
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceA compact chemically driven [2]catenane rotary motor operated through alternate pumping and discharging†
Anquan
Li
a,
Zhenglin
Du
a,
Shilong
Zhang
 b,
Jialin
Xie
c,
Xia
Li
a,
Qing
Chen
a,
Yisong
Tang
a,
Jiawen
Chen
b and
Kelong
Zhu
*a
b,
Jialin
Xie
c,
Xia
Li
a,
Qing
Chen
a,
Yisong
Tang
a,
Jiawen
Chen
b and
Kelong
Zhu
*a
aSchool of Chemistry, Sun Yat-Sen University, Guangzhou, 510275, China. E-mail: zhukelong@mail.sysu.edu.cn
bSchool of South China Academy of Advanced Optoelectronics, South China Normal University, Guangzhou, 510006, China
cSchool of Chemistry and Chemical Engineering, Hainan University, Haikou, 570228, China
First published on 13th August 2024
Abstract
Here we present a compact and precise [2]catenane rotary motor that functions with a single recognition site, capable of achieving a 360° directional rotation powered by chemical fuels. The motor is propelled by an acid–base fueled benzimidazolium pumping cassette and deemed the smallest (molecular weight ∼ 994 Da) catenane rotary motor to date. It can effectively undergo a 180° rotation by transitioning the [24]crown-6 ether (24C6) from the benzimidazolium site to the less favorable alkyl moiety through sequential deprotonation, slipping, and re-protonation operations, generating a meta stable co-conformer. Subsequently, a discharging phase, triggered by de-benzylation and re-benzylation, facilitates the other half-rotation of the motor, returning the 24C6 to its initial position and completing the full directional rotation of the [2]catenane rotary motor within 18 hours. The precision of the motor's operation enables further advances in artificial molecular machines.
Introduction
In nature, a myriad of micro-scale machines endowed with motor functions, known as molecular motors, abound. These molecular motors fulfill diverse functions within organisms, playing integral roles in an array of life processes, ranging from DNA replication to cellular mitosis and beyond.1–4 Drawing inspiration from these natural wonders, chemists have undertaken the endeavor to create artificial molecular motors capable of controlled directional motion. These synthetic machines are designed to harness various external energy sources, such as photons,5–9 electricity,10–15 and chemical fuels,16–20 in pursuit of mimicking the remarkable capabilities observed in their biological counterparts.21,22Catenanes23,24 are a class of mechanically interlocked molecular architectures characterized by the topological entanglement of rings. Attributed to their dynamic behavior, which allows for reversible interconversion between different topological isomers in response to external stimuli, catenanes hold significant promise in the field of artificial molecular machines (AMMs),25–27 particularly rotary motors. In 2003, Leigh and colleagues showcased the groundbreaking achievement of the synchronous unidirectional rotation in a [3]catenane rotary motor, completing a 360-degree revolution of two smaller rings along a larger circular track, marking the inception of catenane rotary motors.7 Subsequently, a [2]catenane rotary motor capable of selective rotation in either direction was developed.8 Building upon these advancements, in 2016, an autonomous [2]catenane rotary motor was introduced, operating through an innovative information ratchet mechanism.17 Additionally, Leigh's group reported a pioneering chemical-fueled [2]catenane rotary motor, driven by acid–base oscillations.18 More recently, a milestone was reached by Stoddart et al. with the development of an electricity-powered [3]catenane rotary motor, showcasing the unidirectional circumrotation of two CBPQT4+ rings around a 50-membered loop incorporated with a molecular pumping cassette.15 Despite these notable accomplishments, ongoing research efforts are focused on exploring catenane rotary motors with novel function mechanisms, aiming for heightened efficiency and precision in operation, as well as improved scalability to facilitate future applications.12
Recently, the benzimidazolium-based molecular pumping cassette demonstrates its capability in directionally collecting crown ether rings. Combining such a controlled molecular transportation with the clipping synthesis has led to precise radial catenanes (Fig. 1).28 Encouragingly, we envision that the molecular pumping cassette could be further developed into a rotary motor operated via a flashing energy rachet mechanism through alternate pumping and discharging. Herein, we present the molecular design, synthesis and precise operation of a compact [2]catenane rotary motor that is deemed the smallest (molecular weight ∼ 994 Da) catenane rotary motor to date.
