
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceDegradable polyolefins prepared by integration of disulfides into metathesis polymerizations with 3,6-dihydro-1,2-dithiine†
Hong-Gyu
Seong
a,
Thomas P.
Russell
 ab and
Todd
Emrick
*a
ab and
Todd
Emrick
*a
aPolymer Science & Engineering Department, Conte Center for Polymer Research, University of Massachusetts, 120 Governors Drive, Amherst, Massachusetts 01003, USA. E-mail: tsemrick@mail.pse.umass.edu
bMaterials Sciences Division, Lawrence Berkeley National Laboratory, 1 Cyclotron Road, Berkeley, California 94720, USA
First published on 26th September 2024
Abstract
Disulfide-containing polyolefins were synthesized by ring-opening metathesis polymerization (ROMP) of the 6-membered disulfide-containing cyclic olefin, 3,6-dihydro-1,2-dithiine, which was prepared by ring-closing metathesis of diallyl disulfide. This approach facilitated the production of disulfide-containing unsaturated polyolefins as copolymers with disulfide monomer units embedded within a poly(cyclooctene) or poly(norbornene) framework. The incorporation of disulfides into the polymer backbone imparts redox responsiveness and enables polymer degradation via chemical reduction or thiol-disulfide exchange. This ROMP copolymerization strategy yielded both linear polyolefins, as well as bottlebrush polymers, with degradable segments, thereby broadening the scope of responsive polymer architectures synthesized by ROMP.
Introduction
The exceptional utility of polyolefins, such as polyethylene, polypropylene, and poly(cyclooctene), has led to their successful performance in a myriad of important commercial polyolefin-based products of societal benefit, spanning applications from construction-grade materials to plastic medical devices used in healthcare.1,2 While the chemical stability of polyolefins enables these applications, the drawbacks of long-term macromolecular durability are increasingly noted in the context of undesirable waste accumulation and pollution resulting from improper plastics disposal.3,4 As such, research activity targeting innovation in polymer degradation, recycling, and upcycling grows rapidly, with polyolefins representing a particularly challenging target.5–8We are interested in synthetic methods that effectively introduce chemical subunits into polymers to serve as handles to promote degradation, recognition, or responsive features. Specific to polyolefins, we previously described ring-opening metathesis polymerization (ROMP) of an eight-membered disulfide-containing cyclic olefin, (Z)-3,4,7,8-tetrahydro-1,2-dithiocine, finding that its copolymerization with other cyclic olefins successfully integrated disulfide units within the polymer backbone (Fig. 1).9 This ROMP copolymerization strategy gave access to a considerable breadth of materials with variable extents of incorporated disulfides which, in turn, allowed for the use of mild reagents to perform reductive degradation and thereby achieve tunable degrees of molecular weight reduction. However, the multistep synthesis required to obtain this particular cyclic olefin monomer imposes limits on its ultimate utility.
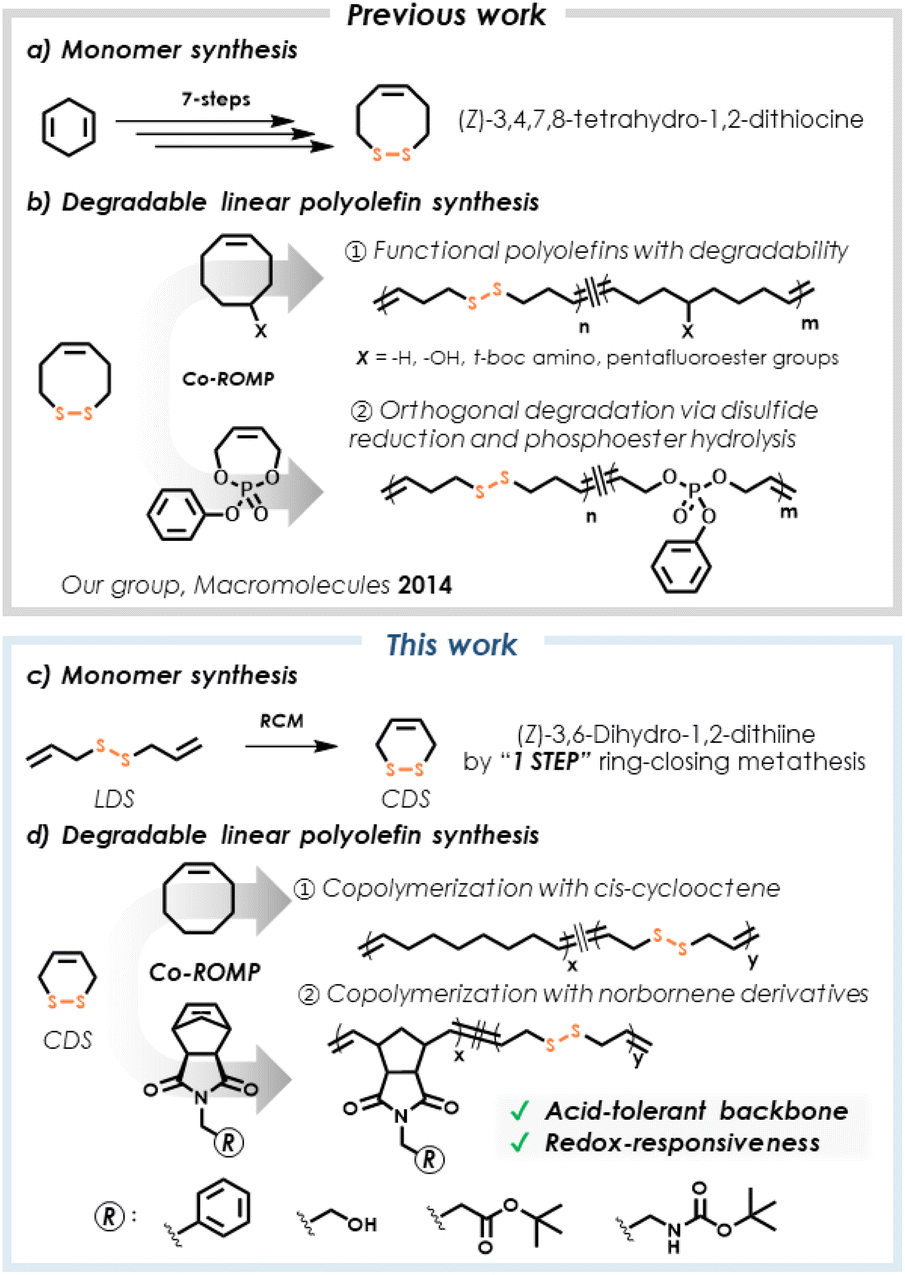 | ||
| Fig. 1 Synthesis of disulfide-containing polyolefins by ROMP: (a) prior multistep synthesis of the 8-membered disulfide-containing cyclic olefin, (Z)-3,4,7,8-tetrahydro-1,2-dithiocine and (b) its copolymerization with cyclic olefins; (c) ring-closing metathesis (RCM) of LDS affords (Z)-3,6-dihydro-1,2-dithiine (CDS); (d) CDS copolymerization with cis-cyclooctene or norbornene derivatives gives entry into disulfide-containing polyolefins by ROMP. | ||
