
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceConsecutive chirality transfer: efficient synthesis of chiral C,O-chelated BINOL/gold(III) complexes for asymmetric catalysis and chiral resolution of disubstituted BINOLs†
Kwok-Heung Aries
Chan
 a,
Wa-Yi
O
a,
Jia-Jun
Jiang
b,
Jian-Fang
Cui
*c and
Man-Kin
Wong
*a
a,
Wa-Yi
O
a,
Jia-Jun
Jiang
b,
Jian-Fang
Cui
*c and
Man-Kin
Wong
*a
aState Key Laboratory of Chemical Biology and Drug Discovery, Research Institute for Future Food, Department of Food Science and Nutrition, The Hong Kong Polytechnic University, Hung Hom, Hong Kong, China. E-mail: mankin.wong@polyu.edu.hk
bDepartment of Applied Biology and Chemical Technology, The Hong Kong Polytechnic University, Hung Hom, Hong Kong, China
cSchool of Science and Engineering, The Chinese University of Hong Kong, Shenzhen, Guangdong 518172, China. E-mail: cuijf@cuhk.edu.cn
First published on 12th September 2024
Abstract
A novel approach for efficient synthesis of chiral C,O-chelated BINOL/gold(III) complexes by diastereomeric resolution using enantiopure BINOL as a chiral resolving agent was demonstrated. The BINOL/gold(III) diastereomers with different solubility were separated by simple filtration, providing optically pure BINOL/gold(III) complexes with up to >99![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 dr. By combining this with an efficient BINOL ligand dissociation process, a simple and column-free method for chiral resolution of racemic gold(III) dichloride complexes on a gram scale was established, affording their enantiopure forms in good yields. Conversely, the resolved enantiopure gold(III) dichloride complexes could serve as chiral resolving agents to resolve disubstituted BINOL derivatives, achieving both BINOLs and gold(III) complexes in good to excellent yields (overall 77–96% and 76–95%, respectively) with a high optical purity of up to 99% ee. Through a consecutive chirality transfer process, the chiral information from an inexpensive chiral source was transferred to highly valuable gold(III) complexes, followed by sterically bulky BINOL derivatives. This work would open a new synthetic strategy facilitating the development of structurally diverse chiral gold(III) complexes and gold(III)-mediated chiral resolution of BINOL derivatives. In addition, this new class of C,O-chelated BINOL/gold(III) complexes achieved asymmetric carboalkoxylation of ortho-alkynylbenzaldehydes with an excellent enantioselectivity of up to 99% ee.
:1 dr. By combining this with an efficient BINOL ligand dissociation process, a simple and column-free method for chiral resolution of racemic gold(III) dichloride complexes on a gram scale was established, affording their enantiopure forms in good yields. Conversely, the resolved enantiopure gold(III) dichloride complexes could serve as chiral resolving agents to resolve disubstituted BINOL derivatives, achieving both BINOLs and gold(III) complexes in good to excellent yields (overall 77–96% and 76–95%, respectively) with a high optical purity of up to 99% ee. Through a consecutive chirality transfer process, the chiral information from an inexpensive chiral source was transferred to highly valuable gold(III) complexes, followed by sterically bulky BINOL derivatives. This work would open a new synthetic strategy facilitating the development of structurally diverse chiral gold(III) complexes and gold(III)-mediated chiral resolution of BINOL derivatives. In addition, this new class of C,O-chelated BINOL/gold(III) complexes achieved asymmetric carboalkoxylation of ortho-alkynylbenzaldehydes with an excellent enantioselectivity of up to 99% ee.
Introduction
Gold catalysis has been rapidly developed in the past few decades because of the exceptional π-Lewis acidity of gold complexes and the outstanding reactivity in a wide range of organic transformations with selective functionalization of C–C multiple bonds.1 In contrast, the initial development of asymmetric gold catalysis was limited due to the linear geometry of chiral gold(I) complexes leading to a long distance between the chiral ligand and active site causing inefficient chiral induction.1d,g,2 Over the past few years, new strategies involving novel chiral phosphine and N-heterocyclic carbene ligands for chiral gold(I) complexes have been developed.2,3 The use of axially chiral digold complexes, chiral counterions, bulky chiral ligands or helical ligands, and merging gold with organocatalysts have overcome the challenges by extending the chiral pocket surrounding the active site to achieve high enantioselectivity.4Compared to the extensively studied asymmetric gold(I) catalysis, the development of asymmetric gold(III) catalysis and gold(I)/gold(III) redox catalysis is still in its early stages.5 Asymmetric gold(III) catalysis takes advantage of the square planar geometry of gold(III) complexes allowing high efficiency in chiral induction by positioning the chiral ligands and substrates in close proximity. However, the high oxidation potential of gold(III) species leads to instability towards reductive processes. Therefore, the development of stable and reactive gold(III) complexes is still challenging.6 Several pioneering studies have demonstrated the potential of using chiral gold(III) complexes in asymmetric catalysis, including the work by Toste's group and our group (Scheme 1a).7,8 Despite these advances, enantioselectivity above 90% ee was rarely achieved in most of the examples.5,6b Thus, it is of ongoing interest to develop novel chiral gold(III) complexes for achieving high enantioselectivity in asymmetric catalysis. On the other hand, Patil and Shi independently reported the examples of ligand-enabled enantioselective redox gold(I)/gold(III) catalysis using chiral hemilabile (P,N)-ligands, which demonstrated potential applications in enantioselective 1,2-difunctionalization of alkenes with 99% ee and 98% ee respectively.9,10
