
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceCompetitive photooxidation of small colorless organics controlled by oxygen vacancies under visible light†
Jianghui
Sun‡
ab,
Xiyang
Ge‡
a,
Yixuan
Gao
a,
Min
Zhang
a,
Qi
Zhao
a,
Guohua
Hou
 a,
Xiaoni
Wang
a,
Yiyan
Yin
a,
Jin
Ouyang
c and
Na
Na
*a
a,
Xiaoni
Wang
a,
Yiyan
Yin
a,
Jin
Ouyang
c and
Na
Na
*a
aKey Laboratory of Radiopharmaceuticals, Ministry of Education, College of Chemistry, Beijing Normal University, Beijing 100875, China. E-mail: nana@bnu.edu.cn
bState Key Laboratory of Bioactive Substance and Function of Natural Medicines, Institute of Materia Medica, Chinese Academy of Medical Sciences and Peking Union Medical College, Beijing 100050, China
cDepartment of Chemistry, College of Arts and Sciences, Beijing Normal University, Zhuhai 519087, China
First published on 23rd September 2024
Abstract
Visible-light photooxidation sensitized by surface attachment of small colorless organics on semiconductor photocatalysts has emerged as an economical method for photocatalytic synthesis or degradation. In particular, heteroatom (X = N and Cl)-containing substrates could undergo either C–N coupling or dechlorination degradation via sensitizing TiO2, but the mechanism in conducting the competitive visible-light sensitized photooxidations is still vague. Herein, the visible-light photooxidation of colorless 4-chlorobenzene-1,2-diamine (o-CAN) on TiO2 was revealed, contributing to selective C–N coupling rather than dechlorination. Oxygen vacancies (OVs) were in situ generated on the TiO2 surface, which could be dominant in weakening the Cl–Ti adsorption of o-CAN and regulating the activation of O2 for selective C–N coupling. The C–N coupling product, functionalized as the sensitizer, further promoted the visible-light photooxidation upon N–Ti and Cl–Ti coordination. This process was then confirmed by on-line mass spectrometric analysis, and the intermediates as well as their kinetics were determined. Thereby, theoretical calculations were employed to verify the roles of OVs in competitive photooxidation and lowering the energy barriers as well. Based on the comprehensive characterizations of both the catalysts and intermediates, this work has provided insights into competitive photooxidations under visible light.
Introduction
Visible light catalysis stimulated by surface attachment of small colorless organics on semiconductor photocatalysts has become an economical and environmental route for chemical transformations.1–4 The easy adsorption of heteroatom (X = O, S, N or Cl)-containing organics on TiO2 greatly promotes the visible-light photooxidation because the ensuing hybrid interfaces allow electron injection from electron rich heteroatoms to the TiO2 conduction band.5–7 With O2 as the terminal oxidant rather than undesirable sacrificial electron donors (like TEMPO),8,9 different oxidation processes would be induced via regenerating the surface-bound sensitizer molecules.10 For example, colorless benzylic amines can be oxidized into the corresponding imines via the formation of a surface complex.11 Meanwhile, the degradation of chlorinated pollutants can also be facilitated by themselves or stable photocatalytic sensitizers.12 Nevertheless, most reports normally focus on developing catalysts or sensitizers for selective photooxidation, and the competitive mechanism between C–N coupling and dechlorination degradation on sensitized catalysts is still vague. Consequently, clear insights into the photooxidations are required, including competitive adsorption of substrates, activation of oxidants (like O2), interactions of the sensitizer with the catalysts and the process of molecular transformation.Notably, native oxygen vacancies (OV) on the surface of metal oxides is an important defect to mediate catalytic reactions via ameliorating photocatalysts.13–15 For example, OVs could be reactive sites to increase charge separation/migration and regulate the adsorption of substrates. In addition, OVs also facilitate the decrease of the reaction barrier to promote substrate activation,16 intermediate formation17 or byproduct suppression.15 In particular, OVs are conducive to the adsorption and activation of O2, which could either be reduced to superoxide (O2˙−) or incorporated into the product for oxofunctionalization.18 Significantly, the generated O2˙− could induce amine oxidation through two different reaction pathways: (1) participating in the oxidative dehydrogenation to afford hydrogen peroxide (H2O2) during C–N coupling.19,20 (2) Forming hydroxyl radicals (˙OH) to initiate degradations for obtaining dechlorination products or even smaller molecules (CO2 and H2O).21,22 Consequently, strategies for creating more OVs on semiconductors have boomed, including doping, reduction and calcination. However, a report revealing the roles of OVs in visible-light sensitized competitive photooxidations is still lacking. Therefore, detailed studies of OV-controllable competitive photooxidation are required, which would be associated to the regulation of molecular adsorption and O2 activation on sensitized semiconductor photocatalysts.
Herein, the visible-light photooxidation of colorless 4-chlorobenzene-1,2-diamine (o-CAN) on a TiO2 surface was comprehensively examined by both off-line and on-line examinations. Interestingly, even in the presence of –Cl (a potential degrading groups),12o-CAN still exhibited selective C–N coupling to generate 7-chlorophenazine-2,3-diamine (7-Cl-Phz) under mild conditions,23 without necessary substrate pre-fuctionalizations.24–27 With the aid of in situ generated OVs on TiO2, the produced 7-Cl-Phz acted as the TiO2 photosensitizer to further facilitate C–N coupling rather than degradation. The selective C–N coupling was attributed to OVs via promoting interfacial electron transfer, regulating o-CAN adsorption and suppressing ˙OH generation. Furthermore, online mass spectrometry analysis was employed for structural identification and dynamic monitoring of intermediates, being conducive to revealing the roles of OVs. As demonstrated by experimental and theoretical examinations, selective C–N coupling was obtained by manipulating substrate adsorption, O2 activation and energy barriers by OVs. This work would not only promote the development of controllable and selective visible-light photooxidation, but also inspire a deeper understanding of nanocatalysis.