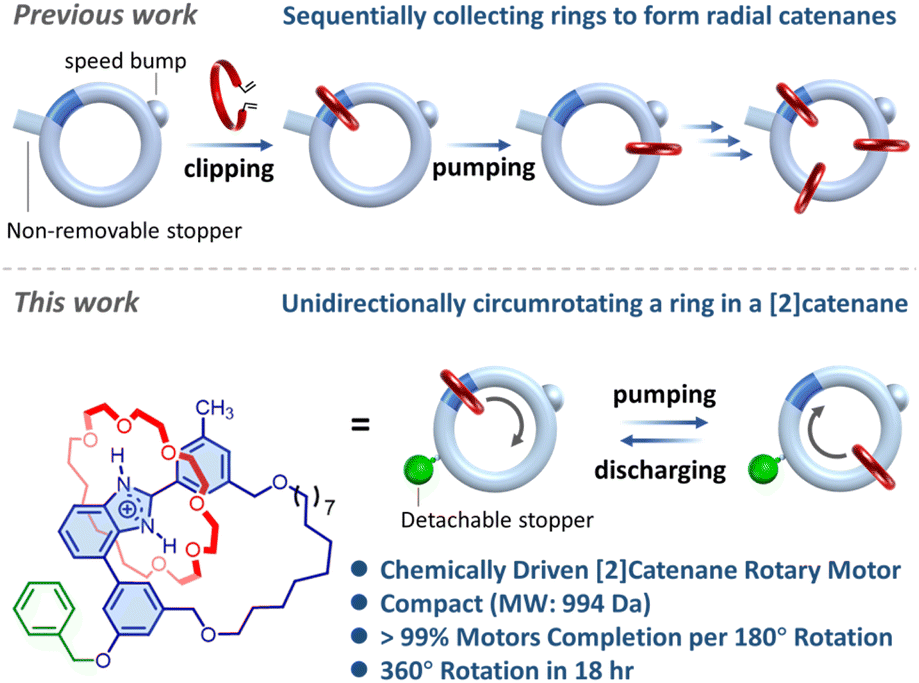 | ||
| Fig. 1 Harnessing the benzimidazolium-based molecular pumping cassette to precisely construct radial catenanes (previous work) and to directionally operate a compact [2]catenane rotary motor via a flashing energy rachet mechanism through alternate pumping and discharging (current work). | ||
Results and discussion
As illustrated in Fig. 1, the [2]catenane rotary motor comprises a [24]crown-6 ether (24C6) wheel (red ring), a loop track molecule incorporating an L-shaped benzimidazolium pumping cassette (blue module within the large loop), and a detachable stopper (green sphere) facilitating controllable translocation of the 24C6 wheel upon chemical transformations. To power the motor, (i) the benzimidazolium is initially deprotonated to generate a thermo-dynamic driving force for shuttling the wheel towards the collecting moiety (phase A to B in Fig. 2); (ii) subsequent acidification progresses the motor to phase C, completing the pumping operation; (iii) cleavage of the stopper triggers a transition from phase C to D, relocating the wheel to the benzimidazolium site; and (iv) re-stoppering finalizes the discharging operation (phase C to A) and resets the motor. Thus, by harnessing the net energy from acid–base and stoppering–detaching reactions, a unidirectional 360-degree rotation of the motor could be ultimately achieved through a flashing energy rachet mechanism.8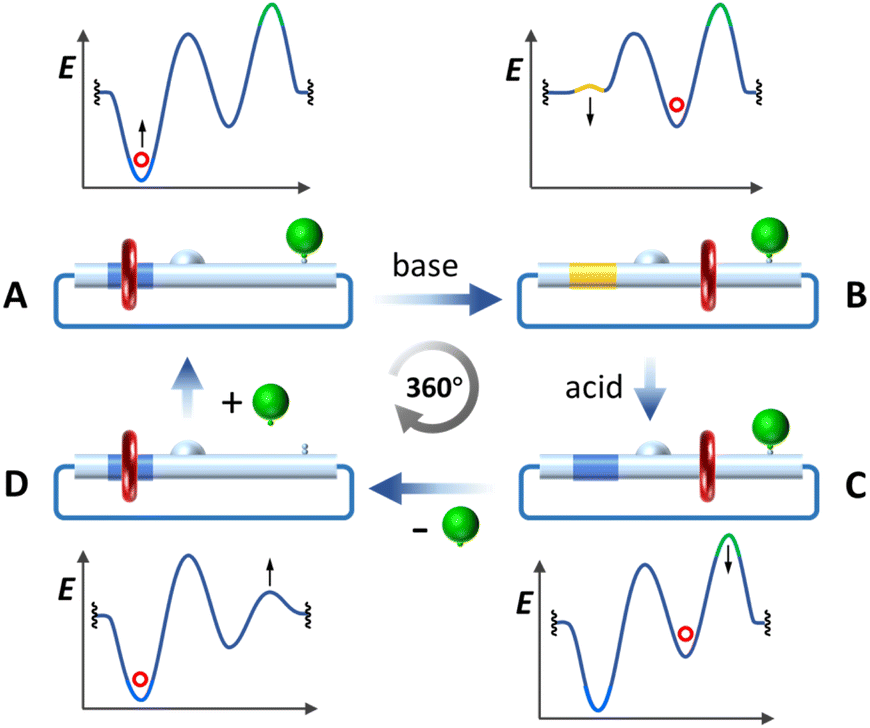 | ||
| Fig. 2 Energy profiles for stages A–D through one 360-degree rotation of the [2]catenane rotary motor. | ||
Accordingly, to ensure successful unidirectional translocation of the wheel, it's imperative to determine if the 24C6 can thread through the L-shaped benzimidazoliums with or without a stoppering group.29,30 Consequently, [1a-H2]+ and [1b-H2]+ were initially synthesized and assessed for their complexation with 24C6 (see ESI† and Fig. 3a). No discernible association was observed between [1a-H2]+ and 24C6 under testing conditions (Fig. S1†), confirming that the 3-methoxyl-5-methylphenyl moiety acts as a stopper for 24C6. Conversely, 1H NMR analysis of the mixture of [1b-H2]+ and 24C6 revealed a complexation with slow binding kinetics (Fig. 3b and S2†). Threading of 24C6 occurs upon heating at 338 K indicating that the 3-hydroxyl-5-methylphenyl group is a slippage