Along similar conceptual lines, several researchers have described methods to merge metathesis polymerization with degradable functionality. For example, Schlaad and coworkers synthesized a cysteine-derived macrocycle by ring-closing metathesis (RCM), which was used subsequently in ROMP to yield polyolefins containing esters, protected amines, and disulfides within the same copolymer backbone.10 Strategies that allow insertion of acid-sensitive groups into polyolefins are particularly prominent, with recent example including acetals,11 carbonates,12 esters,13 enol ethers,14 phosphoesters,15 phosphoramidates,16 and silyl ethers.17 Notably, the acid-tolerance of disulfides is advantageous for advancing the scope of multifunctional materials, allowing chemical modification of disulfide-containing polymers that could not be tolerated by acid-labile functionality.18 Moreover, the orthogonality of disulfide cleavage in comparison to other functional groups may drive chemically selective degradation.
Here, we describe the use of the commercially available diallyl disulfide (denoted here as linear disulfide, or LDS), a disulfide-containing diene, for RCM, followed by ROMP of the corresponding cyclic olefin. Recent attempts to use LDS in polymers in different capacities involved generating materials intended for heavy metal removal,19 adhesives,20,21 and degradable structures.22 We describe the RCM of LDS to produce (Z)-3,6-dihydro-1,2-dithiine (denoted here as cyclic disulfide, or CDS, Fig. 1c) under conditions that bias the metathesis equilibrium towards the cyclic olefin (Fig. 2a). This approach represents a one-step synthesis of a disulfide-containing cyclic olefin as a 6-membered ring (Fig. 1c) for studies in ROMP copolymerization with other cyclic olefin comonomers (Fig. 1d), ultimately embedding disulfides within polyolefin backbones, including linear and bottlebrush architectures, via a chain-growth ring-opening mechanism, with subsequent chemically induced degradation via disulfide cleavage. Use of ring-opening chemistry is key, since prior studies using LDS and cis-cyclooctene in mixed chain/step-growth polymerization are limited with respect to achievable molecular weights.23
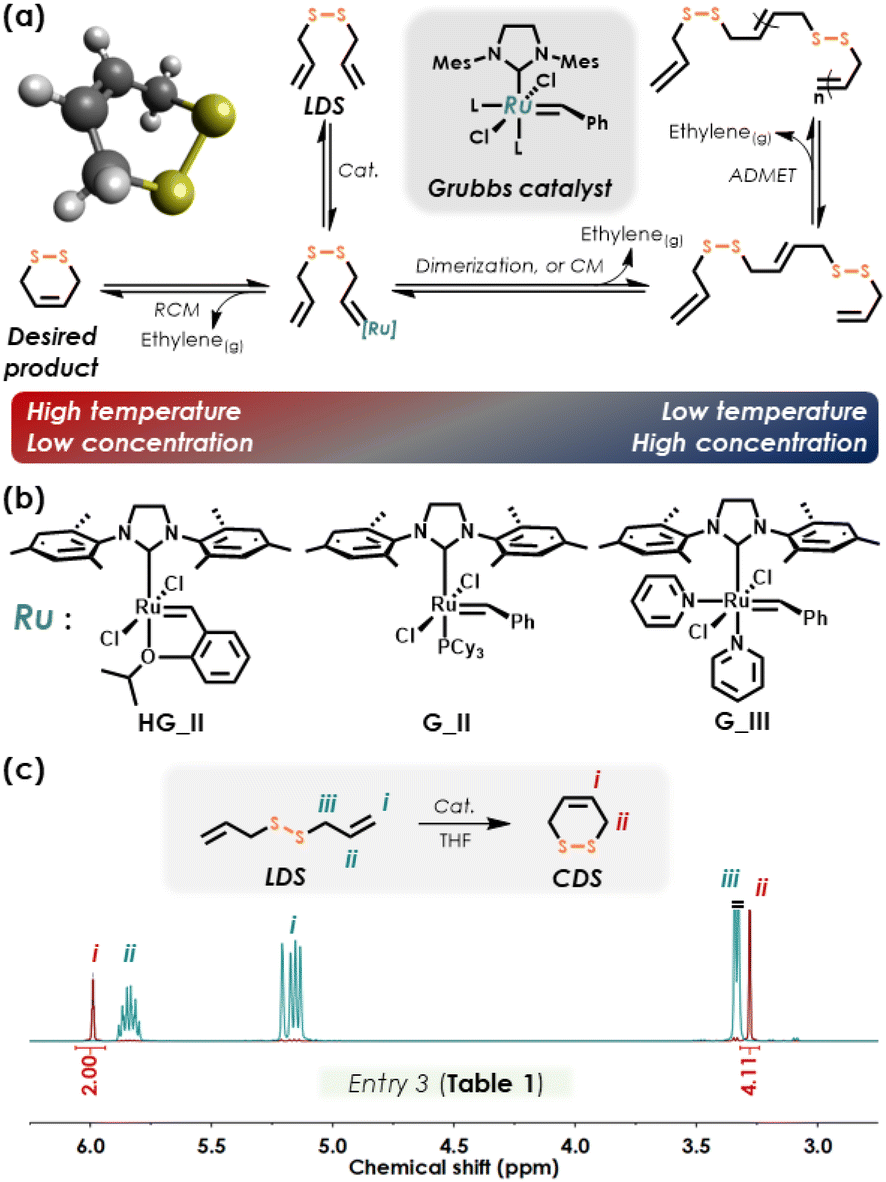 | ||
| Fig. 2 (a) Metathesis CDS–LDS equilibria ; (b) chemical structures of the ruthenium benzylidene catalysts employed in this work; (c) 1H NMR spectra of LDS and CDS (from Entry 3 in Table 1). | ||
Results and discussion
RCM of diallyl disulfide
Due to the reversible nature of metal-mediated metathesis, a number of reaction conditions influence the equilibrium position of the metathesis product, including monomer concentration and temperature (Fig. 2a).24 In RCM, enthalpic contributions to the metathesis equilibrium arise from ring-strain of the cyclic olefin, where the type and number of bonds are the same for the cyclic olefin plus ethylene and that of acyclic diene.25 Entropic contributions to the equilibrium include translational mobility (favoring small molecules and sensitive to monomer concentration and solution viscosity) and conformational motion (favoring polymer).25 Consequently, conducting RCM at low diene concentration and high temperature effectively shifts the equilibrium toward the desired cyclic product, with removal of ethylene from the reaction mixture further driving equilibrium to ring-closed product.For RCM of LDS, anhydrous THF was selected for its higher reflux temperature relative to the commonly used DCM and its tendency to deactivate metathesis catalysts,26 which is advantageous in workup protocols.27 The catalysts employed in this study are shown in Fig. 2b. In a typical RCM experiment, 1H nuclear magnetic resonance (NMR) spectroscopy was used to quantify reaction conversion by comparing the allylic proton of diallyl disulfide (5.90–5.78 ppm for 2H and 5.24–5.10 ppm for 4H) to the vinyl protons of CDS (5.99 ppm for 2H) (Fig. 2c). For example, at a 