Results and discussion
Evaluation of o-CAN visible-light photooxidation
The photooxidation of N, Cl-containing o-CAN catalyzed by TiO2 was selected as the model for examinations. In the reaction, commercial P25–TiO2 was added into the o-CAN solution, following by the visible-light irradiation under mild conditions. Under visible-light irradiation, the colorless o-CAN (with –NH2 and –Cl groups) could be oxidized into 7-Cl-Phz via C–N coupling, or degraded into radical or 3,4-diaminophenol (DAP) in the presence of ˙OH (Fig. 1A). The reaction system was coupled with the multiphase flow of extractive electrospray ionization mass spectrometry (MF-EESI) for reaction monitoring.28 As shown in Fig. 1B, the reaction solution was suspended in a double-decker quartz beaker equipped with a temperature controller. The reaction system (RS) was then coupled to the ionization system (IS) for on-line reaction monitoring. According to our previous studies,23,29 analytes would be extracted by the cooperation of inserted capillary and high-speed N2. Simultaneously, the concentric-spray of methanol and a high DC voltage can facilitate the ionization upon decreasing the interference of TiO2.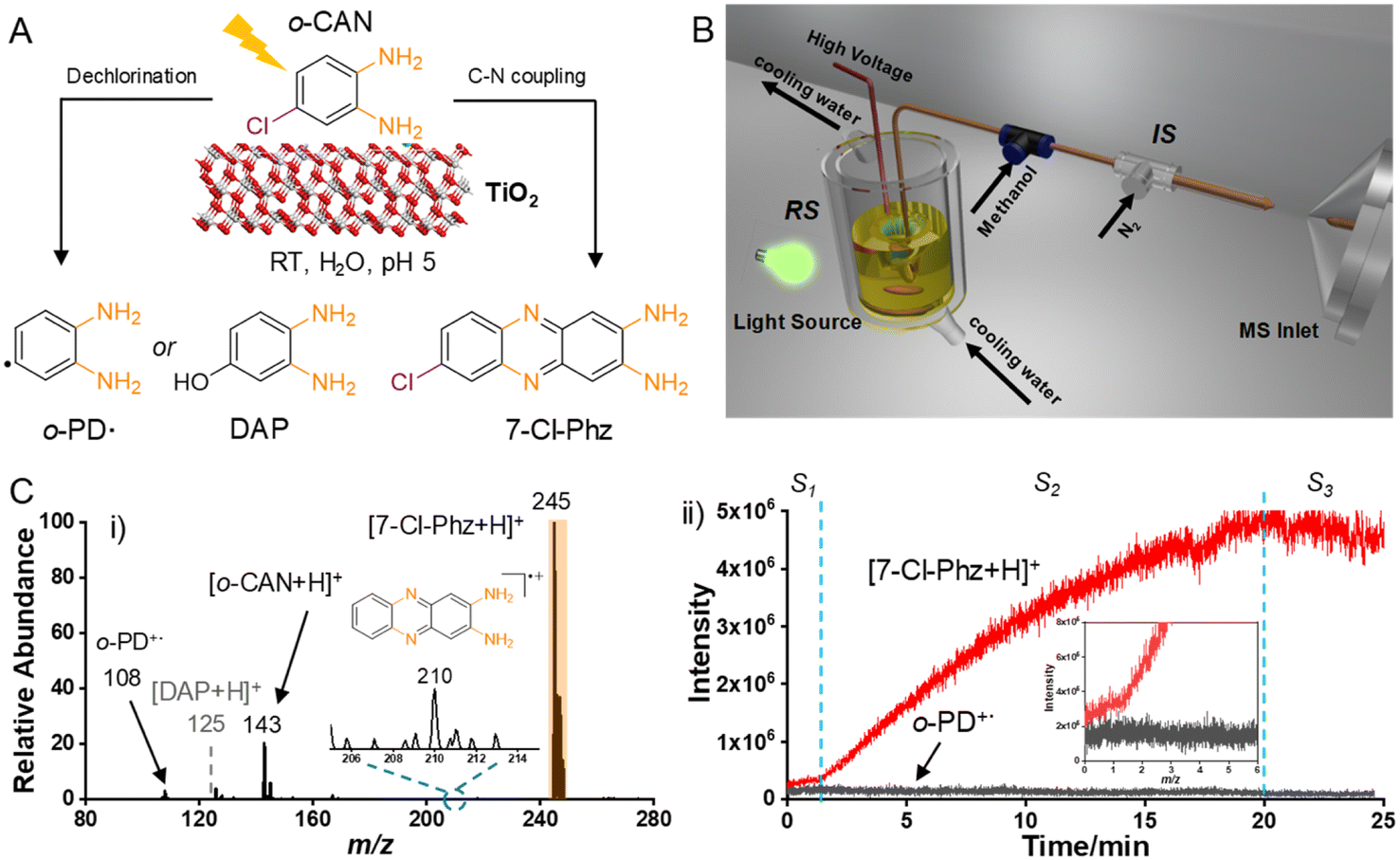 | ||
| Fig. 1 Schematic diagram and reaction evaluation via online mass spectrometry. (A) Schematic diagram of photocatalytic C–N coupling and degradation on TiO2. (B) Schematic diagram of the online reaction monitoring system by coupling the reaction system with an ionization system.28 (C) (i) Mass spectra of the reaction system after visible-light irradiation for 25 min in air. Reaction conditions: o-CAN (0.5 mM), TiO2 (1 mg mL−1), H2O (20 mL, pH = 5), and a xenon lamp (simulated sunlight, AM 1.5). (ii) EICs of the C–N coupling product of [7-Cl-Phz + H]+ and the degradation product of o-PD˙+. | ||
As demonstrated (Fig. 1C(i)), after irradiating for 25 min, the main ion peak was ascribed to the C–N coupling product of [7-Cl-Phz + H]+ at m/z 245, which was further confirmed by collision-induced dissociation (CID) (Fig. S1†) and 1H NMR (Fig. S2†) analyses. On the contrary, no significant ions associated with the degradation were observed, and nor was the dechlorinated ion of o-PD˙+ at m/z 108 (the structure was identified by MS/MS analysis in Fig. S3†) or the ˙OH-related degradation product of DAP in both the positive (m/z 125, the dashed) and negative mode (m/z 123, Fig. S4†).30 It should be noted that to avoid decomposition of the active PD˙+ before detection, the distance from the spray tip to the MS inlet was set at 0.5 cm (Fig. S5†). Besides, a relatively low abundance of reactant ion ([o-CAN + H]+ at m/z 143) was exhibited, indicating the efficient and selective C–N coupling reaction. Consequently, upon photosensitization of TiO2 under visible-light irradiation, the colorless o-CAN preferred to undergo selective C–N coupling rather than degradation.
