are present. We assessed the distribution of syn- and anti-isomers in the active species by measuring the coupling constants between the β-H and γ-H at the “living” chain end using in situ1H-NMR. To eliminate the interference from α-H and δ-H in the coupling of β-H and γ-H, we synthesized 1,1,4,4-tetradeuterobutadiene and initiated its polymerization into “living” selectively deuterated polybutadiene. The coupling constants between β-H and γ-H for the syn- and anti-isomers are approximately 9 and 14 Hz, respectively, and the syn–anti ratio calculated from the integration areas of these signals is presented in Fig. 8b. It becomes evident that ligands with larger volumes and conformational constraints lead to a higher proportion of syn-active species, resulting in the formation of trans-1,4-polybutadiene units. This aligns with the findings derived from 13C-NMR techniques, as illustrated in Fig. S2.†
 | ||
| Fig. 8 (a) The mechanism by which bulky Lewis bases increase the trans proportion in 1,4-units. (b) Preparation of selectively deuterated polybutadienyllithium for observing the ratio of anti/syn active species isomers. (c) 2H-NMR spectra of selectively deuterated polybutadienyllithium after the addition of different Lewis bases. | ||
This work elucidates the decisive role of steric hindrance and conformational constraints in anionic polymerization. However, it is undeniable that other factors also influence the polymer's microstructure, such as polymerization temperature20,29 and the coordinating ability of Lewis bases.41 The influence of temperature on selectivity is well-established, whereas the coordination capabilities of Lewis bases require further exploration. Some scholars have correlated the coordination capacity of Lewis bases to their structural regulatory ability.41 Our study posits that the active center is essentially a lithium complex, with the Lewis base acting as the ligand. The functionality of the ligand relies on its coordination with the central metal. However, excessive steric hindrance, unsuitable chelation angles, or inadequate coordination sites may lead to a weaker coordination force constant, which can impede effective coordination with lithium. Therefore, in the practical application of this theory, it is essential to ensure that the ligand can form a stable and effective complex with allyllithium. This is straightforward: Lewis bases function as ligands, and if they fail to form complexes with Li+, they naturally have no effect.
Conclusions
The quest to unravel the regioselectivity mechanisms in anionic polymerization has been a progressive journey, much like a detective story. Over the years, various theories have emerged and evolved, including “solvent polarity theory”, “active species conformation theory”, and “electronic effect theory”. Each theory, built upon the efforts of previous scholars, has played a crucial role in gradually uncovering the mechanisms of butadiene anionic polymerization regioselectivity.Despite the longstanding belief that Lewis bases primarily modify the regioselectivity of diene anion polymerization through electronic effects, our studies indicate that steric hindrance also plays a decisive role. Utilizing X-ray single-crystal diffraction, we identified the structure of the active species—an allyllithium-Lewis base complex in a tetrahedral configuration. The mechanism by which Lewis bases enhance 1,2-regioselectivity has been clarified. In addition to altering the charge distribution of the chain-end allyl anion, steric effects play a decisive role in increasing the 1,2-selectivity of the polymerization. The distance between butadiene and the Lewis base ligand during 1,4-addition is considerably closer than during 1,2-addition, leading to significant steric hindrance. Therefore, 1,4-addition is hindered, and the polymerization predominantly occurs via 1,2-addition. Our theory does not exclude the electronic effect theory and does not imply that electronic effects are entirely insignificant in this process; on the contrary, in situ NMR quantitative studies reveal that the electronic effects of Lewis bases contribute to 30–40% of the selectivity for 1,2-addition.
Lewis bases characterized by “conformational constraints”, such as Lewis bases 6, 8, 10, and 11, exhibit enhanced structural regulation, resulting in superior 1,2-selectivity. This enhanced selectivity can be attributed to the high rigidity of these conformationally constrained ligands, which restricts their ability to adjust their configurations to accommodate the spatial demands of 1,4-addition.
Moreover, our approach offers the first compelling explanation for the distribution of cis and trans 1,4-units in anionic polybutadiene when Lewis bases are present, an effect attributed to steric hindrance. The enhanced trans-1,4 selectivity attributed to Lewis bases is due to syn-allyl active species (leading to trans-1,4 units) requiring less space than anti-allyl active species (leading to cis-1,4 units). Therefore, Lewis bases with significant steric hindrance not only induce an increase in 1,2-selectivity but also lead to an enhanced trans-1,4-selectivity.
