
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceVisible light-induced Mallory reaction of tertiary benzanilides via iminium intermediates†
Xiaoqiang
Ma‡
,
Si
Wang‡
,
Zhanyong
Tang
,
Jialin
Huang
,
Tianhao
Jia
,
Xingda
Zhao
and
Depeng
Zhao
 *
*
State Key Laboratory of Anti-infective Drug Discovery and Development, School of Pharmaceutical Sciences, Sun Yat-sen University, Guangzhou, 510006, China. E-mail: zhaodp5@mail.sysu.edu.cn
First published on 19th September 2024
Abstract
The Mallory reaction, which involves the photocyclization of stilbenes/diarylethenes and their analogues into polycyclic aromatics, is of significant synthetic importance. However, its application to tertiary benzanilides has not been explored to date. Besides, most of the reported Mallory reactions require ultraviolet irradiation. In this study, we show the first Mallory reaction of tertiary benzanilides promoted by visible light via iminium intermediates formed in situ from tertiary benzanilide, Tf2O (triflic anhydride) and pyridine. UV/vis absorption spectroscopy combined with density functional theory (DFT) calculations revealed that the formation of the iminium intermediate decreased the HOMO–LUMO energy gap, thereby enhancing visible light absorption. This study provides a rapid and practical approach for the preparation of the phenanthridinone skeleton and provides a new idea for the design of new visible light photoswitches.
Introduction
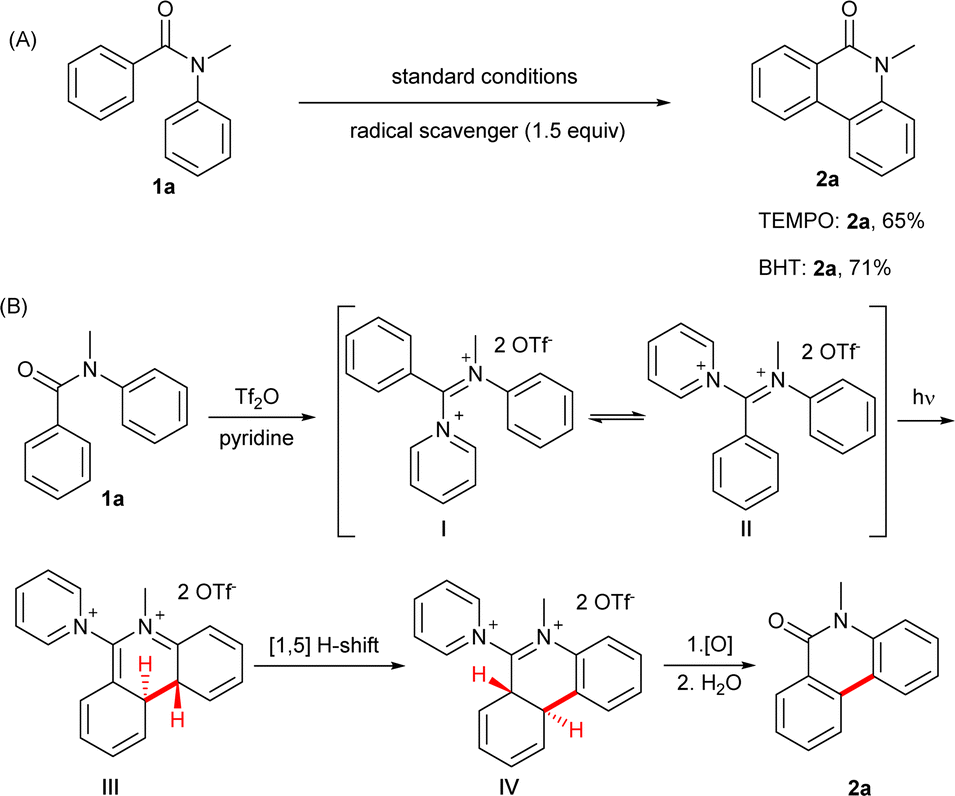
Since its discovery in 1964,1,2 the Mallory reaction has become a powerful synthetic tool for the construction of polycyclic aromatic hydrocarbons and heterocyclic analogues, from simple phenanthrenes and their heterocyclic analogues to complex polycyclic molecules (coronenes, helicenes, etc.) and nanographenes/graphene nanoribbons.3–9 The typical examples of Mallory reactions are shown in Fig. 1B. Under UV irradiation, stilbene or its derivatives undergo 6π-electrocyclization and further oxidation or elimination, resulting in the formation of phenanthrene derivatives.10–12 Though the Mallory-type reaction of benzamides (ArCONHAr) was first reported as early as 1967,13–16 the Mallory-type reaction of tertiary benzanilides remains undeveloped with no clear reason given thus far (Fig. 1B). | ||
| Fig. 1 Background and design for the visible light-promoted cyclization. | ||
Phenanthridinone skeletons are important subunits found extensively in natural products and pharmaceutical compounds (Fig. 1A).17–23 For example, they have been widely used as the PARP inhibitor for anticancer therapies and as neurotrophin activity enhancers for the treatment of nerve diseases.24–28 Over the past several decades, many approaches with the prefunctionalization of the aryl groups of tertiary benzanilides have been developed.20,29–36 Recently, two fascinating examples of C–C cross-coupling reactions involving benzanilides with two unactivated sp2 hybridized C–H bonds were reported (Fig. 1C).37 However, in the first example of Pd-catalysed intramolecular oxidative ortho-arylation of benzanilides, the substrate scope is limited and, in particular, halogens are incompatible. In the latter case of electrochemical intramolecular cross-coupling,38 the scope of the benzoyl moiety is strictly limited to electron-rich aryls with two or more alkoxy groups (Fig. 1C). Given such significance, the development of concise and versatile methods to obtain these high-value skeletons remains a topic of general interest.