Furthermore, the dynamic changes of the dechlorination product of o-PD˙ and C–N coupling product of 7-Cl-Phz were on-line monitored by MF-EESI. The online extracted ion chromatograms (EICs) of o-PD˙+ (m/z 108) and [7-Cl-Phz + H]+ (m/z 245) were collected for comparison. As demonstrated (Fig. 1C(ii)), the photooxidation could be divided into three stages. Before approximately 1.5 min (S1), quite low ion signals were recorded. Although a slightly higher signal of [7-Cl-Phz + H]+ than o-PD˙+ was recorded, either the degradation or C–N coupling was non-selective. Thereafter (S2), [7-Cl-Phz + H]+ dramatically increased along with the gradual decrease of o-PD˙+, followed by the kinetic equilibrium of both signals at ∼20 min. After 20 min (S3), [7-Cl-Phz + H]+ decreased slightly along with the generation of dechlorinated Phz˙+ (m/z 210, the inset of Fig. 1C(i) and S6†), while no significant change of o-PD˙+ was observed. These results indicate two possible facts: (1) the easier absorption of the N,Cl-containing sensitizer of 7-Cl-Phz on TiO2 enhanced photoinduced electron injection, while weaker conjugation of o-CAN or o-PD˙ with TiO2 disfavored the photosensitization and further degradation. (2) Degradation of o-CAN was further blocked by the accumulation of the 7-Cl-Phz sensitizer/product on TiO2.
The photooxidation of o-CAN catalyzed by TiO2 was further evaluated based on the significant UV-vis absorption of 7-Cl-Phz in the visible light region (∼445 nm, Fig. S7-A†). Compared to the absorption of the present o-CAN photooxidation (Fig. S7-B,† entry 1), a similarly high response was obtained by extra addition of 7-Cl-Phz to support photosensitization (Fig. S7-B,† entry 2). Besides, a series of control experiments were carried out to examine the efficient and selective photocatalytic C–N coupling. As demonstrated, the reaction was hardly carried out in the absence of either TiO2 or light irradiation (Fig. S7-B,† entries 3 and 4). Notably, a low yield of 7-Cl-Phz was produced without O2 (Fig. S7-B,† entry 5), indicating that O2 could be the terminal oxidant for o-CAN photooxidation.
Furthermore, to examine the intrinsic effect of the substrate for selectivity, a scope investigation with diverse structures has been carried out. As demonstrated, without a –Cl group, only the C–N coupling product was observed when using o-phenylenediamine or 4-methyl-o-phenylenediamine as the substrate (Fig. S8a and b†). Without –NH2 groups, the chlorobenzene was degraded into ketone or aldehyde (Fig. S8c†), indicating the feasibility of dechlorination degradation. Besides, both the C–N coupling product ions and corresponding degradation product ions were detected for other substrates with –Cl at different positions and –Br substitution, demonstrating the occurrence of competitive reactions (Fig. S8d and e†). When increasing the number of –Cl, the dechlorination degradation was promoted, but a small amount of C–N product ion (m/z 313) was still observed (Fig. S8f†), especially considering the steric hindrance of the –Cl groups. This demonstrated the negative role of –Cl in the C–N coupling reaction, which could be promoted by in situ generated OVs.
Consequently, the generated 7-Cl-Phz can induce dye/self-sensitization of TiO2 to facilitate the competitive photooxidation of o-CAN, which turned out to further promote the C–N coupling. However, this is different from the reports, which demonstrated that the sensitization-based visible-light catalysis of TiO2 is non-selective and could also facilitate degradation.31,32 Consequently, more detailed mechanism studies are required to reveal the competitive photooxidation mechanism in conducting the selective C–N coupling, rather than degradation.
Surface engineering of TiO2
Chemical changes of TiO2 after the photooxidation were characterized. Demonstrated by high resolution transmission electron microscopy (HR-TEM) (Fig. S9†), the particle size of TiO2 increased after the reaction, along with a decreased Brunauer–Emmett–Teller (BET) surface area (SBET). The visible light absorptions of TiO2 before and after the reaction were further examined by diffuse reflectance UV-vis (DR-UV-vis) spectra analysis. As demonstrated (Fig. 2A), a broader and extended absorption of TiO2 (from 400 to 700 nm) was recorded after the reaction. The above results indicated the adsorption of 7-Cl-Phz on TiO2 (Fig. S7-A†), which could act as the photosensitizer to broaden the spectral responses of semiconductors with a wide-band gap. This was also consistent with the reported easy absorption of heteroatom-containing dyes (or substrates) onto the TiO2 surface.33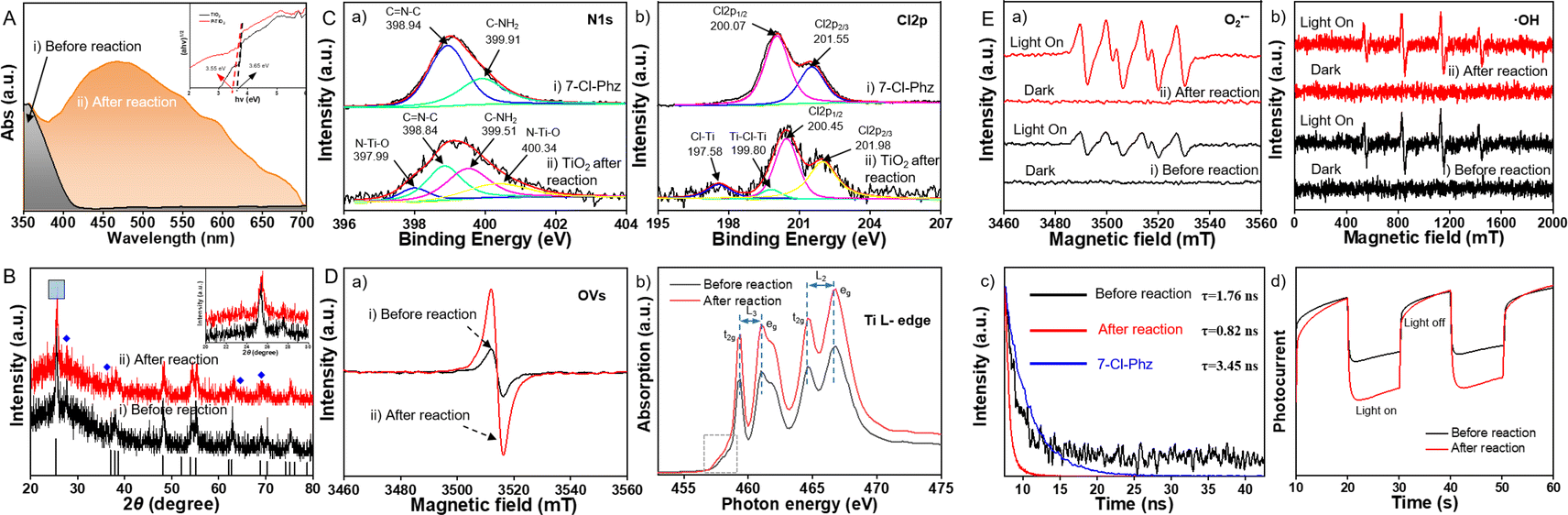 | ||