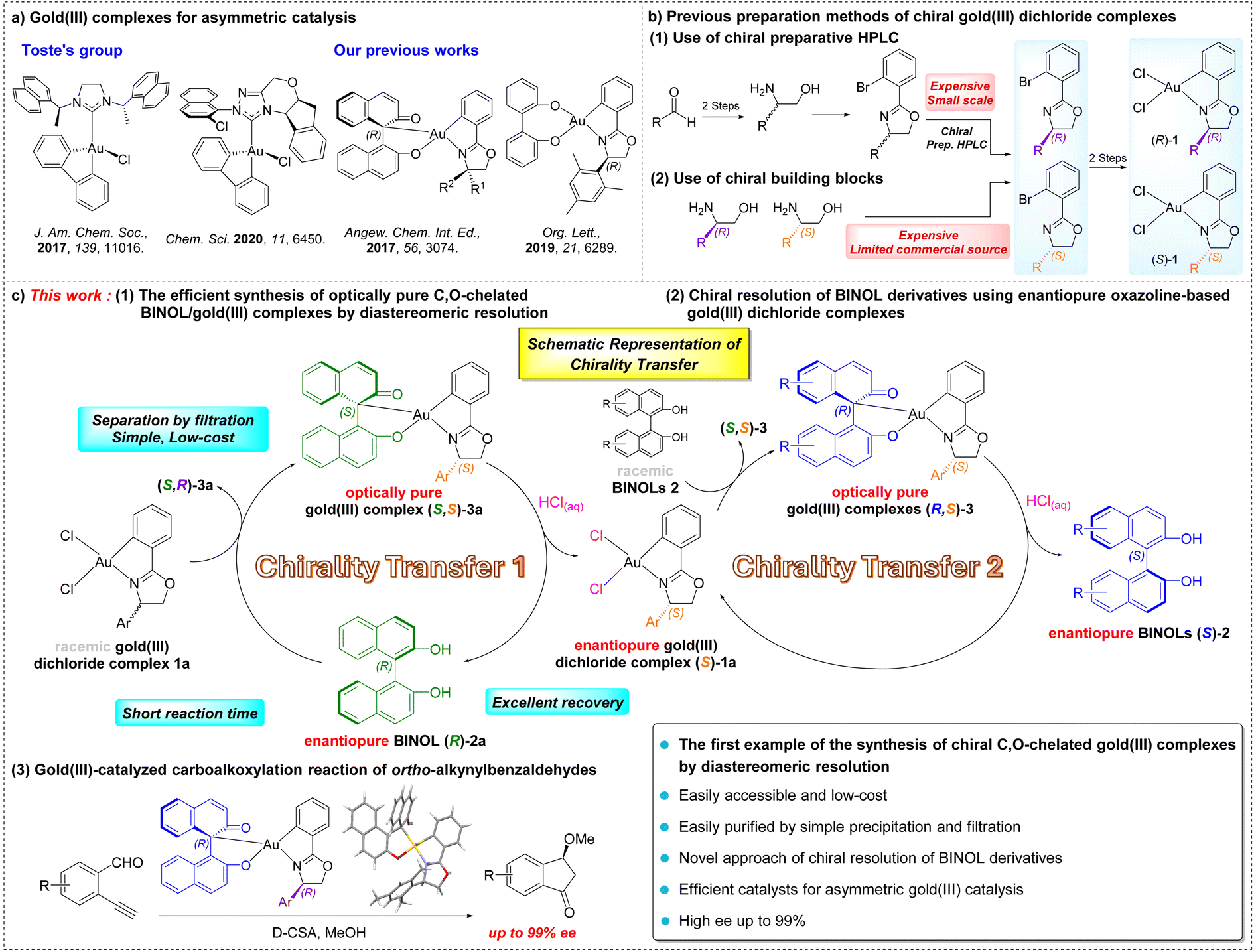 | ||
| Scheme 1 (a) Gold(III) complexes for asymmetric catalysis; (b) previous preparation methods of chiral gold(III) dichloride complexes; (c) this work: (1) the efficient synthesis of optically pure C,O-chelated BINOL/gold(III) complexes by diastereomeric resolution, (2) chiral resolution of BINOL derivatives using enantiopure oxazoline-based gold(III) dichloride complexes, and (3) gold(III)-catalyzed carboalkoxylation reaction of ortho-alkynylbenzaldehydes. | ||
Over the years, we have developed a series of chiral C,O-chelated and O,O′-chelated cyclometalated gold(III) complexes for asymmetric catalysis (Scheme 1a). In 2017, we first developed a series of novel chiral C,O-chelated BINOL/gold(III) cyclometalated complexes providing 41% ee in enantioselective carboalkoxylation of 2-ethnylbenzaldehyde.11 In 2019, we reported a class of 4,4′-biphenol derived O,O′-chelated chiral oxazoline gold(III) complexes achieving a high enantioselectivity of up to 90% ee in asymmetric carboalkoxylation of alkynes.8 In our studies of asymmetric catalysis using O,O′-chelated chiral gold(III) complexes, we found that the gold(III) complex with a 2,4,6-trimethylphenyl group on the chiral oxazoline moiety afforded the product with the highest enantioselectivity (90% ee). This result indicated that the sterically bulky substituent on the chiral oxazoline moiety was important for chiral induction. In the preparation of these cyclometalated chiral oxazoline gold(III) complexes, the chiral oxazoline ligands were obtained either using chiral preparative HPLC for enantiomeric separation of racemates or using chiral amino alcohols as the starting materials (Scheme 1b). However, both methods are expensive and time-consuming due to the use of high-cost HPLC grade solvent and chiral reagents that limit large-scale synthesis. To further explore the potential of this class of cyclometalated chiral gold(III) complexes in asymmetric catalysis, the development of an easily accessible synthetic pathway is of importance.
1,1′-Binaphthalene-2,2′-diol (BINOL) and its derivatives are an essential class of chiral compounds with their representative axial chirality and have been studied extensively as O,O′-ligands in asymmetric catalysis.12,13 To obtain chiral BINOL, various approaches for chiral resolution of racemic BINOL have been developed, such as enzymatic resolution, kinetic resolution, and chemical resolution by forming a variety of diastereomeric salts with chiral auxiliaries.14,15 In contrast, the reverse process of using chiral BINOL in enantioselective separation is rarely reported, in which the literature examples are only limited to the enantioselective ammonium recognition by enantiopure BINOLs for producing chiral ammonium cations.16,17 The use of enantiopure BINOLs for chiral resolution of racemic gold(III) complexes by separating diastereomeric complexes remains unexplored.
Given the potential application of enantiopure BINOLs in chiral resolution, we envision that the combination of enantiopure BINOL and racemic gold(III) dichloride complexes bearing sterically bulky substituents would provide a novel synthetic strategy achieving optically pure C,O-chelated BINOL/gold(III) complexes for asymmetric catalysis. As shown in Scheme 1c, we report herein (1) the efficient synthesis of optically pure C,O-chelated BINOL/gold(III) complexes by diastereomeric resolution. Treatment of the racemic oxazoline-based gold(III) dichloride complexes (rac)-1a with the enantiopure BINOL 2a, an inexpensive chiral source, in the presence of Cs2CO3 in MeOH afforded a diastereomeric gold(III) mixture. Notably, the diastereomers were resolved through simple precipitation and filtration due to their different solubility in MeOH, giving optically pure BINOL/gold(III) complexes 3a in good yields. To our knowledge, we are the first to synthesize chiral C,O-chelated gold(III) complexes by diastereomeric resolution of racemic gold(III) complexes using enantiopure BINOLs. Moreover, enantiopure chiral oxazoline-based gold(III) dichloride complexes, for example (S)-1a, could be obtained in good yields upon acid treatment. With this novel resolution strategy, an efficient and column-free process for chiral resolution of (rac)-1a on a gram scale accessing optically pure gold(III) complexes 1a and 3 was developed (Chirality Transfer 1). (2) Chiral resolution of BINOL derivatives using enantiopure (S)-1a as a chiral resolving agent provided optically active BINOLs in good to excellent yields (overall 77–96%) with a high optical purity of up to 99% ee (Chirality Transfer 2). (3) In addition, asymmetric carboalkoxylation of ortho-alkynylbenzaldehydes using this new class of chiral C,O-chelated BINOL/gold(III) complexes afforded an excellent enantioselectivity of up to 99% ee.