Although previous studies,39 including our NMR results, indicate that the N-methylbenzanilide mainly exists in cis-conformation which should facilitate its photocyclization, attempts to induce its photocyclization under UV light have remained unsuccessful. We inferred that the observed phenomena can be attributed to the resonance of tertiary benzanilides. Although tertiary benzanilide can be described as a resonance between neutral and zwitterionic forms,40,41 the zwitterionic form (which is a good 6π system) makes less contribution to the whole structure (Fig. 1D), resulting in poor photocyclization upon light irradiation. We reasoned that increasing the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N double bond character would enhance the feasibility of 6π photocyclization. As tertiary benzanilides are known to form the oxyiminium ion upon treating with Tf2O, our initial study focused on the Tf2O-mediated42 photocyclization of N-methylbenzanilide. To our delight, the desired product 2a was obtained in 60% 1H NMR yield under 310 nm UV light (see ESI† page 14 for more details). Notably, due to the wide band gaps between the valence and conduction bands, Mallory-type reactions are constrained by their reliance on photoactivity primarily within the UV region.43–45 Based on the previous reports from our group46,47 and others on iminium salts48–52 in photochemical reactions, we planned to investigate the photochemical reactivities of iminium salts of tertiary benzanilide promoted by visible light. We were delighted to find that the iminium intermediate formed in situ from N-methylbenzanilide 1a, Tf2O and Py (pyridine) exhibited a yellow colour, indicating its absorption of visible light. We hypothesized that the iminium intermediate might undergo a 6π cyclization under visible light as a result of the small energy gap between the highest occupied molecular orbital (HOMO) and lowest unoccupied molecular orbital (LUMO), resulting in the formation of 2a upon oxidative aromatization. Herein, we demonstrate the first Mallory-type reaction of tertiary benzanilide to construct phenanthridinone scaffolds via iminium intermediates triggered by visible light. This protocol was successfully applied to the concise total synthesis of a natural alkaloid anhydrolycorinone 2v and an AChE inhibitor N-methylcrinasiadine 2d.53,54 Density functional theory (DFT) calculation studies revealed that a difference in the delocalization contributes to the decrease in the HOMO–LUMO energy gap of the iminium formed in situ.
N double bond character would enhance the feasibility of 6π photocyclization. As tertiary benzanilides are known to form the oxyiminium ion upon treating with Tf2O, our initial study focused on the Tf2O-mediated42 photocyclization of N-methylbenzanilide. To our delight, the desired product 2a was obtained in 60% 1H NMR yield under 310 nm UV light (see ESI† page 14 for more details). Notably, due to the wide band gaps between the valence and conduction bands, Mallory-type reactions are constrained by their reliance on photoactivity primarily within the UV region.43–45 Based on the previous reports from our group46,47 and others on iminium salts48–52 in photochemical reactions, we planned to investigate the photochemical reactivities of iminium salts of tertiary benzanilide promoted by visible light. We were delighted to find that the iminium intermediate formed in situ from N-methylbenzanilide 1a, Tf2O and Py (pyridine) exhibited a yellow colour, indicating its absorption of visible light. We hypothesized that the iminium intermediate might undergo a 6π cyclization under visible light as a result of the small energy gap between the highest occupied molecular orbital (HOMO) and lowest unoccupied molecular orbital (LUMO), resulting in the formation of 2a upon oxidative aromatization. Herein, we demonstrate the first Mallory-type reaction of tertiary benzanilide to construct phenanthridinone scaffolds via iminium intermediates triggered by visible light. This protocol was successfully applied to the concise total synthesis of a natural alkaloid anhydrolycorinone 2v and an AChE inhibitor N-methylcrinasiadine 2d.53,54 Density functional theory (DFT) calculation studies revealed that a difference in the delocalization contributes to the decrease in the HOMO–LUMO energy gap of the iminium formed in situ.