4![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 molar ratio of LDS (0.01 M) to Grubbs 3rd generation (G_III) catalyst, RCM at 60 °C reached 86% after 5 hours (Entry 1, Table 1 and Fig. S1†). Reducing the catalyst loading to LDS:G_III = 19:1 resulted in a lower RCM yield of 78% (Entry 2, Table 1). Notably, increasing the reaction temperature to 70 °C increased the RCM yield of CDS to 96% (Entry 3, Table 1), in accord with metathesis equilibrium principles. We note that acyclic dienes generally favor cyclization for 5-to-7-membered rings at low concentration and high temperature.25 Attempted RCM of LDS at 80 °C in THF did not improve product yield (Entry 4, Table 1), which we attribute to effects of catalyst degradation at this higher temperature.28
:1 molar ratio of LDS (0.01 M) to Grubbs 3rd generation (G_III) catalyst, RCM at 60 °C reached 86% after 5 hours (Entry 1, Table 1 and Fig. S1†). Reducing the catalyst loading to LDS:G_III = 19:1 resulted in a lower RCM yield of 78% (Entry 2, Table 1). Notably, increasing the reaction temperature to 70 °C increased the RCM yield of CDS to 96% (Entry 3, Table 1), in accord with metathesis equilibrium principles. We note that acyclic dienes generally favor cyclization for 5-to-7-membered rings at low concentration and high temperature.25 Attempted RCM of LDS at 80 °C in THF did not improve product yield (Entry 4, Table 1), which we attribute to effects of catalyst degradation at this higher temperature.28
| Entry | [LDS] (M) | Catalyst | Cat. Loading (mol%) | Temp. (°C) | Time (h) | RCM yield (%) |
|---|---|---|---|---|---|---|
| 1 | 0.01 | G_III | 20 | 60 | 5 | 86 |
| 2 | 0.01 | G_III | 5 | 60 | 5 | 78 |
| 3 | 0.01 | G_III | 5 | 70 | 5 | 96 |
| 4 | 0.01 | G_III | 5 | 80 | 5 | 93 |
| 5 | 0.05 | G_III | 5 | 80 | 5 | 84 |
| 6 | 0.10 | G_III | 5 | 80 | 5 | <1 |
| 7 | 0.10 | G_III | 5 | 80 | 24 | 13 |
| 8 | 0.01 | HG_II | 5 | 80 | 5 | <1 |
| 9 | 0.01 | G_II | 5 | 80 | 5 | 90 |
The impact of LDS concentration on RCM was examined from 0.01–0.05 M, with higher concentration resulting in lower yield (Entry 5, Table 1). Further increasing LDS concentration to 0.1 M failed to yield CDS (<1%, Entry 6, Table 1) and instead produced oligomers via acyclic diene metathesis polymerization (ADMET) (Fig. 2a). Other catalysts gave widely variable success in LDS-to-CDS conversion: Hoveyda-Grubbs 2nd generation (HG_II) catalyst produced very low yields (Entry 8, Table 1), while yields obtained when using Grubbs 2nd Generation catalyst (G_II) were good (90%, Entry 9, Table 1). Based on screening experiments in Table 1, Entry 3 was selected as optimal; increasing the scale to 0.5 mL of LDS yielded 95% conversion to CDS, obtained as a colorless oil (isolated yield 0.26 g or 64%). Unlike the case of COEs, where both E and Z configurations exist with different ring strains,29 it is noteworthy that CDS as the RCM product exclusively favors the Z configuration, due to constraints of 6-membered rings (Figure 2c, 13C NMR spectrum in Fig. S2† shows a single olefin carbon peak). Ultimately, CDS proved unisolable in 100% purity by distillation or chromatographic methods, with detailed procedures and characterization given in the ESI.†
ROMP copolymerization of CDS with COE
As illustrated in Fig. 3a, copolymerization of CDS with COE yielded polycyclooctene (PCOE) with disulfides distributed along the backbone (denoted as P-(CDS target mol%)). Monomer conversion and the relative mole percentages of CDS and COE incorporated into the polymer product were quantified by 1H NMR spectroscopy (Fig. 3b). The olefinic protons of CDS (5.99 ppm), COE (5.60 ppm), and polymer (5.38 ppm) were sufficiently baseline-separated to assess monomer conversion quantitatively. Initial attempts to integrate CDS into PCOE via ROMP were conducted at a monomer concentration