| Fig. 2 Chemical characterizations of TiO2 before and after the photooxidation. (A) DR-UV-vis spectra. (B) XRD spectra. (C) Core level HR-XPS spectra in the N 1s region (a) and Cl 2p region (b). (D) EPR spectra of OVs (a) and XAS spectra (b). (E) EPR spectra of O2˙− in the dark and under photoirradiation (a) and ˙OH in the dark and under photoirradiation (b); (c) TRPD spectra of 7-Cl-Phz and TiO2 before and after the photooxidation; (d) transient photocurrent responses. Conditions: +0.5 V versus Ag/AgCl at pH 5.0. Irradiation: 300 W Xe lamp of simulated solar light (100 mW cm−2), under chopped illumination with a fixed time interval. | ||
The strong interaction between TiO2 and 7-Cl-Phz was further confirmed by X-ray diffraction (XRD) characterizations. As illustrated in Fig. 2B, the XRD peaks were red-shifted after the photooxidation, which was attributed to a lattice expansion caused by surface modification of heteroatoms (N or Cl) into the unit cell.34 To further examine the surface chemical composition of TiO2, X-ray photoelectron spectroscopy (XPS) characterizations were employed and the corresponding evident peaks were deconvolved in the HR-XPS spectra. As demonstrated, 7-Cl-Phz exhibited the characteristic peaks of C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N–C and C–NH2 at binding energies of 398.94 and 399.91 eV in the N 1s spectrum (Fig. 2C(a)-i).35 Meanwhile, after the reaction (Fig. 2C(a)-ii), the two peaks shifted to a lower binding energy (398.84 and 399.51 eV), along with the recording of the N–Ti–O peaks (397.99 and 400.34 eV).36 Similarly, the original Cl 2p3/2 and Cl 2p1/2 peaks of 7-Cl-Phz (at 200.07 and 201.55 eV in the XPS Cl 2p spectra, Fig. 2C(b)-i) shifted to higher binding energies (at 200.45 and 201.98 eV) along with the formation of Cl–Ti and Ti–Cl–Ti bonds (197.58 and 199.80 eV, Fig. 2C(b)-ii).37,38 Besides, the characteristic peaks of reformed Ti–Cl and Ti–N–O bonds (at 457.66 and 463.17 eV) were recorded after the reaction (Fig. S10†),39,40 which was in agreement with the N 1s and Cl 2p data (Fig. 2C). Consequently, the adsorption of 7-Cl-Phz on the TiO2 surface was further confirmed upon the direct bonding of both hetero atoms (Cl and N) to Ti.
N–C and C–NH2 at binding energies of 398.94 and 399.91 eV in the N 1s spectrum (Fig. 2C(a)-i).35 Meanwhile, after the reaction (Fig. 2C(a)-ii), the two peaks shifted to a lower binding energy (398.84 and 399.51 eV), along with the recording of the N–Ti–O peaks (397.99 and 400.34 eV).36 Similarly, the original Cl 2p3/2 and Cl 2p1/2 peaks of 7-Cl-Phz (at 200.07 and 201.55 eV in the XPS Cl 2p spectra, Fig. 2C(b)-i) shifted to higher binding energies (at 200.45 and 201.98 eV) along with the formation of Cl–Ti and Ti–Cl–Ti bonds (197.58 and 199.80 eV, Fig. 2C(b)-ii).37,38 Besides, the characteristic peaks of reformed Ti–Cl and Ti–N–O bonds (at 457.66 and 463.17 eV) were recorded after the reaction (Fig. S10†),39,40 which was in agreement with the N 1s and Cl 2p data (Fig. 2C). Consequently, the adsorption of 7-Cl-Phz on the TiO2 surface was further confirmed upon the direct bonding of both hetero atoms (Cl and N) to Ti.
Subsequently, the surface defects of oxygen vacancies (OVs) were examined by electron spin resonance (ESR) and soft X-ray absorption fine structure (sXAFS) analysis. After the photocatalytic oxidation (Fig. 2D(a)), the EPR signals of the OVs on TiO2 increased significantly, indicating the in situ formation of OVs along with the absorption of the 7-Cl-Phz sensitizer. The formation of OVs could be facilitated by the abundant surface –OH, acting as Brønsted base sites for reductive deprotonation (oxidation) of aniline.41,42 Furthermore, the local electronic configuration of TiO2 was further studied by Ti L-edge. Upon photooxidation (Fig. 2D(b)), the onset energy position of the Ti L-edge of TiO2 shifted to a lower energy, suggesting a lower Ti valence state empirically.43 As demonstrated, along with the adsorption of the 7-Cl-Phz sensitizer, surface defects of OVs were in situ generated during the photooxidation of o-CAN. In fact, the generation of surface OVs was crucial for the competitive photooxidation, which was confirmed by the increased C–N coupling efficiency with increasing the concentration of OVs on the TiO2 surface (Fig. S11†).
To further examine the role of OVs, characterizations including ESR, time-resolved photoluminescence decay (TRPD) and electrochemical impedance spectroscopy (EIS) were employed. As demonstrated (Fig. 2E(a)), O2˙− was significantly generated on TiO2 after the reaction, induced by the efficient reduction of O2 upon capturing photoelectrons. Meanwhile, no significant increase of ˙OH was observed (Fig. 2E(b)), indicating that no further degradation would be employed after the generation of o-PD˙ via dichlorination of o-CAN (as illustrated in Fig. 1A). In addition to O2˙− and ˙OH, reactive oxygen species of O21 were also detected, while no signal was observed on TiO2 even after the reaction (Fig. S12†). Therefore, the in situ generated OVs could modulate O2 activation and electron transfer to promote the selective C–N coupling.