Results and discussion
We commenced our investigation by irradiating N-methylbenzanilide 1a in 1,2-dichloroethane (DCE) at 25 °C with 425 nm blue light in the presence of Tf2O (2.0 equiv.) and pyridine (2.0 equiv.) for 20 h. We were pleased to find that the desired phenanthridinone product 2a was indeed obtained in 40% yield (Table 1, entry 2). With these preliminary results in hand, we then systematically screened the reaction conditions to improve the yield. A notable enhancement was observed employing 3.0 equiv. of Tf2O and pyridine, resulting in an increase in the yield to 65% (entry 3). To our delight, on using 3.0 equiv. pyridine with 2.0 equiv. Tf2O, the yield increased significantly from 65% to 90% (entry 4). Apart from Tf2O, no desired product was detected when other activators such as trifluoromethanesulfonic acid (entry 5), thionyl chloride (entry 6) or oxalyl chloride (entry 7) were used. Other substituted pyridines were also tested; however, 2-acetylpyridine (entry 8) and 4-methoxypyridine (entry 9) failed to yield the product, and 2-methylpyridine (entry 10) provided 2a in a low yield (25%). Therefore, we chose 2.0 equiv. of Tf2O and 3.0 equiv. of pyridine for further investigations of commonly used solvents, such as dichloromethane (DCM), ethyl acetate (EA), and acetonitrile (CH3CN). However, none of the above solvents could provide higher yields than that of DCE (entries 11–13). Finally, control experiments confirmed that light, Tf2O and pyridine are all essential for this transformation. No product was observed in the absence of these reaction components (entries 14–16). It is worth noting that this reaction also proceeds without an inert gas atmosphere with a slight decrease in yield (84%, entry 17). Unfortunately, reducing the equivalents of Tf2O (1.1 equiv.) and pyridine (1.6 equiv.) resulted in a considerably lower yield of 2a (27%, entry 1).|
|
||||
|---|---|---|---|---|
| Entry | Pyridine (equiv.) | Activator (equiv.) | Solvent | Yieldb (%) |
| a Unless otherwise mentioned, all reactions were conducted with 1a (0.2 mmol), solvent (2.0 mL), 25 °C, N2, 10 W blue LEDs, 20 h. b Isolated yield. c Identical yield was observed for 12 h. d Under dark. e Without an inert gas atmosphere. 1,2-Dichloroethane (DCE), dichloromethane (DCM), acetonitrile (MeCN), ethyl acetate (EA). | ||||
| 1 | Pyridine (1.6) | Tf2O (1.1) | DCE | 27 |
| 2 | Pyridine (2.0) | Tf2O (2.0) | DCE | 40 |
| 3 | Pyridine (3.0) | Tf2O (3.0) | DCE | 65 |
| 4 | Pyridine (3.0) | Tf2O (2.0) | DCE | 90c |
| 5 | Pyridine (3.0) | TfOH (2.0) | DCE | NR |
| 6 | Pyridine (3.0) | SOCl2 (3.0) | DCE | NR |
| 7 | Pyridine (3.0) | (COCl2) (2.0) | DCE | NR |
| 8 | 2-Acetylpyridine (3.0) | Tf2O (2.0) | DCE | NR |
| 9 | 4-Methoxypyridine (3.0) | Tf2O (2.0) | DCE | NR |
| 10 | 2-Methylpyridine (3.0) | Tf2O (2.0) | DCE | 25 |
| 11 | Pyridine (3.0) | Tf2O (2.0) | DCM | 80 |
| 12 | Pyridine (3.0) | Tf2O (2.0) | EA | 34 |
| 13 | Pyridine (3.0) | Tf2O (2.0) | MeCN | 69 |
| 14 | Pyridine (3.0) | — | DCE | NR |
| 15 | — | Tf2O (2.0) | DCE | NR |
| 16d | Pyridine (3.0) | Tf2O (2.0) | DCE | NR |
| 17e | Pyridine (3.0) | Tf2O (2.0) | DCE | 84 |

With the optimal conditions established, the substrate scope was next explored to probe the generality of this reaction in the synthesis of phenanthridinone derivatives (Scheme 1). Initially, various electron-donating substitutions on the benzoyl derivatives were tested in the photocyclization reaction. When substrates with benzoyl derivatives bearing 4-methyl (1b), 4-methoxyl (1c), piperonyl (1d), and 3,5-dimethyl (1e) were subjected to the standard conditions, the cyclization products 2b, 2c, 2d and 2e were afforded in good to excellent yields (71%, 65%, 66%, and 85%, respectively). Substrate (1f) with an electron-withdrawing group (Cl) in the benzoyl moiety was suitable for this transformation, affording 2f in a good yield (75%). Subsequently, substrates with methyl (1g), methoxyl (1h), halogens (1i–1k), cyano (1l), acetyl (1m) and trifluoromethyl (1n) with the aniline moiety were also examined, and moderate to excellent yields were observed (62–95%). Substrates containing fluorine or trifluoromethyl groups, which are commonly used in medicinal chemistry to enhance the lipophilicity, are suitable for this reaction (2i and 2n).55 To our delight, this iminium activation strategy was viable for both benzoyl and aniline-substituted N-methylbenzanilide, affording the annulation target 2o–2q with satisfactory results (yields: 85%, 85% and 75%, respectively). It is evident that the presence of both electron-donating and electron-withdrawing groups on the N-methylbenzanilides does not considerably impact the reaction yield. Encouraged by the above results, we next tested our strategy of polycyclic or heterocyclic-substituted anilide derivatives (1r–1t). Specifically, 2s and 2t were obtained in relatively high yields (65% and 75%), whereas 2r was obtained in 55% yield, which might be due to the instability of the thiophene moiety under the reaction conditions.