of 0.50 M and a 200:1 monomer:G_III molar ratio. Regardless of the mol% CDS employed, the entire set of copolymerizations (Entries 1–5 in Table 2 and Fig. 3c) suggest the occurrence of extensive secondary metathesis, judging from size exclusion chromatography (SEC) analysis and the intensities of low molecular weight signals (from 25–28 mL retention volume) relative to the breadth of the polymer signal. Such oligomeric products were increasingly evident in experiments performed with higher CDS mol%. This may arise from small amounts of unreacted acyclic diene that facilitate cross-metathesis with growing polymer chain-ends, leading to oligomer formation. Further insight obtained from control experiments are detailed later.
 | ||
| Fig. 3 (a) ROMP copolymerization of COE and CDS; (b) 1H NMR spectrum of P-20 from Entry 2 in Table 2; SEC curves eluting with THF with various (c) CDS mol% while fixing [monomer]/[initiator] ratio at 200, (d) total monomer concentration at [COE]:[CDS] = 4:1 with [monomer]/[initiator] ratio at 200, (e) [monomer]/[initiator] at [monomer] = 2 M and [COE]:[CDS] = 4:1, and (f) impact of reaction time following introduction of G_III. | ||
| Entry | [Ma] (M) | [Ma]/[Cat.] | COE:CDSb |
COE conv.c (%) | CDS conv.c (%) | CDS per chainc (%) | M n, theo. (kDa) | M n, SEC (kDa) | PDId |
|---|---|---|---|---|---|---|---|---|---|
| a Total monomer concentration including COE and CDS. b Molar ratio between two monomers. c Calculated based on the peak integration of 1H NMR spectroscopy. d Determined by SEC (THF as a solvent) with polystyrene standards. | |||||||||
| 1 | 0.5 | 200 | 180:20 |
91 | 97 | 10 | 22.2 | 19.7 | 1.41 |
| 2 | 0.5 | 200 | 160:40 |
83 | 97 | 17 | 22.4 | 16.6 | 1.39 |
| 3 | 0.5 | 200 | 140:60 |
72 | 96 | 29 | 22.5 | 6.8 | 1.38 |
| 4 | 0.5 | 200 | 120:80 |
60 | 98 | 42 | 22.7 | 7.7 | 1.34 |
| 5 | 0.5 | 200 | 100:100 |
48 | 87 | 56 | 22.8 | 6.5 | 1.39 |
| 6 | 2 | 200 | 160:40 |
83 | 98 | 17 | 22.4 | 22.0 | 1.55 |
| 7 | 0.1 | 200 | 160:40 |
93 | 85 | 8 | 22.4 | 1.6 | 1.46 |
| 8 | 2 | 50 | 40:10 |
86 | 99 | 16 | 5.6 | 11.2 | 1.59 |
| 9 | 2 | 100 | 80:20 |
86 | 99 | 25 | 11.2 | 17.3 | 1.59 |
| 10 | 2 | 400 | 320:80 |
85 | 97 | 14 | 44.7 | 27.7 | 1.59 |
| 11 | 2 | 200 | 0:200 |
N/A | 97 | 100 | 23.6 | 1.5 | 1.15 |
Considering the relatively low ring-strain of COE (7.4 kcal mol−1),29 higher monomer concentration is needed to drive metathesis equilibria towards polymer. Targeting a monomer-to-initiator ratio ([monomer]:[G_III]) of 200, total monomer concentration ([M]) was varied from 0.1 to 2.0 M (Fig. 3d). At higher [M], the formation of low molecular oligomers was suppressed significantly, and the estimated molecular weight of the polymer product was in good accord with the targeted value (Mn, theo) of 22.4 kDa (Mn, SEC = 22.0 kDa, PDI = 1.55, 17 mol% disulfide incorporation, Entry 6 in Table 2). Thus, by adjusting reaction conditions with suitable catalyst selection, RCM of LDS and ROMP of CDS were performed in accord with ring-chain equilibrium principles.10,30 Molecular weight control was achieved in copolymerizations with 20 mol% CDS by tuning the [monomer]/[G_III] ratio from 50 to 400. Although the peak-average molecular weight (Mp) obtained by SEC analysis shifted to higher values (Fig. 3e), a non-linear dependence of Mn was observed (Fig. S20†), which is attributed to the presence of trace amounts of LDS in the reactions, and/or to S–Ru coordination at the ruthenium alkylidene chain-end (Fig. S21†).9 Such Ru–S interactions were confirmed by 1H NMR spectroscopy, where resonances associated with the protons attached to the ruthenium alkylidene upon COE or CDS insertion were observed at 19.03 and 18.34 ppm, respectively (Fig. S21†). The relative intensities of these resonances closely correlated with the COE:CDS feed ratio.