Besides, the interfacial charge transfer of TiO2 before and after the reaction was evaluated. Firstly, the TRPD spectra (Fig. 2E(c)) were collected to fit the decay traces, which demonstrated a distinctly faster decay of TiO2 after the reaction (τaverage = 0.82 ns) than that before the reaction (τaverage = 1.76 ns).44,45 Consequently, the rapid charge transfer between the absorbed 7-Cl-Phz and the conduction band (CB) of TiO2 was confirmed, which facilitated the subsequent photooxidation under visible light. Besides, the excited-state lifetime of 7-Cl-Phz was 3.45 ns, much longer than that of TiO2 before the reaction (1.76 ns). This indicated that there would be sufficient time for electron transfer from the excited state of 7-Cl-Phz to TiO2 before relaxation. Furthermore, photocurrent and electrochemical impedance were also evaluated. An increased photocurrent density of TiO2 was observed after the reaction (Fig. 2E(d)), indicating a more efficient photogenerated electron–hole separation on the interface of [7-Cl-Phz]-TiO2. The ensuing active holes on the hetero atoms would promote the subsequent photooxidation of o-CAN. Correspondingly, a decreased Rrec value of TiO2 after the reaction (from 2091 Ω to 1976 Ω) was also recorded upon fitting the Nyquist plot (Fig. S13†). Therefore, the adsorption of 7-Cl-Phz on TiO2 could alter the electronic structure and defect states to enhance charge transfer and separation. This property would be highly beneficial for efficient and selective photooxidation on semiconductor catalysts with wide-band gaps.
Theoretical examinations of OV-controlled competitive photooxidation
To explore the roles of OVs in the competition, theoretical calculation was performed by density functional theory (DFT) using the Vienna Ab Initio Simulation Package (VASP).46,47 Firstly, the adsorptions of the reactant (o-CAN) on TiO2 through both Ti–N and Ti–Cl modes without OVs, as well as that on TiO2 with abundant OVs (TiO2-OVs) were evaluated for comparison. Without OVs, o-CAN was stably adsorbed on TiO2 in both the Ti–N and Ti–Cl modes (Fig. 3A(i)). Meanwhile, in the presence of OVs, a higher adsorption energy of Ti–Cl (0.4 eV) than Ti–N (−0.13 eV) was exhibited (Fig. 3A(ii)). This result indicated that OVs could weaken the direct interaction of Ti–Cl to hinder the dichlorination. The differential charge density of o-CAN and *o-CAN on TiO2 in Ti–Cl modes further verified the limitation of Ti–Cl adsorption (Fig. S14†). Compared to the o-CAN molecule, more electron depletion (cyan) of the N atom of *o-CAN in the Ti–Cl mode was observed. This indicated the decreased electron density of the N atom after adsorption in Ti–Cl mode, which could hinder its further oxidation for selective C–N coupling. | ||
| Fig. 3 Theoretical calculation of competitive photooxidation for examining selective C–N coupling. (A) Adsorption energies of o-CAN on TiO2 (i) and TiO2-OVs (ii). (B) (i) Illustration of the dechlorination and dehydrogenation to generate ˙R1 and ˙R2, respectively. (ii) Free energy changes of dechlorination and dehydrogenation on TiO2-OVs. (C) DOS of the metal d orbital before (i) and after adsorption via Ti–N (ii) and Ti–Cl (iii). | ||
Besides, the energy change of the initial dechlorination (for oxidative degradation) and dehydrogenation (for C–N coupling) (Fig. 3B(i)) was further evaluated. As shown in Fig. 3B(ii), the energy change of degradation (to release ˙R1) is much higher (0.07 eV) than that of the C–N coupling (to release ˙R2) (−0.93 eV). Meanwhile, higher energy barriers for both the degradation (2.3 eV) and C–N coupling (2.0 eV) are produced when catalyzed by TiO2 without OVs (Fig. S15†), further confirming the effect of OVs on selectivity. Consequently, promoted by the in situ generated OVs, the oxidative dehydrogenation of o-CAN (for C–N coupling) is more favorable than the dechlorination (for oxidative degradation). Subsequently, to investigate the photoinduced charge transfer (PICT) between the 7-Cl-Phz sensitizer and TiO2, DFT was employed to calculate the electronic density of states (DOS) of TiO2 before and after the photooxidation. As shown, direct bonding of 7-Cl-Phz via both Ti–N (Fig. 3C(ii)) and Ti–Cl (Fig. 3C(iii)) rendered smaller band gaps than that of the crystal counterpart (Fig. 3C(i)). Although the DFT results of semiconductor structures are qualitative assessments, it is still sufficient to predict the enhancement of PICT by the bonding between 7-Cl-Phz and TiO2.
Reaction monitoring and kinetic studies
To explore the molecular changes and the generation of intermediates, the photooxidation was monitored by in situ Raman spectroscopy.48–51 As shown in Fig. 4A, upon 1 min of irradiation, only typical Raman bands (B1g(1), A1g + B1g(2), and Eg(2)) of TiO2 were recorded. This indicated the weak bonding between the substrate (o-CAN) and TiO2 with lower visible-light absorption and inefficient electron transfer. After 3 min irradiation, the peaks of νCCl (1026 cm−1), νNH (1229 cm−1) and νCN (1580 cm−1) increased upon the accumulation of the 7-Cl-Phz product. Thereafter, a new peak at 1518 cm−1 appeared and increased gradually, which was attributed to the formation of quinonoid intermediates. In addition, a shoulder peak of C∼N˙+ (∼ denotes a bond intermediate between a single and double bond) at 1330 cm−1 was also observed, attributed to the stretching vibration of polaronic structures. Significantly, half-oxidized phenazine-like units were also demonstrated by the strong peak at 1402 cm−1, which could be assigned to the free radical precursor of 7-Cl-Phz.52,53 Consequently, the quinonoid intermediates, C∼N˙+ intermediates and half-oxidized phenazine-like radical precursors were preliminarily confirmed to be involved in the photooxidation. | ||
| Fig. 4 Reaction monitoring. (A) In situ Raman spectroscopic monitoring of photooxidation at different times. (B) Mass spectra of the photooxidation reaction at different times via on-line MS monitoring. (C) EICs of [1+H]+ at m/z 143 (i), intermediate ions of 1˙+ (m/z 142) (ii), [2+H]+ (m/z 141) (iii), [3+H]+ (m/z 283) (iv), [4+H]+ (m/z 281) (v), [6+H]+ (m/z 249) (vi), and [5+H]+ (m/z 247) (vii). (D) Gibbs free energy diagrams for 1 → 3 → 4 → 5 → 7 on TiO2 and TiO2-OVs, respectively. | ||
To directly obtain the molecular information during the selective C–N coupling, intermediate structures, dynamics of reactants, intermediates and products were determined by MS detections. As indicated by the MS spectra at the interval times (Fig. 4B(i)), only the reactant ion of o-CAN ([1+H]+) at m/z 143 was observed without light irradiation. Upon visible light irradiation (Fig. 4B(ii)), the product ion of 7-Cl-Phz ([7+H]+) at m/z 245 came to be observed, which would act as the antenna molecule for visible-light absorption. In addition, the multiple ions attributed to the protonated dimers of o-CAN were observed (Fig. 4B(iii–v)), including the ions at m/z 283 ([3+H]+, measured m/z: 283.0517, theoretical m/z 283.0512, error +0.6 ppm), m/z 281 ([4 + H]+, measured m/z: 281.0363, theoretical m/z 281.0355, error +0.79 ppm), m/z 249 ([6+H]+, measured m/z: 249.0910, theoretical m/z 249.0902, error +1.7 ppm), and m/z 247 ([5+H]+, measured m/z: 247.0757, theoretical m/z 247.0745, error +2.6 ppm). The corresponding structures are shown in the insets of Fig. 4B, which were confirmed via CID experiments (Fig. S16 and S17†). The generation of these intermediates was further confirmed by separating the corresponding ion peaks from these isobars (Fig. S18†). Besides, the ions of C∼N˙+ and the quinonoid intermediates at m/z 142 and 141 were also recorded upon light irradiation, in accordance with the Raman data in Fig. 4A. The corresponding structures were further confirmed by CID experiments (Fig. S19†). Significantly, the recorded ions exhibited different dynamic changes with time-dependent ratios of the relative abundance, which required further dynamic examinations by on-line monitoring.