 | ||
| Scheme 1 Substrate scope of the photocyclization. Reaction conditions: 1 (0.2 mmol, 1.0 equiv.), Tf2O (0.4 mmol, 2.0 equiv.), pyridine (0.6 mmol, 3.0 equiv.), DCE (2.0 mL), 25 °C, N2, 10 W blue LEDs (425 nm), 12–20 h, yields of isolated product. a5.0 mmol scale, 36 h. b4.0 equiv. Tf2O and 6.0 equiv. pyridine. | ||
To further explore the generality of this photocyclization, we next examined the compatibility of N-substituents. When benzanilides bearing five- or six-membered cyclic amine moiety were subjected to the standard reaction conditions, the corresponding polycyclic products (2u–2w) were afforded in moderate to high yields (87%, 65% and 63%, respectively). Likewise, the cyclization with benzanilides having linear aliphatic N-substitutions, such as ethyl (1x), ipropyl (1y), nbutyl (1z) and 4-butanoate (1aa), all furnished the corresponding products 2x–2aa in moderate to high yields (85%, 65%, 65% and 60%, respectively), although 4.0 equiv. Tf2O and 6.0 equiv. pyridine were required for 1y–1aa. In addition, we evaluated the performance of N,N-diaryl substituted benzanilides; both 1ab (phenyl) and 1ac (4-methoxyphenyl) proved to be effective for this protocol, affording products 2ab and 2ac in good yields (54% and 72%). Finally, benzyl N-substituted substrate 1ad also worked well under this protocol (51%), albeit with 4.0 equiv. Tf2O and 6.0 equiv. pyridine. To explore the practical utility of this transformation in organic synthesis, a gram-scale experiment of 1a (5.0 mmol, 1.05 g) was performed, which gave rise to a satisfactory yield of the cyclization product 2a (0.86 g, 82%). Notably, many of the annulation products, such as 2d and 2v, are important subunits in biologically and pharmaceutically-active phenanthridinone alkaloids or their analogues.53,54
To probe the reaction mechanism and validate the involvement of visible light in the Mallory reaction process, we conducted UV/vis spectroscopy experiments. The UV/vis absorption spectra of 1a, 1a/Tf2O/pyridine, 1a/Tf2O, 1a/pyridine, and Tf2O/pyridine are shown in Fig. 2A. As expected, a new absorption band at 360 nm appeared for the ternary mixture of 1a/Tf2O/pyridine with the tail absorption extending until 425 nm, indicating the formation of a new species (orange line, Fig. 2A). In contrast, all the reagents or binary mixtures exhibit weak absorption above 350 nm. Furthermore, Fig. 2A (inset) shows the solution colour contrast from colourless to light-yellow, indicating visible light absorption of the iminium intermediate. Note that other iminiums formed in situ from tertiary benzanilides (1c, 1k, 1l, 1s, 1u and 1ac), Tf2O and pyridine also exhibit visible absorbance bands (Fig. 2B, C and Fig. S4†). Additionally, to validate our strategy of iminium-induced visible light absorption, 1H NMR spectroscopy studies were performed to confirm the formation of the iminium intermediate. As shown in Fig. 2D and S6,† when 1a was treated with 2.0 equiv. Tf2O and 3.0 equiv. pyridine in anhydrous CD3CN. To our delight, two new peaks of N-methyl hydrogen were observed (4.15 and 4.43 ppm). Besides, in the aromatic region, four new sets of signals of the iminium intermediate were observed, indicating the formation of the iminium intermediate (7.40–7.85 ppm). To gain an insight into the reaction mechanism, gas chromatography (GC) was employed to analyse the gases in the Schlenk tube of the reaction of 1a under standard conditions. The results indicated that hydrogen was generated during the reaction (see ESI† page 43 for more details). In addition, radical trapping experiments were performed (Fig. 4A). The presence of radical scavengers (TEMPO or BHT) did not drastically inhibit the formation of product 2a, excluding the participation of radical species.