Copolymerization of COE and CDS ([monomer] = 2 M) yielded P-20-k (Mn, SEC ∼25.5 kDa, PDI ∼1.57) rapidly (∼1 min) after introduction of G_III (Fig. 3f). SEC traces revealed predominant polymer signal centered at elution volumes of ∼21 mL, with the consistent appearance of oligomeric product at longer elution volumes (∼26 mL), the intensity of the latter decreasing as the reaction progressed. Over time, lower molecular weight product (ostensibly cyclic metathesis products) appeared to be incorporated into growing polymer (vide supra).25 Though a minor component of the overall product mixture, the ratio between the kinetic (oligomer) and the thermodynamic (polymer) products was seen to decrease from 1.4% at 1 min to 0.7% at 180 min (based on ratio of the peak areas in the SEC traces). However, it is noteworthy that secondary metathesis was suppressed significantly by adjusting monomer concentration (Fig. 3c) and the amount of oligomeric product in P-20-k was marginal, overall providing evidence for successful ROMP copolymerization of COE and CDS.
Role of RCM as precursor to ROMP
As a chain-growth mechanism, ROMP represents a controlled strategy to attain a desired polymer molecular weight with relatively low PDI values, facilitated in part by monomer ring-strain.24 In contrast, the step-growth characteristics of ADMET polymerization yields high MW only at high monomer conversion.31 Thus, the presence of acyclic diene impurity in a ROMP reaction may detach the active ruthenium alkylidene catalyst from the growing chain-end and initiate new chains (Fig. 4a). In our case, since the CDS monomer always contained a small percentage of LDS (optimally <5% of unreacted LDS), the resultant combination of chain- and step-growth propagation mechanisms produces polymers of lower MW relative to what would result from a purely ring-opening (chain-growth) mechanism. | ||
| Fig. 4 (a) Copolymerization of COE/CDS/LDS mixture; (b) SEC (eluting with THF) curves resulting from polymerizations with 0, 10, or 20 mol% LDS; (c) Plot of Mn, SEC and PDI vs. LDS mol%. | ||
To test the impact of mixed chain/step-growth mechanisms, several polymerizations were performed using LDS in conjunction with CDS and COE. In these experiments, the [COE]:[CDS + LDS] ratio was fixed at 4:1, with variation of the CDS:LDS ratio. At 10 mol% LDS (i.e., 1:1 CDS:LDS), a substantial decrease in Mn was observed (22.0 to 4.6 kDa) (Fig. 4b), which declined even further (to 2.8 kDa) using 4:1 COE:LDS (i.e., absent CDS, Fig. 4c). Consequently, when using RCM as a method to prepare cyclic olefin monomers for ROMP, the purity of the cyclic olefin is key for realizing desirable ROMP outcomes.