Results and discussion
Synthesis of optically pure C,O-chelated BINOL/gold(III) complexes from racemic gold(III) dichloride complexes by diastereomeric resolution using chiral BINOL
We initiated the study through the reaction of chiral BINOL with the oxazoline-based gold(III) dichloride complex bearing a 2,4,6-trimethylphenyl group (Scheme 2a). Treatment of the racemic oxazoline-based gold(III) dichloride complexes (rac)-1a with the enantiopure BINOL (S)-2a in the presence of Cs2CO3 in MeOH for 1 h afforded a diastereomeric gold(III) mixture. Interestingly, the diastereomers (R,R)-3a and (R,S)-3a exhibited different solubility in MeOH. (R,S)-3a was highly soluble in MeOH, while (R,R)-3a showed poor solubility in MeOH. Given its poor solubility in MeOH, (R,R)-3a easily formed precipitates that were collected and washed with MeOH and H2O to obtain an optically pure product in 43% yield. The filtrate was collected and purified by column chromatography to obtain optically pure (R,S)-3a in 42% yield. The structure of the C,O-chelated gold(III) complexes was confirmed by 1H NMR, 13C NMR, ESI-MS and circular dichroism (see details in the ESI†). The structure of (R,R)-3a was further confirmed by X-ray crystallography (CCDC 2355500). | ||
| Scheme 2 (a) Synthesis of optically pure C,O-chelated gold(III) complexes by diastereomeric resolution using enantiopure BINOL (S)-2a; (b) 1H NMR analysis for diastereomeric resolution of (R)-1a using BINOL (S)-2a as a chiral resolving agent. | ||
To examine the chiral resolving ability on (rac)-1a, we performed the BINOL ligand dissociation of (R,R)-3a to generate (R)-1a. (R,R)-3a was dissolved in DCM/MeOH, followed by treatment with 6 N HCl for 1 min. An immediate color change of the reaction mixture from red to colorless could be easily observed (Scheme S1†). The resulting solution was dried and subjected to column chromatography, affording (S)-2a and enantiopure oxazoline-based gold(III) dichloride complex (R)-1a in excellent yields of 95% and 91%, respectively. Notably, (S)-2a retained optical purity with >99.5% ee upon the central-to-axial chirality transfer after the acid treatment. This result indicated that the enantiopure BINOL ligand could be recovered without the loss of optical purity throughout the whole process.
The successful synthesis of optically pure C,O-chelated BINOL/gold(III) complexes from a racemic gold(III) source and the regeneration of enantiopure (R)-1a indicated that the enantiopure BINOL was useful for resolving racemic 1a. To provide insights into this novel synthetic strategy, we studied the role of BINOL in separating enantiomers by 1H NMR analysis. As indicated in the 1H NMR spectrum of the crude reaction mixture, a pair of well separated peaks corresponding to the BINOL/gold(III) diastereomers could be easily observed for determining the diastereomeric ratio (dr) of 1:1. By filtration, (R,R)-3a obtained in the precipitate gave dr > 99:1, while (R,S)-3a obtained in the filtrate gave dr = 72:28 in the 1H NMR spectrum (Scheme 2b). The enantiomeric excess (ee) of the chiral gold(III) dichloride complex 1a obtained after the acid treatment was directly reflected by the dr of the resolved BINOL/gold(III) complexes. These results indicated that the BINOL ligand served as a chiral resolving agent for (rac)-1a, making the diastereomers distinguishable in 1H NMR analysis.
Column-free chiral resolution of (rac)-1a on a gram scale to obtain optically pure gold(III) complexes 1a and 3a
Next, we further developed a column-free method for the chiral resolution of (rac)-1a to obtain optically pure gold(III) complexes 1a and 3a (Scheme 3). Initially, the BINOL/gold(III) complex 3a was prepared using (rac)-1a (1.0 g, 1.878 mmol) and (S)-2a (590 mg, 2.066 mmol). By filtration, (R,R)-3a and (R,S)-3a were collected separately and the diastereomeric ratio was determined to be >99:1 and 72:28, respectively, by 1H NMR analysis. (R,R)-3a was then treated with 6 N HCl for 1 min. The resulting solution was concentrated, followed by the addition of MeOH for precipitation. The precipitate was collected, affording enantiopure (R)-1a with 99% ee. Similarly, (R,S)-3a was treated with HCl to afford (S)-1a with 44% ee. To obtain highly optically pure (S)-1a, we performed the reaction of (S)-1a (44% ee) with (R)-2a under standard conditions, affording (S,S)-3a with 99:1 dr and (S,R)-3a with 78:22 dr. Treatment of (S,S)-3a with HCl provided enantiopure (S)-1a with 98% ee. As a result, a total of 70% yield of the enantiopure gold(III) dichloride complexes (R)-1a and (S)-1a was obtained without using column chromatography purification. The used BINOLs (S)-2a and (R)-2a could also be recovered by column purification with >95% yield and no loss of optical purity. With the enantiopure gold(III) dichloride complexes (R)-1a and (S)-1a in hand, the other two gold(III) complexes (S,R)-3a and (R,S)-3a could be synthesized with high optical purity by coupling with the corresponding BINOLs.
 | ||
| Scheme 3 A column-free process for chiral resolution of (rac)-1a to successively obtain optically pure gold(III) complexes 1a and 3a. | ||
Chiral resolution of racemic BINOL derivatives enabled by enantiopure oxazoline-based gold(III) dichloride complexes
Axially chiral biaryls, including BINOL and its derivatives showing the most representative structure with axial chirality, have shown wide application in asymmetric catalysis.12,18 The substitution of BINOL largely affects the steric environment as well as the chiral environment, especially for organocatalysts derived from bulky 3,3′-disubstituted BINOL motifs achieving high enantioselectivity.18,19 Thus, it is of great interest in the preparation of enantiopure substituted BINOL derivatives, including asymmetric oxidative coupling reactions, modification of the enantiopure BINOL scaffold, chiral resolution, enzymatic hydrolysis/esterification and central-to-axial chirality transformations.12a,14,19b,20,21,22 Although a wide range of methods have been developed for the enantioselective resolution of simple BINOLs, efficient approaches to access enantiopure BINOL derivatives are still limited. Most examples demonstrated the chiral resolution of BINOL with substituents at 4- to 7-positions, yet studies on resolving BINOL with substituents at the 3-position remain sparse.14We have previously demonstrated the use of chiral gold(III) complexes for the optical resolution of 1,16-dihydroxytetraphenylene (DHTP).23 The reported strategy was only applicable with benzyl-substituted chiral oxazoline gold(III) complexes, while the use of phenyl-, isopropyl- or tertiary butyl-substituted analogs did not lead to the optical resolution of DHTP. These findings implied that the substituent on the chiral oxazoline ligand was critical for the formation of C,O-chelated gold(III) complexes with different solubility. Therefore, we reasoned that the difference in the solubility of the diastereomers (R,R)-3a and (R,S)-3a originated from the 2,4,6-trimethylphenyl group on the oxazoline ligand. Given the efficient synthesis of 3a using the diastereomeric resolution strategy, we envisioned that the enantiopure gold(III) dichloride complex 1a would be useful for chiral resolution of diverse racemic BINOL derivatives.