Drawing on these insights, our newly developed Lewis base 11 with enhanced conformational constraint effect exhibits unparalleled control over the microstructure of polymers. Not only does it achieve unprecedented 1,2-selectivity (>99.8%) in the polymerization of butadiene at room temperature, but it also maintains superior performance at higher temperatures, surpassing all known Lewis bases.
Data availability
Crystallographic data for complex 6a has been deposited at the Cambridge Crystallographic Data Centre under deposition number 2365399 and can be obtained from https://www.ccdc.cam.ac.uk/structures. The data supporting this article have been included as part of the ESI.†Author contributions
The manuscript was written by Jian Tang. All authors have given approval to the final version of the manuscript. CRediT: Jian Tang B data curation, formal analysis, investigation, conceptualization, methodology, validation, visualization, writing – original draft, and writing – review & editing; Yuan Fu data curation, formal analysis, investigation, methodology; Jing Hua and Zhibo Li formal analysis, funding acquisition, project administration, resources, supervision; Jiahao Zhang and Shuoli Peng data curation, formal analysis, investigation, validation.Conflicts of interest
There are no conflicts to declare.Acknowledgements
Financial support for this work was provided by the National Natural Science Foundation of China (contract grant number 52373010) and Natural Science Foundation of Shandong Province (contract grant number ZR2020ME059). We would like to thank Xueyun Geng, Xiaoju Li and Haiyan Sui from Shandong University Core Facilities for Life and Environmental Sciences for their help with the XRD. We appreciate Mr Zhijie Liu's help in preparing the illustrations.Notes and references
- L. Lei, L. Han, H. Ma, R. Zhang, X. Li, S. Zhang, C. Li, H. Bai and Y. Li, Well-Tailored Dynamic Liquid Crystal Networks with Anionically Polymerized Styrene-Butadiene Rubbers toward Modulating Shape Memory and Self-Healing Capacity, Macromolecules, 2021, 54, 2691–2702 CrossRef CAS.
- K. Ntetsikas, V. Ladelta, S. Bhaumik and N. Hadjichristidis, Quo Vadis Carbanionic Polymerization?, ACS Polym. Au, 2023, 3, 158–181 CrossRef CAS PubMed.
- R. Sáez, C. McArdle, F. Salhi, J. Marquet and R. M. Sebastián, Controlled living anionic polymerization of cyanoacrylates by frustrated Lewis pair based initiators, Chem. Sci., 2019, 10, 3295–3299 RSC.
- O. W. Webster, Living polymerization methods, Science, 1991, 251, 887–893 CrossRef CAS PubMed.
- N. Hadjichristidis, H. Iatrou, M. Pitsikalis and J. Mays, Macromolecular architectures by living and controlled/living polymerizations, Prog. Polym. Sci., 2006, 31, 1068–1132 CrossRef CAS.
- R. Quirk, in Handbook of Polymer Synthesis, Characterization, and Processing, 2013, pp. 127–162, doi: DOI:10.1002/9781118480793.ch7.
- D. T. Gentekos, R. J. Sifri and B. P. Fors, Controlling polymer properties through the shape of the molecular-weight distribution, Nat. Rev. Mater., 2019, 4, 761–774 CrossRef.
- R. Whitfield, N. P. Truong, D. Messmer, K. Parkatzidis, M. Rolland and A. Anastasaki, Tailoring polymer dispersity and shape of molecular weight distributions: methods and applications, Chem. Sci., 2019, 10, 8724–8734 RSC.
- D. J. Walsh, D. A. Schinski, R. A. Schneider and D. Guironnet, General route to design polymer molecular weight distributions through flow chemistry, Nat. Commun., 2020, 11, 3094 CrossRef CAS PubMed.