ROMP copolymerization of CDS with norbornene derivatives
To assess the ability to integrate CDS into copolymerizations with cyclic olefins of higher ring strain, the norbornene dicarboxyimide derivatives shown in Fig. 5a were prepared and used in ROMP copolymerizations with CDS (monomers 1–4, described in ESI†). For example, ROMP of CDS with benzyl-substituted norbornene 1 produced copolymers with ∼10 mol% CDS and molecular weights in the 11–40 kDa range with PDI of 1.2–1.5. Notably, adjusting overall monomer concentration from 0.05–2.0 M (at target DP = 200) showed that lower concentrations led to more narrow molecular weight distributions as a result of smaller shoulders on the high molecular weight side of the SEC traces. At 0.05 M monomer, low MW oligomers were absent (i.e., ∼25–28 mL elution volume in Fig. 5b), contrasting that seen for COE copolymerizations. Variation of polymerization temperature did little to change Mp values, as seen in Fig. 5c, suggesting minimal entropic contributions for the higher ring-strain norbornenes (∼27 kcal mol−1), in line with ring-strain impacts on metathesis equilibria that overwhelm entropic factors.29 | ||
| Fig. 5 (a) ROMP of CDS with norbornene derivatives 1–4; (b) 1H NMR spectrum of P1-20 from Entry 2 in Table 3; SEC curves with variation of (c) total monomer concentration at [1]:[CDS] = 4:1 with [monomer]/[initiator] ratio at 200; (d) temperature; (e) [monomer]/[initiator] at 2 M monomer and [1]:[CDS] = 4:1; and (f) ROMP copolymerization of CDS with norbornene derivatives with containing pendent, hydroxy, t-butyl ester, and t-boc amine groups. | ||
Molecular weight control of copolymers composed of 1 with ∼6–11 mol% CDS was achieved by adjusting the [monomer]:[G_III] ratio from 50–400 (Entries 2 and 6–8 in Table 3 and MW vs. target DP in Fig. S22†). In each copolymerization, high norbornene conversion was achieved, with Mn values of the polymer products increasing to nearly 40 kDa at higher monomer:catalyst ratios. Most copolymerizations involving monomer 1 gave reasonable agreement between target (theoretical) and experimental (SEC-estimated) molecular weights, with the exception of the highest target DP of 400 (Entry 8, Table 3). Further versatility of this disulfide monomer in ROMP was seen in CDS-norbornene copolymerizations conducted with comonomers 2–4 containing hydroxy, t-butyl ester, and t-boc amine pendent groups, respectively. While attempted ROMP of 2 with CDS produced only limited chain growth due to premature precipitation (Table 3, Entry 9), both the t-butyl ester-substituted monomer 3 and t-boc amine-substituted monomer 4 copolymerized cleanly with CDS to yield polymers with Mn, values of >30 kDa and PDI of 1.6-1,7 (Table 3, Entries 10, 11). In all copolymerizations involving norbornene derivatives, CDS incorporation is much lower than for the COE copolymers, which we attribute to multiple interrelated factors, including (1) the substantial ring-strain difference between the two monomers and (2) the relatively fast polymerization of norbornene, even at lower concentrations, that may lead to consecutive CDS units that would exacerbate chelation to the ruthenium catalyst (hypothetical illustration in Fig. S23†). In contrast, the CDS-COE copolymer case allows for higher CDS incorporation due to the more similar ring-strain energies of the two monomers.
| Entry | Comonomer | Temp. (°C) | [Ma] (M) | [Ma]/[Cat.] | NBb:CDS |
NB conv.c (%) | CDS conv.c (%) | CDS per chainc (%) | M n, theo. (kDa) | M n, SEC (kDa) | PDId |
|---|---|---|---|---|---|---|---|---|---|---|---|
| a Total monomer concentration (NB and CDS). b Comonomer used. c Calculated based on peak integration from 1H NMR spectroscopy. d Determined by SEC (THF eluent) with polystyrene calibration standards. e Upon injection of G3 stock solution, the solution became turbid and the product was insoluble in THF and CDCl3. | |||||||||||
| 1 | 1 | 25 | 2 | 200 | 160:40 |
>99 | 78 | 8.8 | 44.1 | 28.8 | 1.51 |
| 2 | 1 | 25 | 0.5 | 200 | 160:40 |
>99 | 72 | 11.5 | 44.1 | 30.9 | 1.35 |
| 3 | 1 | 25 | 0.05 | 200 | 160:40 |
>99 | 0 | 0 | 44.1 | 31.0 | 1.19 |
| 4 | 1 | 0 | 0.5 | 200 | 160:40 |
>99 | 0 | 0 | 44.1 | 27.7 | 1.25 |
| 5 | 1 | 50 | 0.5 | 200 | 160:40 |
>99 | 76 | 8.6 | 44.1 | 29.5 | 1.28 |
| 6 | 1 | 25 | 0.5 | 50 | 40:10 |
>99 | 68 | 5.8 | 11.3 | 10.9 | 1.24 |
| 7 | 1 | 25 | 0.5 | 100 | 80:20 |
>99 | 72 | 6.8 | 22.6 | 19.1 | 1.28 |
| 8 | 1 | 25 | 0.5 | 400 | 320:80 |
>99 | 72 | 8.8 | 85.8 | 39.6 | 1.32 |
| 9e | 2 | 25 | 0.5 | 200 | 160:40 |
— | — | — | 36.7 | — | — |
| 10 | 3 | 25 | 0.5 | 200 | 160:40 |
>99 | 57 | 5.8 | 50.2 | 31.4 | 1.58 |
| 11 | 4 | 25 | 0.5 | 200 | 160:40 |
>99 | 72 | 9.0 | 52.6 | 32.1 | 1.67 |
Degradation of disulfide-containing metathesis polymers