 | ||
| Fig. 2 UV/vis absorption spectra of (A) Tf2O and pyridine; 1a and pyridine (3.0 equiv.); 1a and Tf2O (2.0 equiv.); 1a, Tf2O (2.0 equiv.) and pyridine (3.0 equiv.); 1a in DCE (5 × 10−5 mol L−1); (B): 1c; 1c, Tf2O (2.0 equiv.) and pyridine (3.0 equiv.) in DCE (5 × 10−5 mol L−1); (C) 1ac; 1ac, Tf2O (2.0 equiv.) and pyridine (2.0 equiv.) in DCE (5 × 10−5 mol L−1); inset: colour changes of the corresponding substrate and mixture (5 × 10−3 mol L−1); (D) 1H NMR experiments: 1a (0.05 mmol) in CD3CN (0.5 mL); mixture of 1a (0.05 mmol), Tf2O (2.0 equiv.) and pyridine (3.0 equiv.) in CD3CN (0.5 mL); mixture of 1a (0.05 mmol) and Tf2O (2.0 equiv.) in CD3CN (0.5 mL). | ||
We further employed theoretical calculation to confirm that the formation of the iminium intermediate causes intramolecular cyclization under visible light irradiation (see Computational details in ESI†). The molecular minimized structure of 1a and iminium II are shown in Fig. 3A and S1.† As previously reported, the calculated stilbenes correspond to the optimal cis conformation for the photocyclization reaction.56Fig. 3B shows the frontier molecular orbitals of the cis conformation of 1a and iminium II, where red and green colours indicate two opposite phases of orbitals, i.e., positive (or crest) and negative (or trough) regions of molecular orbitals, respectively. For single 1a, both the HOMO and LUMO are spread over the entire molecule, as illustrated in Fig. 3B. However, in iminium II, the HOMO mainly concentrates on the phenyl and N-methylaniline, while the LUMO is located on the whole iminium II. The evaluated energies of the HOMO–LUMO energy gap of 1a and iminium intermediate II were found to be 5.28 and 3.67 eV, respectively. The observed decrease in the HOMO–LUMO energy gap can be attributed to differential delocalization, where LUMO shows a more significant reduction during the formation of iminium intermediates.57,58 To analyse the spatial distribution of the electron–hole after iminium II excitation, the electron–hole excitation analysis of iminium II was performed using Multiwfn.59,60 The results showed charge transfer (CT) excitation from the aniline (HOMO) to the pyridine moiety (LUMO). This charge transfer state may prolong the excited lifetime.61–63 This small HOMO–LUMO energy gap accounts for the crucial electronic transitions decrease and the absorption wavelength shows the bathochromic effect. Furthermore, the E0,0 of iminium II was also estimated from the wavelength at the intersection point of the normalized UV-vis absorption spectrum and the emission spectrum in solution (367 nm, 3.37 eV, Fig. S5†). Additionally, we speculate that intermolecular interactions might be responsible for the absorption at ∼425 nm, as confirmed by the calculation of the HOMO–LUMO energy gap of the D–A complex 1a/iminium II (Fig. S1†).
 | ||
| Fig. 3 (A) Optimized molecular structure of cis-1a and iminium intermediate II; (B) HOMO and LUMO orbital levels and energy gap of cis-1a and iminium intermediate II, calculated with DFT at the B3LYP-D3(BJ)/def2-TZVP level using Gaussian 16. | ||
Based on the above results and previous work,46,47,64 we proposed a plausible reaction mechanism (Fig. 4B). Firstly, iminium intermediate I was formed in situ.50,65 Subsequently, iminium intermediate I was tautomerized into iminium intermediate II.66,67 Followed by a photochemical 6π-electrocyclization under visible light conditions, iminium intermediate III was generated by ring closure. Then, intermediate III was expected to undergo a suprafacial [1,5]-H shift, affording intermediate IV, which is driven by the aromatization.45,68,69 In the end, intermediate IV was converted to phenanthridinone via oxidation or dehydrogenation and hydrolysis.