Thereafter, the kinetics of the dye/self-sensitized photooxidation on TiO2 were online monitored by MF-EESI MS and the extracted ion chromatograms (EICs) of different ions were collected. As shown in Fig. 4C(i), the reactant ion of [1+H]+ at m/z 143 decreased along with the reaction taking place. Meanwhile, the ions of 1˙+ (m/z 142) and [2+H]+ (m/z 141) increased immediately within 0.4 min (Fig. 4C(ii and iii)), which were generated from the rapid oxidation of 1 to form 1˙+ and then to 4-chlorocyclohexa-3,5-diene-1,2-diimine (2). Subsequently, the ions of 1˙+ and [2+H]+ gradually decreased, which indicated that both ions could be important intermediates of the photooxidation. Interestingly, along with the relatively rapid decrease of [2+H]+ (Fig. 4C(iii)), [3+H]+ (m/z283) increased gradually until 6.5 min (Fig. 4C(iv)). Simultaneously, [4+H]+ (m/z 281) increased dramatically and was rapidly consumed after 2.8 min (Fig. 4C(v)). Both ions of [3+H]+ and [4+H]+ could be assigned as the intermediates converted from 2. Simultaneously, the ion of [6+H]+ (m/z 249) also exhibited a gradual increase and decrease after ∼3 min, indicating another potential pathway with 6 as the intermediate (Fig. 4C(vi)). Along with the generation of [4+H]+ and [6+H]+, [5+H]+ increased dramatically and was rapidly consumed after 3.2 min (Fig. 4C(vii)), which induced the generation of the product. Therefore, dynamic changes of the above intermediates involved in different potential pathways were provided for mechanism examination.
To further study the effect of OVs on the selective C–N coupling, the free energy diagrams of the main route 1 → 3 → 4 → 5 → 7 on TiO2 and TiO2-OVs were calculated. The optimized structures of these intermediates were also determined (Fig. 4D). On TiO2 (the black line in Fig. 4D), for generating 3 upon reaction between the adsorbed o-CAN (1) and intermediate 2, the energy change is −0.92 eV. Subsequently, 4 is obtained via intramolecular dehydrogenation of 3 with a potential barrier of 1.60 eV. Then, electrophilic substitution of -Cl via –NH2 can form intermediate 5 with the energy change of −1.75 eV. Finally, the product of 7-Cl-Phz (7) is obtained by further dehydrogenation of 5 through an exothermic process. While in the presence of OVs, a lower Gibbs free energy for obtaining each intermediate is observed (the red line in Fig. 4D). In particular, the energy barrier of the step from intermediate 3 to intermediate 4 on TiO2-OVs (1.03 eV) is lower than that of TiO2 (1.60 eV), indicating that vacancies can reduce the energy barrier of this step. Therefore, except for promoting electron transfer, regulating molecular adsorption, modulating O2 and substrate activation, the in situ generated OVs can also lower the potential barriers of intermediate formation.
Mechanism of OV-controlled competitive photooxidation
On the basis of both experimental and theoretical examinations, the mechanism of the OV-controlled selective C–N coupling is proposed in three routes (Fig. 5). Initially, the substrate (1) could be adsorbed on the surface, resulting in weak coordinations via N–Ti or Cl–Ti. This allows electron injection from 1 to the conduction band (CB) of TiO2 to give 1˙+ (m/z 142). Notably (in Route I), the intermolecular hole transfer between 1˙+ and neutral 1 would be initiated to afford 7-Cl-Phz (7) along with the in situ generation of OVs. Simultaneously, the heteroatom-containing 7-Cl-Phz product can be easily adsorbed onto TiO2via surface coordination, acting as the sensitizer for the subsequent reactions. This dye-sensitization process would produce a new electron donor level above the VB of TiO2 from the 2p orbital of Cl or N, facilitating broader visible-light absorption than the self-sensitization between o-CAN and TiO2. Upon light irradiation, positive charge (h+) could be left at the heteroatoms after injecting electrons into the CB, which would ultimately transfer to O2 to form O2˙−. Subsequently, the intermolecular hole transfer between 7˙+ and surface adsorbed 1 (mainly by N–Ti) would afford 1˙+ and regenerate the surface-bound sensitizer 7, fulfilling the cyclic dye-sensitization of TiO2. | ||
| Fig. 5 Mechanism of OV-controlled selective C–N coupling. | ||
Thereafter (in Route II), 1˙+ would be oxidized by O2˙− (obtained in Route I) to generate 2 (m/z 141)54 and H2O2. Subsequently, 2 reacts with neutral 1 to generate 3 (m/z 283). Then, in the presence of H2O2, 3 undergoes intramolecular oxidative dehydrogenation to form 4 (m/z 281), followed by cyclization to form intermediate 5 (m/z 247). Further oxidation of 5 would afford the half-oxidized phenazine (Fig. 4A), which acts as the precursor of the product 7.55 In addition (in Route III), 2 could also react with neutral 1 to generate intermediate 6 (m/z 249). Subsequently, 5 would be generated upon the oxidation and cyclization of 6, which supports the obtaining of product 7 as Route II indicated. Therefore, upon the absorption of the substrate and product on OV-containing TiO2, intermolecular hole transfer would be initiated to facilitate the sensitization and step-by-step C–N coupling. This clearly reveals the mechanism of efficient and selective C–N coupling via photooxidation under visible light, even using small colorless organics as the reactant.