As shown in Fig. 6a, polymer degradation experiments were performed using P-20-k (Mn, SEC = 29.1 kDa, PDI = 1.50, ESI†) as a representative example. As anticipated, the CDS-derived disulfide units are easily amenable to reduction, which, when conducted with excess tri-n-butylphosphine (n-TBP), led to a rapid (minutes) and steep decline of molecular weight. Alternatively, thiol/disulfide exchange was equally effective, for example by adding 1-dodecanethiol (DSH) to a DCM solution of P-20-k. Depending on the amount of DSH used, molecular weight reduction proceeded to 17.8 kDa (with 1 equiv. DSH with respect to disulfide, PDI 1.71) or as low as 9.1 kDa (with 5 equiv. DSH, PDI 1.76). The use of CDS to integrate disulfides into macromolecules is not limited to linear polymers, but also proved effective for the preparation of degradable bottlebrush polymers. In a proof-of-concept example, an ω-norbornene dicarboxyimide-terminated polystyrene macromonomer was synthesized (NB-PS, ESI†) and utilized in ROMP with 10 mol% CDS at target DP 100, yielding the desired disulfide-containing bottlebrush structure (PS BP, Fig. 6d). From the SEC curves in Fig. 6d and e, subjecting the degradable bottlebrush polymer to n-TBP led to significant molecular weight degradation, in accord with the extent of CDS incorporation in the backbone. Notably, due to the large molecular weight difference between CDS and the cyclic olefin macromonomer, using very small quantities of CDS in the copolymerizations allows for large molecular weight reduction upon degradation. Although we did not perform a detailed evaluation to yield information as to the microstructure of the CDS-containing bottlebrush, the reduction in molecular weight suggests a likely random or gradient disulfide distribution, as opposed to a diblock polymer, such that even a small amount of disulfide incorporation provides access to triggering a substantial molecular reduction as desired. | ||
| Fig. 6 (a) P-20-k and PS BP-10 degradation by (i) reduction of disulfide or (ii) thiol-disulfide exchange; SEC curves eluting with THF of (b) P-20-k reduction of disulfide with n-TBP, (c) P-20-k thiol-disulfide exchange with 1-dodecanethiol, and (d) PS BP degradation by n-TBP as a reducing agent before and after disulfide reduction. | ||
Conclusions
In summary, we demonstrated the synthesis of a 6-membered, disulfide-containing cyclic olefin, CDS, by a one-step RCM reaction, then used this monomer successfully in ROMP copolymerizations with cyclic olefin comonomers of different ring-strain, while tailoring reaction conditions to bias equilibria towards productive metathesis. Variation of [monomer]:[initiator] ratios allowed for molecular weight control, while kinetics studies showed gradual consumption of a minor oligomeric product population by the macromolecular species. Use of CDS in these polymerizations, as opposed to the linear diallyl disulfide, LDS, was key for preventing cross metathesis and shifting the polymerization to a chain-growth mechanism. Extending further, ROMP of CDS with norbornene derivatives bearing pendent functionality yielded disulfide-rich polyolefins, in which degradation was conducted easily by reducing or exchange conditions. Such redox-responsive systems are of interest for subsequent studies in areas such as polymer blends, interfacial processes, and degradation/repolymerization designs.
Data availability
The data supporting this article have been included as part of the ESI.†Author contributions
H.-G. Seong: conceptualization, synthesis, characterization, writing – original draft, review, editing; T. P. Russell: supervision, review, editing; T. Emrick: conceptualization, supervision, writing, review, editing.Conflicts of interest
The authors declare no competing interests.Acknowledgements
The authors appreciate the support of the University of Massachusetts Amherst and partial support from the National Science Foundation for the preparation of functional polymers and nanoscale structures (NSF-CHE-2203578) and from the Army Research Office under contract W911NF-24-2-0041.References
- N. C. Paxton, M. C. Allenby, P. M. Lewis and M. A. Woodruff, Biomedical applications of polyethylene, Eur. Polym. J., 2019, 118, 412–428 CrossRef CAS.
- D. Jubinville, E. Esmizadeh, S. Saikrishnan, C. Tzoganakis and T. Mekonnen, A comprehensive review of global production and recycling methods of polyolefin (PO) based products and their post-recycling applications, Sustainable Mater. Technol., 2020, 25, e00188 CrossRef CAS.
- Z. O. G. Schyns and M. P. Shaver, Mechanical Recycling of Packaging Plastics: A Review, Macromol. Rapid Commun., 2021, 42, e2000415 CrossRef PubMed.
- S. Yin, R. Tuladhar, F. Shi, R. A. Shanks, M. Combe and T. Collister, Mechanical reprocessing of polyolefin waste: A review, Polym. Eng. Sci., 2015, 55, 2899–2909 CrossRef CAS.
- C. W. S. Yeung, J. Y. Q. Teo, X. J. Loh and J. Y. C. Lim, Polyolefins and Polystyrene as Chemical Resources for a Sustainable Future: Challenges, Advances, and Prospects, ACS Mater. Lett., 2021, 3, 1660–1676 CrossRef CAS.
- A. H. Westlie, E. Y. Chen, C. M. Holland, S. S. Stahl, M. Doyle, S. R. Trenor and K. M. Knauer, Polyolefin Innovations toward Circularity and Sustainable Alternatives, Macromol. Rapid Commun., 2022, 43, e2200492 CrossRef PubMed.
- G. W. Coates and Y. D. Y. L. Getzler, Chemical recycling to monomer for an ideal, circular polymer economy, Nat. Rev. Mater., 2020, 5, 501–516 CrossRef CAS.
- X.-Y. Wang, Y. Gao and Y. Tang, Sustainable developments in polyolefin chemistry: Progress, challenges, and outlook, Prog. Polym. Sci., 2023, 143, 101713 CrossRef CAS.
- C.-C. Chang and T. Emrick, Functional Polyolefins Containing Disulfide and Phosphoester Groups: Synthesis and Orthogonal Degradation, Macromolecules, 2014, 47, 1344–1350 CrossRef CAS.
- F. N. Behrendt and H. Schlaad, Metathesis polymerization of cystine-based macrocycles, Polym. Chem., 2017, 8, 366–369 RSC.
- C. Fraser, M. A. Hillmyer, E. Gutierrez and R. H. Grubbs, Degradable Cyclooctadiene/Acetal Copolymers: Versatile Precursors to 1,4-Hydroxytelechelic Polybutadiene and Hydroxytelechelic Polyethylene, Macromolecules, 1995, 28, 7256–7261 CrossRef CAS.