We used (S)-1a as the chiral resolving agent to resolve several disubstituted BINOL derivatives (Scheme 4). The coupling of enantiopure (S)-1a and racemic BINOL 2b bearing a 4-(2-naphthyl)phenyl group on the 3,3′-position was performed in the presence of Cs2CO3 and MeOH for 1 h, affording a crude diastereomeric mixture of BINOL/gold(III) complex 3b. The diastereomeric pair (R,S)-3b and (S,S)-3b was resolved using a hexane/acetone solvent system and separated by simple filtration. The filtration residue and filtrate were then respectively treated with 6 N HCl, followed by column purification. The resulting BINOL (R)-2b and (S)-2b were afforded in excellent yields (overall 96% with respect to 1 equiv. of racemic 2b) with high optical purity (96% and 99% ee respectively). After the whole chiral resolution process, (S)-1a was successfully recovered in 95% yield.
 | ||
| Scheme 4 Chiral resolution of BINOL derivatives using (S)-1a. | ||
Using a similar approach by changing the solvent system, we further resolved 3,3′-disubstituted BINOL 2c–2e bearing 2-naphthyl, 3,5-bis(trifluoromethyl)phenyl and methyl substituents, respectively (see details in the ESI†). Optically pure (S)-2c, (S)-2d and (S)-2e were obtained in 36–40% yields (with respect to 1 equiv. of racemic 2) with excellent optical purities (85–99% ee), while the corresponding enantiomeric pairs (R)-2c, (R)-2d and (R)-2e were obtained in 40–56% yields with 50–98% ee. The chiral resolving ability of (S)-1a on 6,6′-disubstituted BINOL was also demonstrated. (rac)-2f was resolved to provide (R)-2f and (S)-2f in good yield (overall 90%) with 80% ee and 96% ee, respectively. These results indicated that the difference in solubility of the diastereomeric pairs (R,S)-3 and (S,S)-3 was significantly influenced by the substituents on the BINOL skeleton, especially for BINOL derivatives 2b and 2c bearing bulky 4-(2-naphthyl)phenyl and 2-naphthyl groups on the 3,3′-position providing high optical purity of both R and S forms. Among all the resolution processes, the gold(III) complex (S)-1a as the chiral resolving agent was well recovered with 76–95% yield. To our knowledge, this is the first example of chiral resolution of disubstituted BINOL derivatives using enantiopure gold(III) dichloride complexes. Compared to the literature reported chemical resolution methods for BINOL derivatives, which generally required overnight or even several days,14,19b our method reported herein only required a quite short reaction time (around 1 h). Notably, the chirality of (S)-1a was transferred initially from enantiopure BINOL 2a to gold(III) complex 1a, and finally to BINOL derivatives 2b–2f. This consecutive chirality transfer process was fast and simple with excellent enantioselectivity and yields.
Catalytic activity of the sterically hindered chiral C,O-chelated BINOL/gold(III) complexes
In our previous study on the C,O-chelated BINOL/gold(III) complexes, we have demonstrated that one of the chiral BINOL/gold(III) complexes bearing an isopropyl substituent on the oxazoline ligand could achieve asymmetric catalysis.11 Followed this our further study on the chiral O,O′-chelated gold(III) complexes revealed that the enantioselectivity of the catalytic products was highly affected by the steric bulkiness of the substituents on chiral oxazoline ligands.8 Along this direction, we set out to investigate the ligand effect of the chiral BINOL/gold(III) complexes on enantioselectivity. In line with our previously reported designs of chiral oxazoline ligands, the reactions of enantiopure BINOL (S)-2a with the chiral gold(III) dichloride complexes bearing different bulky substituents on the chiral oxazoline ligands were performed, affording chiral C,O-chelated BINOL/gold(III) complexes (R,R)-3g, (R,S)-3h, (R,R)-3i, and (R,R)-3j in 78–85% yields (see details in the ESI†).With these new chiral catalysts, we studied their catalytic activity for carboalkoxylation of ortho-alkynylbenzaldehydes. Given that the gold(III) complex (R,R)-3a could be easily prepared and purified by simple filtration, it was used as a catalyst for optimizing the reaction conditions (Table 1). 2-Ethynylbenzaldehyde 4a was selected as the model substrate for the reaction. Without the addition of an activator, the catalyst was inactive in the reaction (entry 1). We commenced the initial optimization studies with the catalyst (5 mol%) and by varying the acid (2.5 mol%). No desired product was determined in the presence of acetic acid (entry 2). The desired product 5a was obtained with 22–54% yield and 75–89% ee (entries 3–6). D-Camphorsulfonic acid (D-CSA) was found to be the most efficient acid giving 5a with 89% ee (entry 4) and was selected for further studies.
|
|
||||||
|---|---|---|---|---|---|---|
| Entry | Acid | Catalyst loading (mol%) | Acid loading (mol%) | Catalyst/acid ratio | Yieldb (%) | eec (%) |
| a Reaction conditions: 0.2 mmol of substrate 4a and 2 mL of MeOH. b Isolated yield. c Determined by chiral HPLC. | ||||||
| 1 | — | 5 | 0 | — | n.d. | — |
| 2 | AcOH | 5 | 2.5 | 2:1 |
n.d. | — |
| 3 | L-CSA | 5 | 2.5 | 2:1 |
38 | 80 |
| 4 | D -CSA | 5 | 2.5 |
2:1 |
47 | 89 |
| 5 | TsOH | 5 | 2.5 | 2:1 |
54 | 75 |
| 6 | BINOL-PO2H | 5 | 2.5 | 2:1 |
22 | 75 |
| 7 | D-CSA | 5 | 1.25 | 4:1 |
6 | 80 |
| 8 | D-CSA | 5 | 5 | 1:1 |
45 | 85 |
| 9 | D-CSA | 5 | 7.5 | 1:1.5 |
50 | 83 |
| 10 | D-CSA | 5 | 25 | 1:5 |
14 | 77 |
| 11 | D-CSA | 2.5 | 1.25 | 2:1 |
22 | 80 |
| 12 | D-CSA | 10 | 5 | 2:1 |
37 | 88 |

We examined the effect of acid loading on enantioselectivity by changing the catalyst:acid ratio from 4:1 to 1:5 (Table 1, entries 7–10). When 1.25 mol% D-CSA was added, the desired product 5a was obtained with low yield but high enantioselectivity (6% yield and 80% ee) (entry 7). With 2.5 mol%, 5 mol% and 7.5 mol% of D-CSA, the product yields and enantioselectivities were improved to 45–50% yield and 83–89% ee (entries 4 and 8–9). When the catalyst:acid ratio was further changed to 1:5 (25 mol% D-CSA), the product yield and enantioselectivity dropped to 14% and 77%, respectively (entry 10).