Conclusion
In conclusion, in situ generated OVs on TiO2 significantly facilitate the competitive visible-light photooxidation of colorless 4-chlorobenzene-1,2-diamine for selective C–N coupling rather than dechlorination degradation. By on-line MS examinations, the structural identification and dynamic monitoring of intermediates were successfully employed. This greatly supported the evaluation of interactions between the catalyst and reaction species or generated sensitizer, as well as examinations of energy changes upon molecular adsorption and transformation. The C–N coupling product was easily adsorbed on TiO2, which extended the light absorption via surface coordination. The surface OVs enhanced the interfacial charge transfer to promote selective C–N coupling via regulating the adsorption of the substrate and sensitizer, lowering the energy barrier and modulating O2 activation on TiO2. Based on this work, two general rules of visible light catalysis for photooxidation of small colorless organics could be drawn: (1) the synthetic product containing abundant heteroatoms can act as a better sensitizer than the substrate to exhibit higher reaction efficiency. (2) Oxidation of the substrates could be mediated by regulating the concentration of OVs on the semiconductor catalyst. This work has initiated the comprehensive examination of both the catalyst and reaction species, which would promote the development of visible light catalysis.Data availability
The data supporting this article have been included as part of the ESI.†Author contributions
J.-H. S. and N. N. designed the project. J.-H. S. performed the experiments and wrote the manuscript. X.-Y. G. contributed to the theoretical calculations and the revision of the manuscript. Y.-X. G., M. Z. and Q. Z. participated in the characterizations of the catalyst. G.-H. H., X.-N. W., Y.-Y. Y. and J. O. contributed to the mechanism studies. All authors have given approval to the final version of the manuscript. N. N. directed the research.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We gratefully acknowledge the National Natural Science Foundation of China (NNSFC 22274012, 22474010), the Fundamental Research Funds for the Central Universities (No. 2233300007) and the Key Project of Science and Technology Plan of Beijing Education Commission (No. KZ20231002807).Notes and references
- X. Lang, X. Chen and J. Zhao, Chem. Soc. Rev., 2014, 43, 473–486 RSC.
- S. P. Pitre, J. C. Scaiano and T. P. Yoon, ACS Catal., 2017, 7, 6440–6444 CrossRef CAS.
- X. Xie, Z. Zhang, Y. Hu and H. Cheng, Chem. Eng. J., 2018, 334, 1242–1251 CrossRef CAS.
- D. Shi, R. Zheng, M.-J. Sun, X. Cao, C.-X. Sun, C.-J. Cui, C.-S. Liu, J. Zhao and M. Du, Angew. Chem., Int. Ed., 2017, 56, 14637–14641 CrossRef CAS PubMed.
- J. Zoller, D. C. Fabry and M. Rueping, ACS Catal., 2015, 5, 3900–3904 CrossRef CAS.
- X. Lang, W. Hao, W. R. Leow, S. Li, J. Zhao and X. Chen, Chem. Sci., 2015, 6, 5000–5005 RSC.
- Z. Cao, T. Zhang, P. Ren, D. Cao, Y. Lin, L. Wang, B. Zhang and X. Xiang, Catalysts, 2020, 10, 69 CrossRef CAS.
- M. Zhang, C. Chen, W. Ma and J. Zhao, Angew. Chem., Int. Ed., 2008, 47, 9730–9733 CrossRef CAS PubMed.
- X. Lang and J. Zhao, Chem.–Asian J., 2018, 13, 599–613 CrossRef CAS PubMed.
- X. Lang, W. Ma, C. Chen, H. Ji and J. Zhao, Acc. Chem. Res., 2014, 47, 355–363 CrossRef CAS.
- X. Lang, W. Ma, Y. Zhao, C. Chen, H. Ji and J. Zhao, Chem. Eur J., 2012, 18, 2624–2631 CrossRef CAS PubMed.
- W. Zhao, Y. Sun and F. N. Castellano, J. Am. Chem. Soc., 2008, 130, 12566–12567 CrossRef CAS PubMed.
- C. Mao, H. Cheng, H. Tian, H. Li, W.-J. Xiao, H. Xu, J. Zhao and L. Zhang, Appl. Catal., B, 2018, 228, 87–96 CrossRef CAS.
- W. Gao, G. Li, Q. Wang, L. Zhang, K. Wang, S. Pang, G. Zhang, L. Lv, X. Liu, W. Gao, L. Sun, Y. Xia, Z. Ren and P. Wang, Chem. Eng. J., 2023, 464, 142694 CrossRef CAS.
- X. Wei, X. Wen, Y. Liu, C. Chen, C. Xie, D. Wang, M. Qiu, N. He, P. Zhou, W. Chen, J. Cheng, H. Lin, J. Jia, X.-Z. Fu and S. Wang, J. Am. Chem. Soc., 2022, 144, 11530–11535 CrossRef CAS.
- K. Rajendran, R. Madampadi, S. Shee, R. Subramaniam, T. S. Khan, S. Gupta, M. A. Haider and D. Jagadeesan, Chemcatchem, 2023, 15, e202301048 CrossRef CAS.
- A. Liu, X. Liu, Y. She, X. Hu, M. Hu, Z. Zhang, X. Wang and B. Liu, Green Chem., 2023, 25, 8633–8644 RSC.
- X. Lang, J. Zhao and X. Chen, Angew. Chem., Int. Ed., 2016, 55, 4697–4700 CrossRef CAS PubMed.
- A. Han, J. Sun, H. Zhang, G.-K. Chuah and S. Jaenicke, Chemcatchem, 2019, 11, 6425–6430 CrossRef CAS.
- G. He, Y. Lai, Y. Guo, H. Yin, B. Chang, M. Liu, S. Zhang, B. Yang and J. Wang, ACS Appl. Mater. Interfaces, 2022, 14, 53724–53735 CrossRef CAS.
- M. Chen and W. Chu, Chem. Eng. J., 2014, 248, 273–279 CrossRef CAS.
- B. Szczepanik, P. Slomkiewicz, D. Widel, M. Czaplicka and L. Frydel, Catalysts, 2021, 11, 1548 CrossRef CAS.
- J. Sun, X. Fan, H. Lu, H. Tan, Y. Zhang, Y. Wang, Y. Zhao, J. Ouyang and N. Na, Chem. Commun., 2021, 57, 3921–3924 RSC.