- N. Baulu, M. Langlais, R. Ngo, J. Thuilliez, F. Jean-Baptiste-Dit-Dominique, F. D'Agosto and C. Boisson, Switch from Anionic Polymerization to Coordinative Chain Transfer Polymerization: A Valuable Strategy to Make Olefin Block Copolymers, Angew. Chem., Int. Ed., 2022, 61, e202204249 CrossRef CAS PubMed.
- W. Wang, W. Lu, A. Goodwin, H. Wang, P. Yin, N.-G. Kang, K. Hong and J. W. Mays, Recent advances in thermoplastic elastomers from living polymerizations: macromolecular architectures and supramolecular chemistry, Prog. Polym. Sci., 2019, 95, 1–31 CrossRef CAS.
- S. L. Perry and C. E. Sing, 100th Anniversary of Macromolecular Science Viewpoint: Opportunities in the Physics of Sequence-Defined Polymers, ACS Macro Lett., 2020, 9, 216–225 CrossRef CAS PubMed.
- J. Zhang, P. Zhou, B. Shi, P. Li and G. Wang, High-Efficient Access to Inverse Morphologies via Living Anionic Polymerization-Mediated Polymerization-Induced Cooperative Assembly, Macromolecules, 2023, 56, 5743–5753 CrossRef CAS.
- B. M. Hosford, W. Ramos and J. R. Lamb, Combining photocontrolled-cationic and anionic-group-transfer polymerizations using a universal mediator: enabling access to two- and three-mechanism block copolymers, Chem. Sci., 2024, 15, 13523–13530 RSC.
- F. Xu, K. Li, S. Li, C. Li, C. Yu, Y. Wang, W. Jiang, S. Lin, Y. Mai and Y. Zhou, Self-Assembly of Peapod-like Micrometer Tubes from a Planet-Satellite-type Supramolecular Megamer, Angew. Chem., Int. Ed., 2022, 61, e202213178 CrossRef CAS PubMed.
- G. Polymeropoulos, G. Zapsas, K. Ntetsikas, P. Bilalis, Y. Gnanou and N. Hadjichristidis, 50th Anniversary Perspective: Polymers with Complex Architectures, Macromolecules, 2017, 50, 1253–1290 CrossRef CAS.
- G. Shao, A. Li, Y. Liu, B. Yuan and W. Zhang, Branched Polymers: Synthesis and Application, Macromolecules, 2024, 57, 830–846 CrossRef CAS.
- M. A. Tasdelen, M. U. Kahveci and Y. Yagci, Telechelic polymers by living and controlled/living polymerization methods, Prog. Polym. Sci., 2011, 36, 455–567 CrossRef CAS.
- M. Meier-Merziger, J. Imschweiler, F. Hartmann, B.-J. Niebuur, T. Kraus, M. Gallei and H. Frey, Bifunctional Carbanionic Synthesis of Fully Bio-Based Triblock Structures Derived from β-Farnesene and ll-Dilactide: Thermoplastic Elastomers, Angew. Chem., Int. Ed., 2023, 62, e202310519 CrossRef CAS PubMed.
- A. Forens, K. Roos, C. Dire, B. Gadenne and S. Carlotti, Accessible microstructures of polybutadiene by anionic polymerization, Polymer, 2018, 153, 103–122 CrossRef CAS.
- H. Bai, L. Han, X. Wang, H. Yan, H. Leng, S. Chen and H. Ma, Anion Migrated Ring Opening and Rearrangement in Anionic Polymerization Induced C7 and C8 Polymerizations, Macromolecules, 2022, 55, 9751–9765 CrossRef CAS.
- T. La Porta, R. Perrin, M. Ray, B. Gadenne, C. Dire and S. Carlotti, Calcium–Lithium Systems as Innovative Bimetallic Initiators for the Anionic Polymerization of Butadiene: Toward Control and High 1,4-trans Microstructure, Macromolecules, 2024, 57, 2376–2384 CrossRef CAS.
- C. Hahn, S. Becker, A. H. E. Müller and H. Frey, Anionic polymerization of ferulic acid-derived, substituted styrene monomers, Eur. Polym. J., 2024, 211, 113004 CrossRef CAS.
- A. Halasa, D. Schulz, D. Tate and V. Mochel, in Advances in organometallic chemistry, Elsevier, 1980, vol. 18, pp. 55–97 Search PubMed.