 | ||
| Fig. 4 (A) Radical trapping and (B) plausible reaction mechanism. | ||
Conclusions
In summary, we report the first Mallory reaction of tertiary benzanilides promoted by visible light via an iminium intermediate. Experimental and theoretical analyses suggested that the iminium intermediate formed in situ could considerably decrease the HOMO–LUMO energy gap, resulting in visible light-induced photocyclization. A wide range of substrates, including benzoyl-substituted, aniline-substituted, N-substituted and heterocyclic benzanilide derivatives are compatible with this method, giving the products in good to excellent yields. In addition, this visible light-induced Mallory reaction was successfully applied to synthesize a natural alkaloid anhydrolycorinone 2v and an AChE inhibitor N-methylcrinasiadine 2d.Data availability
All the data related to the above-mentioned manuscript are available in the ESI.†Author contributions
D. Z., X. M. and S. W. conceived and designed the experiments and mechanism studies. D. Z. guided the project. X. M. performed DFT calculations. S. W., Z. T., J. H., T. J. and X. Z. performed the experiments. D. Z. and X. M. wrote the manuscript. All the authors discussed the results and commented on the manuscript.Conflicts of interest
The authors declare no conflict of interest.Acknowledgements
We are grateful for the support of this work by the National Natural Science Foundation of China (22371307, 21971267), and the program for Guangdong Introducing Innovative and Entrepreneurial Teams (2017ZT07C069). The Fundamental Research Funds for the Central Universities, Sun Yat-sen University (24qnpy058).Notes and references
- F. B. Mallory, C. S. Wood and J. T. Gordon, J. Am. Chem. Soc., 1964, 86, 3094–3102 CrossRef CAS.
- A. G. Lvov, J. Org. Chem., 2020, 85, 8749–8759 CrossRef CAS PubMed.
- A. Ghosh, D. Csókás, M. Budanović, R. D. Webster, I. Pápai and M. C. Stuparu, Chem. Sci., 2021, 12, 3977–3983 RSC.
- N. Hoffmann, Chem. Rev., 2008, 108, 1052–1103 CrossRef CAS PubMed.
- T. Moriguchi, M. Higashi, D. Yakeya, V. Jalli, A. Tsuge, T. Okauchi, S. Nagamatsu and W. Takashima, J. Mol. Struct., 2017, 1127, 413–418 CrossRef CAS.
- M. K. Weclawski, M. Jakesová, M. Charyton, N. Demitri, B. Koszarna, K. Oppelt, S. Sariciftci, D. T. Gryko and E. D. Glowacki, J. Mater. Chem. A, 2017, 5, 20780–20788 RSC.
- D. Zanetti, O. Matuszewska, G. Giorgianni, C. Pezzetta, N. Demitri and D. Bonifazi, JACS Au, 2023, 3, 3045–3054 CrossRef CAS PubMed.
- A. Jolly, D. Miao, M. Daigle and J. F. Morin, Angew. Chem., Int. Ed., 2019, 59, 4624–4633 CrossRef PubMed.
- H. Y. Li, X. Y. Niu, S. S. Zhang, S. Shen, Q. Y. Meng and X. L. Yang, Org. Lett., 2024, 26, 5364–5369 CrossRef PubMed.
- J. Bao and P. M. Weber, J. Am. Chem. Soc., 2011, 133, 4164–4167 CrossRef CAS PubMed.
- E. B. Hulley and E. L. Clennan, J. Am. Chem. Soc., 2024, 146, 1122–1131 CrossRef CAS PubMed.
- K. B. Jørgensen, Molecules, 2010, 15, 4334–4358 CrossRef PubMed.
- B. S. Thyagarajan, N. Kharasch, H. B. Lewis and W. Wolf, Chem. Commun., 1967, 614–615 RSC.
- N. Wang, D. Wang, Y. He, J. Xi, T. Wang, Y. Liang and Z. Zhang, Adv. Synth. Catal., 2022, 364, 1150–1155 CrossRef CAS.
- Y. Kanaoka and K. Itoh, J. Chem. Soc., Chem. Commun., 1973, 647–648 RSC.
- J. Grimshaw and A. P. d. Silva, J. Chem. Soc., Chem. Commun., 1980, 302–303 RSC.
- N. Aravindan, V. Vinayagam and M. Jeganmohan, Org. Lett., 2022, 24, 5260–5265 CrossRef CAS PubMed.
- S. K. Bera and P. Mal, J. Org. Chem., 2021, 86, 14144–14159 CrossRef CAS PubMed.
- D. Ferraris, Y. S. Ko, T. Pahutski, R. P. Ficco, L. Serdyuk, C. Alemu, C. Bradford, T. Chiou, R. Hoover, S. Huang, S. Lautar, S. Liang, Q. A. Lin, M. X. C. Lu, M. Mooney, L. Morgan, Y. Z. Qian, S. Tran, L. R. Williams, Q. Y. Wu, J. Zhang, Y. N. Zou and V. Kalish, J. Med. Chem., 2003, 46, 3138–3151 CrossRef CAS PubMed.