- I. J. Pachter and M. C. Kloetzel, J. Am. Chem. Soc., 1951, 73, 4958–4961 CrossRef CAS.
- I. J. Pachter and M. C. Kloetzel, J. Am. Chem. Soc., 1952, 74, 971–973 CrossRef CAS.
- Y. Lian, J. R. Hummel, R. G. Bergman and J. A. Ellman, J. Am. Chem. Soc., 2013, 135, 12548–12551 CrossRef CAS PubMed.
- Y. Xiao, X. Wu, H. Wang, S. Sun, J.-T. Yu and J. Cheng, Org. Lett., 2019, 21, 2565–2568 CrossRef CAS.
- Y. Wang, M. Sun, J. Qiao, J. Ouyang and N. Na, Chem. Sci., 2018, 9, 594–599 RSC.
- W. Li, J. Sun, Y. Wang, J. Qiao, L. He, J. Ouyang and N. Na, Chem. Commun., 2021, 57, 2955–2958 RSC.
- H. Lu, Y. Yin, J. Sun, W. Li, X. Shen, X. Feng, J. Ouyang and N. Na, Chin. Chem. Lett., 2021, 32, 3457–3462 CrossRef CAS.
- L. Yu, X. Zhang, G. Li, Y. Cao, Y. Shao and D. Li, Appl. Catal., B, 2016, 187, 301–309 CrossRef CAS.
- Y.-H. Chiu, T.-F. M. Chang, C.-Y. Chen, M. Sone and Y.-J. Hsu, Catalysts, 2019, 9, 430 CrossRef CAS.
- S. Mathew, A. Yella, P. Gao, R. Humphry-Baker, B. F. E. Curchod, N. Ashari-Astani, I. Tavernelli, U. Rothlisberger, M. K. Nazeeruddin and M. Graetzel, Nat. Chem., 2014, 6, 242–247 CrossRef CAS.
- X. Liu, L. Yin, D. Ren, L. Wang, Y. Ren, W. Xu, S. Lapidus, H. Wang, X. He, Z. Chen, G.-L. Xu, M. Ouyang and K. Amine, Nat. Commun., 2021, 12, 4235 CrossRef CAS PubMed.
- P. Kumar, E. Vahidzadeh, W. K. Thakur, P. Kar, K. M. Alam, A. Goswami, N. Mandi, K. Cui, G. M. Bernard, V. K. Michaelis and K. Shankar, J. Am. Chem. Soc., 2019, 141, 5415–5436 CrossRef CAS.
- F. Dong, W. Zhao, Z. Wu and S. Guo, J. Hazard. Mater., 2009, 162, 763–770 CrossRef CAS.
- P. Huang, Z. Cheng, L. Zeng, J. Yu, L. Tan, P. Mohapatra, L.-S. Fan and Y. Zhu, ACS Catal., 2020, 10, 14928–14935 CrossRef CAS.
- L. P. Delgado, M. Z. Figueroa-Torres, M. C. Ceballos-Chuc, R. Garcia-Rodriguez, J. J. Alvarado-Gil, G. Oskam and G. Rodriguez-Gattorno, Mater. Sci. Eng., C, 2020, 117, 111290 CrossRef CAS.
- S. Hu, F. Li, Z. Fan and J. Gui, Chem. Eng. J., 2014, 236, 285–292 CrossRef CAS.
- L. Yan, G. Chen, S. Tan, M. Zhou, G. Zou, S. Deng, S. Smirnov and H. Luo, ACS Appl. Mater. Interfaces, 2015, 7, 24212–24217 CrossRef CAS.
- C. Liu, W. Guo, J. Chen, J. Zou, Z. Wang and L. Wu, Catal. Sci. Technol., 2021, 11, 162–170 RSC.
- Y. Song, H. Wang, S. Liang, Y. Yu, L. Li and L. Wu, J. Catal., 2018, 361, 105–115 CrossRef CAS.
- G. Jia, Y. Wang, X. Cui, H. Zhang, J. Zhao, L. H. Li, L. Gu, Q. Zhang, L. Zheng, J. Wu, Q. Wu, D. J. Singh, W. Li, L. Zhang and W. Zheng, Matter, 2022, 5, 206–218 CrossRef CAS.
- J. Yang, J. Jing, W. Li and Y. Zhu, Adv. Sci., 2022, 9, 2201134 CrossRef CAS PubMed.
- Y. Gao, X. Su, J. Zhang, H. Tan, J. Sun, J. Ouyang and N. Na, Small, 2021, 17, 2103773 CrossRef CAS.
- G. Kresse and J. Furthmuller, Comput. Mater. Sci., 1996, 6, 15–50 CrossRef CAS.
- Q. Gao, X. Zhuang, S. Hu and Z. Hu, J. Phys. Chem. C, 2020, 124, 4644–4651 CrossRef CAS.
- M. Radoicic, Z. Saponjic, I. A. Jankovic, G. Ciric-Marjanovic, S. P. Ahrenkiel and M. I. Comora, Appl. Catal., B, 2013, 136, 133–139 CrossRef.
- M. Radoicic, Z. Saponjic, J. Nedeljkovic, G. Ciric-Marjanovic and J. Stejskal, Synth. Met., 2010, 160, 1325–1334 CrossRef CAS.
- M. Radoicic, Z. Saponjic, G. Ciric-Marjanovic, Z. Konstantinovic, M. Mitric and J. Nedeljkovic, Polym. Compos., 2012, 33, 1482–1493 CrossRef CAS.
- R. C. Quintanilha, E. S. Orth, A. Grein-Iankovski, I. C. Riegel-Vidotti and M. Vidotti, J. Colloid Interface Sci., 2014, 434, 18–27 CrossRef CAS.
- Z. Yu, Y. Park, L. Chen, B. Zhao, Y. M. Jung and Q. Cong, ACS Appl. Mater. Interfaces, 2015, 7, 23472–23480 CrossRef CAS PubMed.
- A. Malinauskas, M. Bron and R. Holze, Synth. Met., 1998, 92, 127–137 CrossRef CAS.
- T. Nogami, T. Hishida, M. Yamada, H. Mikawa and Y. Shirota, Bull. Chem. Soc. Jpn., 1975, 48, 3709–3714 CrossRef CAS.
- R. Khattar, A. Yadav and P. Mathur, Spectrochim. Acta, Part A, 2015, 142, 375–381 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4sc04531a |
| ‡ These authors contributed equally. |
| This journal is © The Royal Society of Chemistry 2024 |