- T. M. McGuire, C. Perale, R. Castaing, G. Kociok-Kohn and A. Buchard, Divergent Catalytic Strategies for the Cis/Trans Stereoselective Ring-Opening Polymerization of a Dual Cyclic Carbonate/Olefin Monomer, J. Am. Chem. Soc., 2019, 141, 13301–13305 CrossRef CAS PubMed.
- R. M. Weiss, A. L. Short and T. Y. Meyer, Sequence-Controlled Copolymers Prepared via Entropy-Driven Ring-Opening Metathesis Polymerization, ACS Macro Lett., 2015, 4, 1039–1043 CrossRef CAS PubMed.
- J. D. Feist and Y. Xia, Enol Ethers Are Effective Monomers for Ring-Opening Metathesis Polymerization: Synthesis of Degradable and Depolymerizable Poly(2,3-dihydrofuran), J. Am. Chem. Soc., 2020, 142, 1186–1189 CrossRef CAS PubMed.
- T. Steinbach, E. M. Alexandrino and F. R. Wurm, Unsaturated poly(phosphoester)s via ring-opening metathesis polymerization, Polym. Chem., 2013, 4, 3800–3806 RSC.
- Y. Liang, H. Sun, W. Cao, M. P. Thompson and N. C. Gianneschi, Degradable Polyphosphoramidate via Ring-Opening Metathesis Polymerization, ACS Macro Lett., 2020, 9, 1417–1422 CrossRef CAS PubMed.
- P. Shieh, H. V. Nguyen and J. A. Johnson, Tailored silyl ether monomers enable backbone-degradable polynorbornene-based linear, bottlebrush and star copolymers through ROMP, Nat. Chem., 2019, 11, 1124–1132 CrossRef CAS PubMed.
- M. Pięta, V. B. Purohit, J. Pietrasik and C. M. Plummer, Disulfide-containing monomers in chain-growth polymerization, Polym. Chem., 2023, 14, 7–31 RSC.
- S. Kim, K. Tang, T.-H. Kim and Y. Hwang, Selective removal of cationic organic pollutants using disulfide-linked polymer, Sep. Purif. Technol., 2022, 288, 120522 CrossRef CAS.
- K. R. Albanese, Y. Okayama, P. T. Morris, M. Gerst, R. Gupta, J. C. Speros, C. J. Hawker, C. Choi, J. R. de Alaniz and C. M. Bates, Building Tunable Degradation into High-Performance Poly(acrylate) Pressure-Sensitive Adhesives, ACS Macro Lett., 2023, 12, 787–793 CrossRef CAS PubMed.
- Z. Tang, Z. Liu, M. You, H. Yin, H. Yu, X. Shi, J. Yang, G. Qin, L. Zhu and Q. Chen, Dynamic Disulfide Bond Regulated Tough Adhesion and On-Demand Debonding of the Albumin-Based Double Network Hydrogel to Diverse Substrates, ACS Appl. Polym. Mater., 2024, 6, 330–340 CrossRef CAS.
- K. R. Albanese, P. T. Morris, J. Read de Alaniz, C. M. Bates and C. J. Hawker, Controlled-Radical Polymerization of alpha-Lipoic Acid: A General Route to Degradable Vinyl Copolymers, J. Am. Chem. Soc., 2023, 145, 22728–22734 CrossRef CAS PubMed.
- Y. Xia, F. Zhou, W. Hao and S. Tang, Synthesis of Degradable Polyolefins Bearing Disulfide Units via Metathesis Copolymerization, Polymers, 2023, 15, 3101 CrossRef CAS PubMed.
- C. W. Bielawski and R. H. Grubbs, Living ring-opening metathesis polymerization, Prog. Polym. Sci., 2007, 32, 1–29 CrossRef CAS.
- S. Monfette and D. E. Fogg, Equilibrium Ring-Closing Metathesis, Chem. Rev., 2009, 109, 3783–3816 CrossRef CAS PubMed.
- S. E. Blosch, M. Alaboalirat, C. B. Eades, S. J. Scannelli and J. B. Matson, Solvent Effects in Grafting-through Ring-Opening Metathesis Polymerization, Macromolecules, 2022, 55, 3522–3532 CrossRef CAS.
- A. B. Smith III, K. Basu and T. Baosanac, Total Synthesis of (-)-Okilactomycin, J. Am. Chem. Soc., 2007, 129, 14872–14874 CrossRef PubMed.
- J. Louie and R. H. Grubbs, Metathesis of Electron-Rich Olefins: Structure and Reactivity of Electron-Rich Carbene Complexes, Organometallics, 2002, 21, 2153–2164 CrossRef CAS.
- R. Walter, R. M. Conrad and R. H. Grubbs, The Living ROMP of trans-Cyclooctene, Macromolecules, 2009, 42, 599–605 CrossRef PubMed.
- M. J. Marsella, H. D. Maynard and R. H. Grubbs, Template-Directed Ring-Closing Metathesis: Synthesis and Polymerization of Unsaturated Crown Ether Analogs, Angew. Chem., Int. Ed., 1997, 36, 1101–1103 CrossRef CAS.
- L. Caire da Silva, G. Rojas, M. D. Schulz and K. B. Wagener, Acyclic diene metathesis polymerization: History, methods and applications, Prog. Polym. Sci., 2017, 69, 79–107 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4sc04468a |
| This journal is © The Royal Society of Chemistry 2024 |