Keeping the catalyst:acid ratio at 2:1 (which afforded the highest enantioselectivity of 89% ee), the effect of varying the catalyst loading was also investigated (entries 11 and 12). Reducing the catalyst loading to 2.5 mol% led to a decrease in yield and enantioselectivity (22% yield and 80% ee) (entry 11). Further increase of catalyst loading to 10 mol% did not improve either the yield or enantioselectivity (37% yield and 88% ee) (entry 12). These results indicated that the D-CSA loading slightly affected the enantioselectivity but significantly influenced the product yield. The study of the effect of the chirality of CSA on the reaction enantioselectivity is given in ESI.† Overall, the use of 5 mol% catalyst and 2.5 mol% acid with a 2:1 catalyst:acid ratio gave the highest enantioselectivity (89% ee, entry 4), which would be optimal for further investigations.
To understand the reaction progress and the product formation, we performed a kinetic profile study of the carboalkoxylation of 4a by 1H NMR analysis (Scheme 5). As indicated in the 1H NMR spectrum of the reaction mixture of 4a with D-CSA (2.5 mol%) in CD3OD, a trace amount (<1%) of 4a was observed while its acetal form 4a′ was the major species in the reaction mixture (>99%) (Scheme 5a). Notably, only 11% of 4a′ was found in CDCl3, which implied that 4a′ was in situ generated in CD3OD with D-CSA. With an assumption that 4a was fully converted to its acetal form 4a′ in the catalytic reaction, we set 4a′ as 100% at t = 0 in the kinetic profile. After the addition of catalyst (R,R)-3a, the conversion of 4a′ and the formation of product 5a were monitored over time. The result showed that 4a′ was almost completely consumed after 4 h. The competing reaction (i.e., the formation of hydration product 6a) limited the formation of 5a, leading to low yield in the reaction (Scheme 5b).
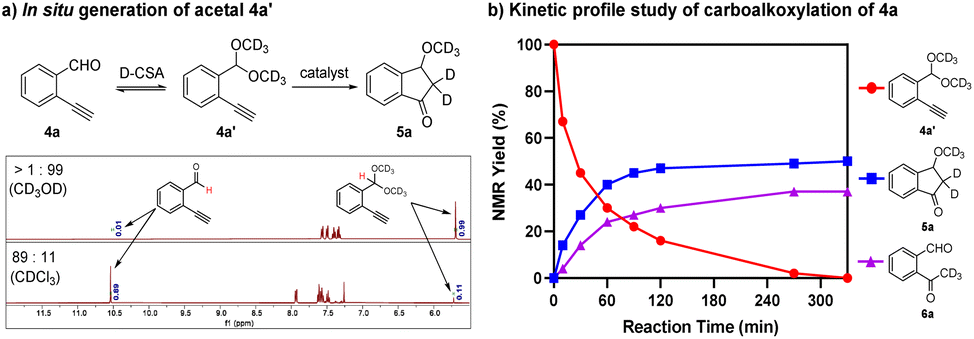 | ||
| Scheme 5 (a) In situ generation of acetal 4a′; (b) kinetic profile study of carboalkoxylation of 4a. | ||
With the optimized reaction conditions, we investigated the ligand effect on the product enantioselectivity (Table 2). We first used 5 mol% of the four enantiomers/diastereomers (R,R)-3a, (R,S)-3a, (S,R)-3a and (S,S)-3a with 2.5 mol% D-CSA to catalyze the reactions at room temperature. The desired products were obtained with high enantioselectivity (89% ee, −88% ee, 80% ee and −89% ee, respectively) (entries 1–4). Notably, the gold(III) complexes (R,R)-3a and (R,S)-3a with opposite configurations on the oxazoline moiety gave similar but opposite ee values (89% ee and −88% ee, respectively) (entries 1 and 2). Similarly, this opposite chiral induction was also observed by (S,R)-3a and (S,S)-3a, which afforded 80% ee and −89% ee respectively (entries 3 and 4). These results indicated that the chiral induction of the reaction was dominant by the chiral oxazoline ligand of the gold(III) catalysts, while the configuration of the BINOL ligand showed little effect on the enantioselectivity. Using the four enantiomers/diastereomers as catalysts, substrate 4a was completely consumed after stirring for 16 h as observed by TLC, giving 25–47% product yields (entries 1–4). The 1H NMR yield analysis of these results is shown in Table S2.† To gain more insights into the structural effects of the BINOL moiety, we selected gold(III) catalysts (R,S)-3b, (S,S)-3b, (R,S)-3c, (S,S)-3c, (R,S)-3d, and (R,S)-3f, which were obtained from the chiral resolution of racemic BINOL derivatives with high dr, for further study (entries 5–10). Compared with (R,S)-3a and (S,S)-3a, the activities of these catalysts were relatively low. Particularly, (R,S)-3d showed fast colour change of the reaction mixture within 10 min under optimized conditions, giving the product 5a with 38% yield and −83% ee (entry 9). To improve the activation efficiency, we increased the acid loading for the rest of the catalysts (entries 5–8 and 10). With increased acid loading to 10 mol% D-CSA and reaction time to 40 h, (R,S)-3b was the most unreactive catalyst that gave low conversion of the reaction, affording the product with a low yield and ee (11% yield and −35% ee) (entry 5). This result may be caused by the extremely poor solubility of (R,S)-3b in MeOH. Full conversions were observed using catalysts (S,S)-3b, (R,S)-3c, (S,S)-3c, and (R,S)-3f in the presence of 5 mol% D-CSA, providing product 5a with moderate yields and high ee values (27–43% yield and −75 to −90% ee) (entries 6–8 and 10). Despite the unreactive (R,S)-3b, these results further implied that the substituents and configuration of the BINOL moiety did not have a large effect on the enantioselectivity, but had a significant impact on the activation process of the catalysts.