- D. E. Anderson, A. Tortajada and E. Hevia, New Frontiers in Organosodium Chemistry as Sustainable Alternatives to Organolithium Reagents, Angew. Chem., Int. Ed., 2024, 63, e202313556 CrossRef CAS PubMed.
- M. Morton and L. J. Fetters, Anionic Polymerization of Vinyl Monomers, Rubber Chem. Technol., 1975, 48, 359–409 CrossRef CAS.
- M. Steube, T. Johann, H. Hübner, M. Koch, T. Dinh, M. Gallei, G. Floudas, H. Frey and A. H. E. Müller, Tetrahydrofuran: More than a “Randomizer” in the Living Anionic Copolymerization of Styrene and Isoprene: Kinetics, Microstructures, Morphologies, and Mechanical Properties, Macromolecules, 2020, 53, 5512–5527 CrossRef CAS.
- A. F. Halasa, D. Lohr and J. Hall, Anionic polymerization to high vinyl polybutadiene, J. Polym. Sci., Polym. Chem. Ed., 1981, 19, 1357–1360 CrossRef CAS.
- T. A. Antkowiak, A. E. Oberster, A. F. Halasa and D. P. Tate, Temperature and concentration effects on polar-modified alkyllithium polymerizations and copolymerizations, J. Polym. Sci., Part A-1: Polym. Chem., 1972, 10, 1319–1334 CrossRef CAS.
- C. Wu, B. Liu, F. Lin, M. Wang and D. Cui, Cis-1,4-Selective Copolymerization of Ethylene and Butadiene: A Compromise between Two Mechanisms, Angew. Chem., Int. Ed., 2017, 56, 6975–6979 CrossRef CAS PubMed.
- D. Liu, M. Wang, Y. Chai, X. Wan and D. Cui, Self-Activated Coordination Polymerization of Alkoxystyrenes by a Yttrium Precursor: Stereocontrol and Mechanism, ACS Catal., 2019, 9, 2618–2625 CrossRef CAS.
- Y. Zhang, Y. Zhang, X. Hu, C. Wang and Z. Jian, Advances on Controlled Chain Walking and Suppression of Chain Transfer in Catalytic Olefin Polymerization, ACS Catal., 2022, 12, 14304–14320 CrossRef CAS.
- G. Ricci, G. Pampaloni, A. Sommazzi and F. Masi, Dienes Polymerization: Where We Are and What Lies Ahead, Macromolecules, 2021, 54, 5879–5914 CrossRef CAS.
- C. Chen, Designing catalysts for olefin polymerization and copolymerization: beyond electronic and steric tuning, Nat. Rev. Chem, 2018, 2, 6–14 CrossRef CAS.
- H. Mu, L. Pan, D. Song and Y. Li, Neutral Nickel Catalysts for Olefin Homo- and Copolymerization: Relationships between Catalyst Structures and Catalytic Properties, Chem. Rev., 2015, 115, 12091–12137 CrossRef CAS PubMed.
- L. Porri, A. Giarrusso and G. Ricci, Recent views on the mechanism of diolefin polymerization with transition metal initiator systems, Prog. Polym. Sci., 1991, 16, 405–441 CrossRef CAS.
- D. Hesterwerth, D. Beckelmann and F. Bandermann, Classification of polar additives with respect to their influence on the microstructure in anionic polymerization of butadiene with butyllithium by transition energy measurements, J. Appl. Polym. Sci., 1999, 73, 1521–1532 CrossRef CAS.
- I. Kuntz and A. Gerber, The butyllithium-initiated polymerization of 1,3-butadiene, J. Polym. Sci., 1960, 42, 299–308 CrossRef CAS.
- R. Milner and R. N. Young, 1H NMR spectroscopy of the complexation of polybutadienyllithium by N,N,N′,N′-tetramethylethylenediamine, Polymer, 1982, 23, 1636–1640 CrossRef CAS.
- H. Hsieh and R. P. Quirk, Anionic polymerization: principles and practical applications, CRC Press, 1996 Search PubMed.