- T. S. Manikandan, R. Ramesh and D. Semeril, Organometallics, 2018, 38, 319–328 CrossRef.
- Y. Nishiyama, S. Mori, M. Makishima, S. Fujii, H. Kagechika, Y. Hashimoto and M. Ishikawa, ACS Med. Chem. Lett., 2018, 9, 641–645 CrossRef CAS PubMed.
- A. L. Ruchelman, P. J. Houghton, N. Zhou, A. Liu, L. F. Liu and E. J. LaVoie, J. Med. Chem., 2005, 48, 792–804 CrossRef CAS PubMed.
- F. Rafiee, Appl. Organomet. Chem., 2017, 31, e3820 CrossRef.
- D. Bondar, O. Bragina, J. Y. Lee, I. Semenyuta, I. Järving, V. Brovarets, P. Wipf, I. Bahar and Y. Karpichev, Helv. Chim. Acta, 2023, 106, e202300133 CrossRef CAS.
- M. Cohen-Armon, Cancers, 2020, 12, 1628 CrossRef CAS PubMed.
- Y. Q. Wang, P. Y. Wang, Y. T. Wang, G. F. Yang, A. Zhang and Z. H. Miao, J. Med. Chem., 2016, 59, 9575–9598 CrossRef CAS PubMed.
- M. Wan, L. Zhang, Y. Chen, Q. Li, W. Fan, Q. Xue, F. Yan and W. Song, Front. Oncol., 2019, 9, 274 CrossRef PubMed.
- W. E. Campbell, J. J. Nair, D. W. Gammon, C. Codina, J. Bastida, F. Viladomat, P. J. Smith and C. F. Albrecht, Phytochemistry, 2000, 53, 587–591 CrossRef CAS PubMed.
- N. Conde, F. Churruca, R. SanMartin, M. T. Herrero and E. Domínguez, Adv. Synth. Catal., 2015, 357, 1525–1531 CrossRef CAS.
- M. Feng, B. Tang, H. X. Xu and X. Jiang, Org. Lett., 2016, 18, 4352–4355 CrossRef CAS PubMed.
- X. Abel-Snape, A. Whyte and M. Lautens, Org. Lett., 2020, 22, 7920–7925 CrossRef CAS PubMed.
- L. Donati, P. Leproux, E. Prost, S. Michel, F. Tillequin, V. Gandon and F. H. Porée, Chem.–Eur. J., 2011, 17, 12809–12819 CrossRef CAS PubMed.
- C. S. Nervig, P. J. Waller and D. Kalyani, Org. Lett., 2012, 14, 4838–4841 CrossRef CAS PubMed.
- X. Geng, H. He, A. Shatskiy, E. V. Stepanova, G. R. Alvey, J. Q. Liu, M. D. Kärkäs and X. S. Wang, J. Org. Chem., 2023, 88, 12738–12743 CrossRef CAS PubMed.
- Y. Fanga and G. K. Tranmer, Med. Chem. Commun., 2016, 7, 720–724 RSC.
- K. Q. Chen, B. B. Zhang, Z. X. Wang and X. Y. Chen, Org. Lett., 2022, 24, 4598–4602 CrossRef CAS PubMed.
- C. S. Yeung, X. Zhao, N. Borduas and V. M. Dong, Chem. Sci., 2010, 1, 331–336 RSC.
- K. Okamoto, N. Shida, H. Morizumi, Y. Kitano and K. Chiba, Angew. Chem., Int. Ed., 2022, 61, e202206064 CrossRef CAS PubMed.
- A. Itai, Y. Toriumi, N. Tomioka, H. Kagechika, I. Azumaya and K. Shudo, Tetrahedron Lett., 1989, 30, 6177–6180 CrossRef CAS.
- C. R. Kemnitz and M. J. Loewen, J. Am. Chem. Soc., 2007, 129, 2521–2528 CrossRef CAS PubMed.
- M. Szostak and J. Aubé, Chem. Rev., 2013, 113, 5701–5765 CrossRef CAS PubMed.
- K. L. White, M. Mewald and M. Movassaghi, J. Org. Chem., 2015, 80, 7403–7411 CrossRef CAS PubMed.
- H. Meier, Angew. Chem., Int. Ed., 1992, 31, 1399–1420 CrossRef.
- D. Drelinkiewicz, S. T. Alston, T. Durand and R. J. Whitby, React. Chem. Eng., 2023, 8, 2134–2140 RSC.
- B. C. Baciu, J. A. Verges and A. Guijarro, J. Org. Chem., 2021, 86, 5668–5679 CrossRef CAS PubMed.