|
|
|||||
|---|---|---|---|---|---|
| Entry | Catalyst | Temp (°C) | Reaction time (h) | Yieldb (%) | eec (%) |
| a Reaction conditions: 5 mol% of different catalysts, 2.5 mol% of D-CSA, 0.2 mmol of substrate 4a, and 2 mL of MeOH. b Isolated yield. c Determined by chiral HPLC. d 10 mol% of D-CSA. e 5 mol% of D-CSA. | |||||
| 1 | (R,R)-3a | r.t. | 16 | 47 | 89 |
| 2 | (R,S)-3a | r.t. | 16 | 37 | −88 |
| 3 | (S,R)-3a | r.t. | 16 | 25 | 80 |
| 4 | (S,S)-3a | r.t. | 16 | 41 | −89 |
| 5d | (R,S)-3b | r.t. | 40 | 11 | −35 |
| 6e | (S,S)-3b | r.t. | 16 | 43 | −87 |
| 7e | (R,S)-3c | r.t. | 16 | 43 | −89 |
| 8e | (S,S)-3c | r.t. | 16 | 35 | −90 |
| 9 | (R,S)-3d | r.t. | 16 | 38 | −83 |
| 10e | (R,S)-3f | r.t. | 16 | 27 | −75 |
| 11 | (R,R)-3g | r.t. | 16 | 34 | 92 |
| 12 | (R,S)-3h | r.t. | 16 | 32 | −40 |
| 13 | (R,R)-3i | r.t. | 16 | 46 | 13 |
| 14 | (R,R)-3j | r.t. | 16 | 21 | 9 |
| 15 | (R,R)-3a | 4 | 16 | 49 | 92 |
| 16 | (R,R)-3a | −20 | 40 | 36 | 96 |
| 17 | (R,S)-3a | −20 | 40 | 30 | −96 |
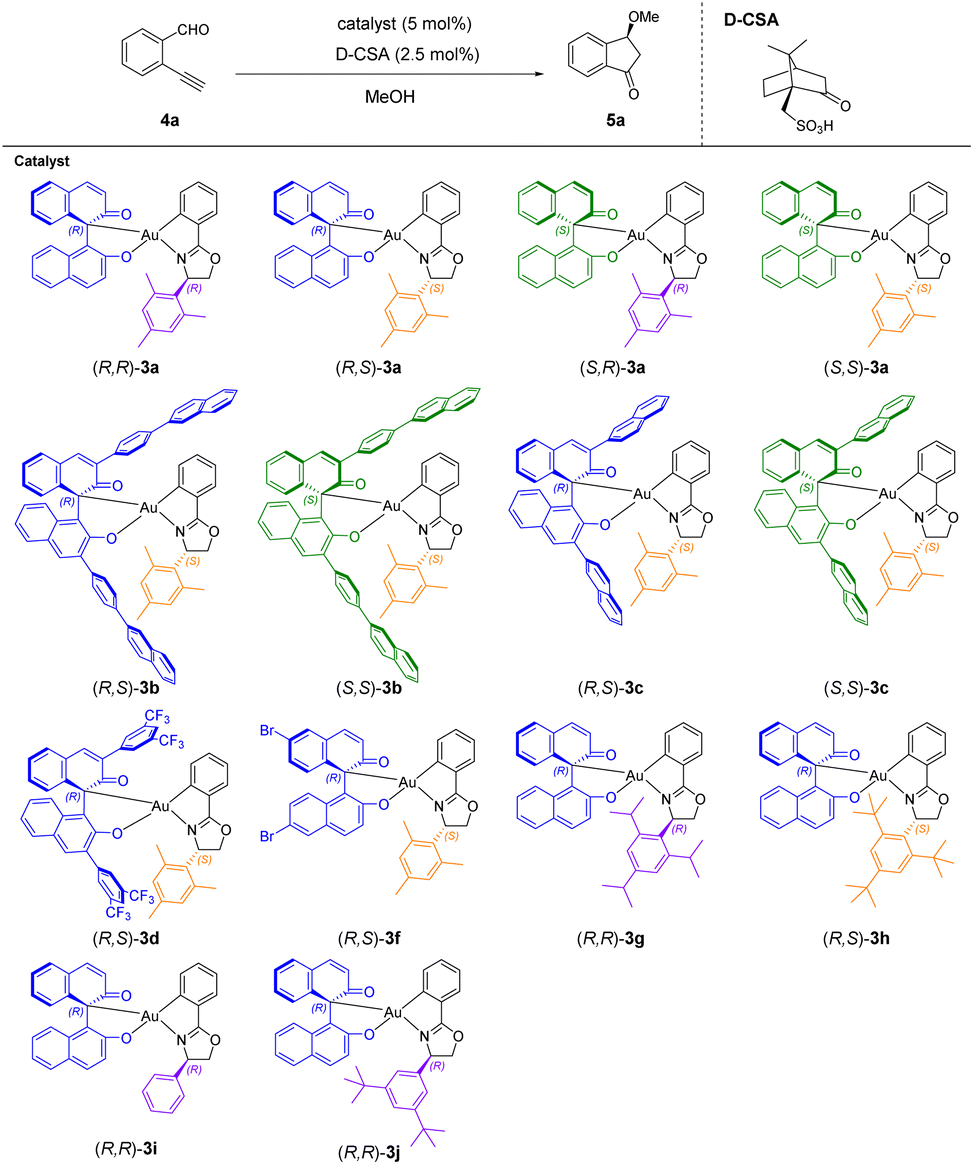
In view of the fact that the configuration of the oxazoline ligand determined the optical rotation direction of the product (Table 2, entries 1–4), we further examined the steric effects of the oxazoline moiety on enantioselectivity by modifying the substituents of the aryl group attached to the chiral oxazoline ring (entries 11–14). With the increased steric bulkiness of the ortho- and para-substituents of the aryl group on the oxazoline moiety, (R,R)-3g with a 2,4,6-triisopropylphenyl group afforded an improved enantioselectivity of 92% ee under the optimized conditions (entry 11). Compared with (R,S)-3a providing −88% ee, (R,S)-3h with a more bulky tert-butyl substituent significantly reduced the enantioselectivity to −40% ee (entry 12). (R,S)-3i with a simple phenyl group on the oxazoline moiety gave a low enantioselectivity of 13% ee (entry 13). These results implied that the chiral induction could be improved by increasing the steric bulkiness of the substituents on the aryl group, but the enantioselectivity started to level off with exceptionally bulky substituents. In addition, the steric effect of the meta-substituent of the aryl group was also studied. (R,R)-3j with a tert-butyl substituent in the meta-position provided a poor enantioselectivity of 9% ee (entry 14), which was comparable with the 13% ee of (R,R)-3i (entry 13). This result indicated that the meta-substituent was possibly too far away for interacting with the incoming substrate in the enantiodetermining step during the reaction. As slight changes in the ortho-substituent significantly affected enantioselectivity, the ortho-substituent of the aryl group was possibly the key component influencing the enantioselectivity (entries 1 and 11–12). Next, we examined the temperature effect on the enantioselectivity using catalyst (R,R)-3a under optimized conditions (entries 15 and 16). When the reaction was cooled to 4 °C, a similar yield was obtained while the enantioselectivity was slightly improved (49% yield and 92% ee) (entry 15). We further reduced the reaction temperature to −20 °C. Incomplete conversion of the substrate 4a was observed by TLC after 16 h due to the low reaction temperature. The reaction time was extended to 40 h, which afforded the desired product 5a with 36% yield and 96% ee (entry 16). Using catalyst (R,S)-3a for the reaction at −20 °C also gave an enhanced enantioselectivity of −96% ee (entry 17), indicating that the product enantioselectivity was temperature-dependent.