- R. Kozak and M. Matlengiewicz, Effect of enthalpy of polar modifiers interaction with n-butyllithium on the reaction enthalpy, kinetics and chain microstructure during anionic polymerization of 1,3-butadiene, Polym. Test., 2017, 64, 20–37 CrossRef CAS.
- G. Fraenkel, A. F. Halasa, V. Mochel, R. Stumpe and D. Tate, Remarks on the chameleon behavior of an allyllithium: carbon-13 and lithium-6 NMR, J. Org. Chem., 1985, 50, 4563–4565 CrossRef CAS.
- S. Bywater, Structure and mechanism in anionic polymerization, Prog. Polym. Sci., 1994, 19, 287–316 CrossRef CAS.
- S. Bywater and D. J. Worsfold, Charge distribution in substituted allyl-alkali metal compounds by 13C NMR, J. Organomet. Chem., 1978, 159, 229–235 CrossRef CAS.
- J. Tang, T. Xie, Y. Yuan, J. Hua, T. Zhuang, Y. Luo and J. Geng, Degradation of Polydienes Induced by Alkyllithium: Characterization and Reaction Mechanism, Macromolecules, 2021, 54, 1147–1158 CrossRef CAS.
- D. Seyferth and M. A. Weiner, Preparation of Organolithium Compounds by the Transmetalation Reaction. III. Allyllithium and Methallyllithium 1, 2a, J. Org. Chem., 1961, 26, 4797–4800 CrossRef CAS.
- J. Tang, Z. Xu, Z. Liu, Y. Fu and J. Hua, Pyridine-amido aluminum catalyst precursors for 1,3-butadiene transition-metal-free stereospecific polymerization, Polym. Chem., 2023, 14, 980–989 RSC.
- B. Liu, X. Wang, Y. Pan, F. Lin, C. Wu, J. Qu, Y. Luo and D. Cui, Unprecedented 3,4-Isoprene and cis-1,4-Butadiene Copolymers with Controlled Sequence Distribution by Single Yttrium Cationic Species, Macromolecules, 2014, 47, 8524–8530 CrossRef CAS.
- A. R. O'Connor, P. S. White and M. Brookhart, The Mechanism of Polymerization of Butadiene by “Ligand-Free” Nickel(II) Complexes, J. Am. Chem. Soc., 2007, 129, 4142–4143 CrossRef PubMed.
- M. Frisch, G. Trucks, H. Schlegel, G. Scuseria, M. Robb, J. Cheeseman, G. Scalmani, V. Barone, B. Mennucci and G. Petersson, in Gaussian 09, Gaussian Inc Wallingford, 2009 Search PubMed.
- F. Neese, Software update: the ORCA program system, version 4.0, WIRES Comput. Mol. Sci., 2018, 8, e1327 CrossRef.
- J. A. Berson, Kinetics, Thermodynamics, and the Problem of Selectivity: The Maturation of an Idea, Angew. Chem., Int. Ed., 2006, 45, 4724–4729 CrossRef CAS PubMed.
- J. Hay, J. McCabe and J. Robb, Kinetics of reaction of metal alkyls with alkenes. Part 7.—n-butyl lithium and N: N: N′: N′-tetramethyl ethylene diamine with butadiene, J. Chem. Soc., Faraday Trans. 1, 1972, 68, 1–9 RSC.
- M. Morton, E. E. Bostick, R. A. Livigni and L. J. Fetters, Homogeneous anionic polymerization. IV. Kinetics of butadiene and isoprene polymerization with butyllithium, J. Polym. Sci., Part A: Gen. Pap., 1963, 1, 1735–1747 CrossRef.
- L. Falivene, Z. Cao, A. Petta, L. Serra, A. Poater, R. Oliva, V. Scarano and L. Cavallo, Towards the online computer-aided design of catalytic pockets, Nat. Chem., 2019, 11, 872–879 CrossRef CAS PubMed.
- J.-D. Chai and M. Head-Gordon, Long-range corrected double-hybrid density functionals, J. Chem. Phys., 2009, 131, 174105 CrossRef PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Additional experimental and computational details, materials, characterization methods, supplementary tables and figures, and spectra for synthetic compounds and polymers. CCDC 2365399. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d4sc05144k |
| This journal is © The Royal Society of Chemistry 2024 |