- Z. Tang, K. Mo, X. Ma, J. Huang and D. Zhao, Angew. Chem., Int. Ed., 2022, 61, e202208089 CrossRef CAS PubMed.
- Z. Chang, J. Huang, S. Wang, G. Chen, H. Zhao, R. Wang and D. Zhao, Nat. Commun., 2021, 12, 4342 CrossRef CAS PubMed.
- J. W. Beatty and C. R. J. Stephenson, Acc. Chem. Res., 2015, 48, 1474–1484 CrossRef CAS PubMed.
- Z. Y. Cao, T. Ghosh and P. Melchiorre, Nat. Commun., 2018, 9, 3274 CrossRef PubMed.
- M. Silvi, C. Verrier, Y. P. Rey, L. Buzzetti and P. Melchiorre, Nat. Chem., 2017, 9, 868–873 CrossRef CAS PubMed.
- C. R. Gonçalves, M. Lemmerer, C. J. Teskey, P. Adler, D. Kaiser, B. Maryasin, L. González and N. Maulide, J. Am. Chem. Soc., 2019, 141, 18437–18443 CrossRef PubMed.
- Q. He, J. L. Ye, F. F. Xu, H. Geng, T. T. Chen, H. Chen and P. Q. Huang, J. Org. Chem., 2021, 86, 16300–16314 CrossRef CAS PubMed.
- Z. Luo, F. Wang, J. Zhang, X. Li, M. Zhang, X. Hao, Y. Xue, Y. Li, F. D. Horgen, G. Yao and Y. Zhang, J. Nat. Prod., 2012, 75, 2113–2120 CrossRef CAS PubMed.
- G. Zhan, B. Gao, J. Zhou, T. Liu, G. Zheng, Z. Jin and G. Yao, Phytochemistry, 2023, 207, 113564 CrossRef CAS PubMed.
- B. Jeffries, Z. Wang, H. R. Felstead, J. Y. L. Questel, J. S. Scott, E. Chiarparin, J. Graton and B. Linclau, J. Med. Chem., 2020, 63, 1002–1031 CrossRef CAS PubMed.
- K. Martin, C. Melan, T. Cauchy and N. Avarvari, ChemPhotoChem, 2022, 6, e202100215 CrossRef CAS.
- D. Hashemi, X. Ma, R. Ansari, J. Kim and J. Kieffer, Phys. Chem. Chem. Phys., 2019, 21, 789–799 RSC.
- N. Herrero-García, I. Fernández and J. O. Barcina, Chem.–Eur. J., 2011, 17, 7327–7335 CrossRef PubMed.
- T. Lu and F. Chen, J. Comput. Chem., 2011, 33, 580–592 CrossRef PubMed.
- Z. Liu, T. Lu and Q. Chen, Carbon, 2020, 165, 461–467 CrossRef CAS.
- G.-J. Huang, M. A. Harris, M. D. Krzyaniak, E. A. Margulies, S. M. Dyar, R. J. Lindquist, Y. Wu, V. V. Roznyatovskiy, Y.-L. Wu, R. M. Young and M. R. Wasielewski, J. Phys. Chem. B, 2016, 120, 756–765 CrossRef CAS PubMed.
- N. A. Garcia and T. Kowalczyk, J. Phys. Chem. C, 2017, 121, 20673–20679 CrossRef CAS.
- X. Zhang, X. Chen and J. Zhao, Dalton Trans., 2021, 50, 59–67 RSC.
- J. L. Garnett, M. L. Long and R. F. W. Vining, J. Am. Chem. Soc., 1972, 94, 5914–5915 CrossRef.
- Y. P. Liu, C. J. Zhu, C. C. Yu, A. E. Wang and P. Q. Huang, Eur. J. Org Chem., 2019, 7169–7174 CrossRef CAS.
- J. J. Murphy, D. Bastida, S. Paria, M. Fagnoni and P. Melchiorre, Nature, 2016, 532, 218–222 CrossRef CAS PubMed.
- J. J. Zhao, H. H. Zhang, X. Shen and S. Yu, Org. Lett., 2019, 21, 913–916 CrossRef CAS PubMed.
- B. A. Jones, P. Solon, M. V. Popescu, J. Y. Du, R. Paton and M. D. Smith, J. Am. Chem. Soc., 2023, 145, 171–178 CrossRef CAS PubMed.
- Y. G. Shi, S. K. Mellerup, K. Yuan, G. F. Hu, F. Sauriol, T. Peng, N. Wang, P. Chen and S. Wang, Chem. Sci., 2018, 9, 3844–3855 RSC.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4sc03907f |
| ‡ These authors made equal contributions to this work. |
| This journal is © The Royal Society of Chemistry 2024 |