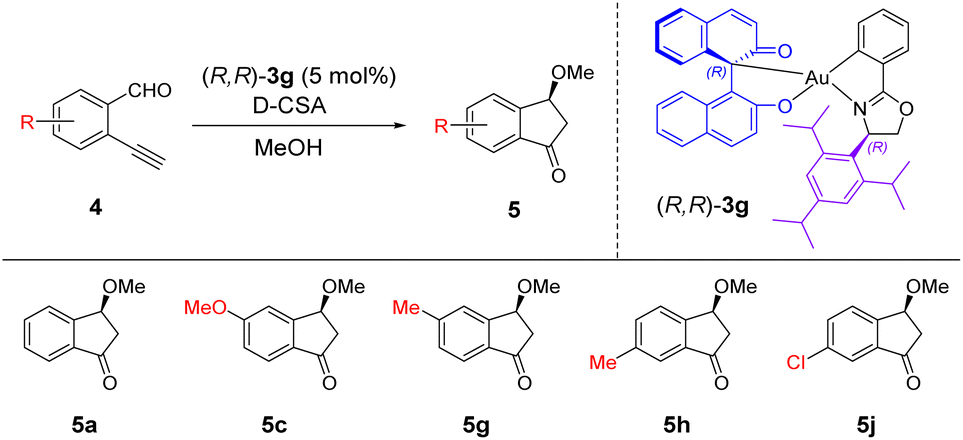
As (R,R)-3a exhibited high catalytic activity in the carboalkoxylation of 4a, it was selected as the catalyst for the expansion of substrate scope (Table 3). Under the optimized conditions, we first performed the carboalkoxylation of mono-substituted 2-ethynylbenzaldehyde 4b–4i with an electron donating group affording the corresponding products 5b–5i with 23–72% isolated yield and 11–85% ee. These results were comparable with those of the NMR yield analysis, in which products were obtained in good yields up to 83% (yields in parentheses). The wide range of ee values indicated that the enantioselectivity was significantly affected by the positions of the substituents on the substrates. Products 5b, 5e, 5f and 5i, with substituents on 3- and 6-positions, were obtained with low enantioselectivity (11–43% ee). The enantioselectivity was improved when the reaction was performed at −20 °C, giving products with up to 72% ee. In contrast, products 5c, 5d, 5g and 5h, with substituents on 4- and 5-positions, were afforded with moderate to high enantioselectivity of 50–85% ee under optimized conditions, and excellent enantioselectivity of 82–96% ee at a reaction temperature of −20 °C. These results suggested that the substituents adjacent to the aldehyde and ethynyl groups of the substrates reduced the ee values of products significantly, while the para-substituents gave relatively higher ee. Considering that the 4-substituted products provided relatively high yields and enantioselectivities, we screened substrates 4j–4k bearing an electron withdrawing group at the 4-position. Product 5j was obtained in low yield (10%) with high enantioselectivity (80% ee), while product 5k was not determined, revealing that the electron withdrawing group on the substrate significantly affected the reaction. The disubstituted substrates 4l and 4m were also screened for the reaction, giving moderate yields and poor enantioselectivities (62–84% yield and 7–33% ee). When EtOH was used as the solvent, the corresponding products 5n and 5o were obtained with low yields and moderate enantioselectivities (20–32% yield and 63–67% ee). The explanation of the product yields is given in the ESI.†
| a Reaction conditions: 5 mol% of (R,R)-3a, 2.5 mol% of D-CSA, 0.2 mmol of different substrates, and 2 mL of R2OH. b Isolated yield. c % ee values of products were determined by chiral HPLC. d Yields were determined by 1H NMR spectroscopy with 1,3,5-trimethoxybenzene as an internal standard. e −20 °C, and 40 h. |
|---|

|
With the above promising results, we screened the substrates using (R,R)-3g as the catalyst for further improving the enantioselectivity. As (R,R)-3g was less active in the carboalkoxylation reaction, we only selected substrates 4a, 4c, 4g, 4h and 4j for the reaction to examine its catalytic performance (Table 4). We first conducted the reaction using 4a with 5 mol% of (R,R)-3g and 2.5 mol% of D-CSA at −20 °C, but the desired product was not determined (entry 1). Surprisingly, product 5a was obtained with an excellent enantioselectivity of 99% ee when the acid loading was increased to 5 mol% (entry 2). Using the same reaction conditions, products 5c, 5g and 5h were obtained in low to moderate yields with excellent enantioselectivities (16–52% yield and 92–99% ee, entries 3–5), while product 5j bearing an electron withdrawing group was not determined (entry 6). We performed the reaction at room temperature for substrate 4j and obtained the product with low yield and excellent enantioselectivity (17% yield and 92% ee, entry 7). These results suggested that the oxazoline ligand of the gold(III) catalyst with a more bulky isopropyl substituent on the aryl ring possibly reduced the product yield but improved the enantioselectivity.
|
|
||||||
|---|---|---|---|---|---|---|
| Entry | Product | D-CSA (mol%) | Temp (°C) | Reaction time (h) | Yieldb (%) | eec (%) |
| a Reaction conditions: 5 mol% of (R,R)-3g, 0.2 mmol of different substrates, and 2 mL of MeOH. b Isolated yield. c Determined by chiral HPLC. | ||||||
| 1 | 5a | 2.5 | −20 | 72 | n.d. | — |
| 2 | 5a | 5 | −20 | 72 | 12 | 99 |
| 3 | 5c | 5 | −20 | 72 | 16 | 97 |
| 4 | 5g | 5 | −20 | 72 | 52 | 99 |
| 5 | 5h | 5 | −20 | 72 | 43 | 92 |
| 6 | 5j | 5 | −20 | 72 | n.d. | — |
| 7 | 5j | 5 | rt | 16 | 17 | 92 |
In our previous work, we have developed a series of chiral C,O-chelated and O,O′-chelated cyclometalated gold(III) complexes for enantioselective carboalkoxylation of 2-ethynylbenzaldehyde and its acetal form providing 41% and 90% ee, respectively.8,11 In this work, we continued to develop more sterically hindered C,O-chelated gold(III) complexes by modifying the substituents of the aryl group attached to the chiral oxazoline moiety. The enantioselectivity was increased from 9% to 92% by changing the steric size and position of the substituents on the aryl group, revealing that the enantioselectivity was highly sensitive to the structural change of the chiral ligands. The enantioselectivity was further improved by decreasing the reaction temperature, providing a high enantioselectivity of up to 99% ee (Scheme 6). Our findings demonstrated the great potential of chiral gold(III) complexes in asymmetric catalysis.
 | ||
| Scheme 6 Summary of our research on asymmetric gold(III) catalysis. | ||
Conclusions
In summary, we have developed a novel approach for the synthesis of optically pure C,O-chelated BINOL/gold(III) complexes by diastereomeric resolution. With the aid of a successful BINOL ligand dissociation process, we have established a simple, efficient and column-free method for chiral resolution of racemic oxazoline-based gold(III) dichloride complexes on a gram scale. Meanwhile, the resolved enantiopure gold(III) dichloride complexes were capable of acting as chiral resolving agents to resolve disubstituted BINOL derivatives. These results open new avenues in the development of chiral gold(III) complexes and gold(III)-mediated chiral resolution of BINOL derivatives. The outstanding performance of a consecutive chirality transfer process was demonstrated, in which the chiral information was transferred initially from the inexpensive chiral source BINOL to highly valuable gold(III) complexes, and finally to BINOL derivatives. Furthermore, the catalytic activity of this new class of C,O-chelated BINOL/gold(III) complexes was studied by asymmetric carboalkoxylation of ortho-alkynylbenzaldehydes, giving products with an excellent enantioselectivity of up to 99% ee. Despite the limited examples of gold(III)-catalyzed asymmetric transformations, the high potential of chiral gold(III) complexes in asymmetric catalysis was demonstrated. We envision that this work would enrich the applications of chiral gold(III) chemistry in more research fields.Data availability
The data supporting this article have been included as part of the ESI.†Author contributions
Kwok-Heung Aries Chan: writing – original draft preparation, methodology, data curation, formal analysis, and investigation. Wa-Yi O: writing – original draft preparation, data curation, formal analysis, and investigation. Jia-Jun Jiang: methodology, data curation, formal analysis, and investigation. Jian-Fang Cui: writing – review & editing, supervision, data curation, and investigation. Man-Kin Wong: writing – review & editing, conceptualization, supervision, project administration, and funding acquisition.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We gratefully acknowledge the financial support by the Research Grants Council of the Hong Kong Special Administrative Region, China (PolyU15300019 and PolyU15302723), the State Key Laboratory of Chemical Biology and Drug Discovery, The Hong Kong Polytechnic University (P0043816: WZ0Y), and the University Research Facility in Life Sciences (ULS) of PolyU.References
-
(a) A. S. K. Hashmi, Gold Bull., 2003, 36, 3–9 CrossRef CAS
; (b) A. S. K. Hashmi, Angew. Chem., Int. Ed., 2005, 44, 6990–6993 CrossRef CAS PubMed
- Y.-M. Wang, A. D. Lackner and F. D. Toste, Acc. Chem. Res., 2014, 47, 889–901 CrossRef CAS PubMed
-
(a) R. A. Widenhoefer, Chem.–Eur. J., 2008, 14, 5382–5391 CrossRef CAS PubMed
-
(a) G. L. Hamilton, E. J. Kang, M. Mba and F. D. Toste, Science, 2007, 317, 496–499 CrossRef CAS
-
(a) J.-J. Jiang and M.-K. Wong, Chem.–Asian J., 2021, 16, 364–377 CrossRef CAS PubMed
-
(a) J. Rodriguez and D. Bourissou, Angew. Chem., Int. Ed., 2018, 57, 386–388 CrossRef CAS
-
(a) P. T. Bohan and F. D. Toste, J. Am. Chem. Soc., 2017, 139, 11016–11019 CrossRef CAS
- J.-J. Jiang, J.-F. Cui, B. Yang, Y. Ning, N. C.-H. Lai and M.-K. Wong, Org. Lett., 2019, 21, 6289–6294 CrossRef CAS PubMed
- C. C. Chintawar, V. W. Bhoyare, M. V. Mane and N. T. Patil, J. Am. Chem. Soc., 2022, 144, 7089–7095 CrossRef CAS PubMed
- X. Ye, C. Wang, S. Zhang, Q. Tang, L. Wojtas, M. Li and X. Shi, Chem.–Eur. J., 2022, 28, e202201018 CrossRef CAS PubMed
- J.-F. Cui, H.-M. Ko, K.-P. Shing, J.-R. Deng, N. C.-H. Lai and M.-K. Wong, Angew. Chem., Int. Ed., 2017, 56, 3074–3079 CrossRef CAS PubMed
-
(a) Y. Chen, S. Yekta and A. K. Yudin, Chem. Rev., 2003, 103, 3155–3212 CrossRef CAS PubMed
- Y.-Q. Han, Y. Ding, T. Zhou, S.-Y. Yan, H. Song and B.-F. Shi, J. Am. Chem. Soc., 2019, 141, 4558–4563 CrossRef CAS PubMed
-
(a) H. Aoyama, M. Tokunaga, J. Kiyosu, T. Iwasawa, Y. Obora and Y. Tsuji, J. Am. Chem. Soc., 2005, 127, 10474–10475 CrossRef CAS PubMed
-
(a) K. Tanaka, T. Okada and F. Toda, Angew. Chem., Int. Ed., 1993, 32, 1147–1148 CrossRef
-
(a) E. Tayama and H. Tanaka, Tetrahedron Lett., 2007, 48, 4183–4185 CrossRef CAS
- M. P. Walsh, J. M. Phelps, M. E. Lennon, D. S. Yufit and M. O. Kitching, Nature, 2021, 597, 70–76 CrossRef CAS PubMed
- Y.-B. Wang and B. Tan, Acc. Chem. Res., 2018, 51, 534–547 CrossRef CAS PubMed
-
(a) T. Akiyama and K. Mori, Chem. Rev., 2015, 115, 9277–9306 CrossRef CAS
-
(a) H. Wang, Chirality, 2010, 22, 827–837 CrossRef CAS
-
(a) M. Juárez-Hernandez, D. V. Johnson, H. L. Holland, J. McNulty and A. Capretta, Tetrahedron: Asymmetry, 2003, 14, 289–291 CrossRef
-
(a) J. D. Jolliffe, R. J. Armstrong and M. D. Smith, Nat. Chem., 2017, 9, 558–562 CrossRef CAS PubMed
-
(a) J. Guo, W.-B. Xiong, H.-R. Ma, L. Fan, Y.-Y. Zhou, H. N. C. Wong and J.-F. Cui, Chem. Sci., 2022, 13, 4608–4615 RSC
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4sc04221b |
| This journal is © The Royal Society of Chemistry 2024 |