stopper for 24C6. An association constant of 5.9 × 103 M−1 in CD3NO2 was determined for the [2]pseudorotaxane complex [1b-H2⊂24C6]+ (Fig. S3†), whose structure was unambiguously elucidated by X-ray crystal structure analysis (Fig. 3c and Table S3†). These results strongly affirm the ability of the L-shaped benzimidazolium featuring a 3-hydroxyl-5-methylphenyl moiety to translocate 24C6 in a directional manner when integrated into a [2]catenane rotary motor.
 | ||
| Fig. 3 (a) Schematic representation of threading 24C6 through L-shaped benzimidazoliums. (b) Partial 1HNMR (400 MHz, 298 K, CD3NO2) spectra of an equimolar (2.0 mM) mixture of [1b-H2]+ and 24C6 before and after heating at 65 °C for 5 h. (c) Crystal structure of the complex [1b-H2⊂24C6][BF4]. Hydrogen bonds: green dashed line. | ||
Subsequently, a loop molecule [6-H2]+, incorporating L-shaped benzimidazolium and a 14-membered aliphatic ring collecting moiety, was synthesized (see ESI† and Scheme 1). The hydroxyl [2]catenane [M-H2]+ (where M denotes the motor) was then generated via Ru-catalyzed ring-closure metathesis (RCM), utilizing the bis-olefin precursor 24C6-pre on [6-H2]+, followed by hydrogenation wherein de-benzylation occurred simultaneously. Acidification with HBF4 finally afforded [M-H2]+ (40% yield in 3 steps). The interlocked structure of [M-H2]+ was confirmed by observing resonance signals for both cyclic components with a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 integral ratio in 1H NMR analysis (Fig. 4a and S33–S36†). The downfield chemical shifts for the NH protons (13.24 and 12.88 ppm corresponding to Hm and Hn, respectively) validate the presence of 24C6 at the benzimidazolium site primarily stabilized by N–H⋯O hydrogen bonds. This observation is consistent with the X-ray crystal structure of its neutral counterpart M-H resulting from deprotonation during crystallization (Fig. 5 and Table S4†).
:1 integral ratio in 1H NMR analysis (Fig. 4a and S33–S36†). The downfield chemical shifts for the NH protons (13.24 and 12.88 ppm corresponding to Hm and Hn, respectively) validate the presence of 24C6 at the benzimidazolium site primarily stabilized by N–H⋯O hydrogen bonds. This observation is consistent with the X-ray crystal structure of its neutral counterpart M-H resulting from deprotonation during crystallization (Fig. 5 and Table S4†).
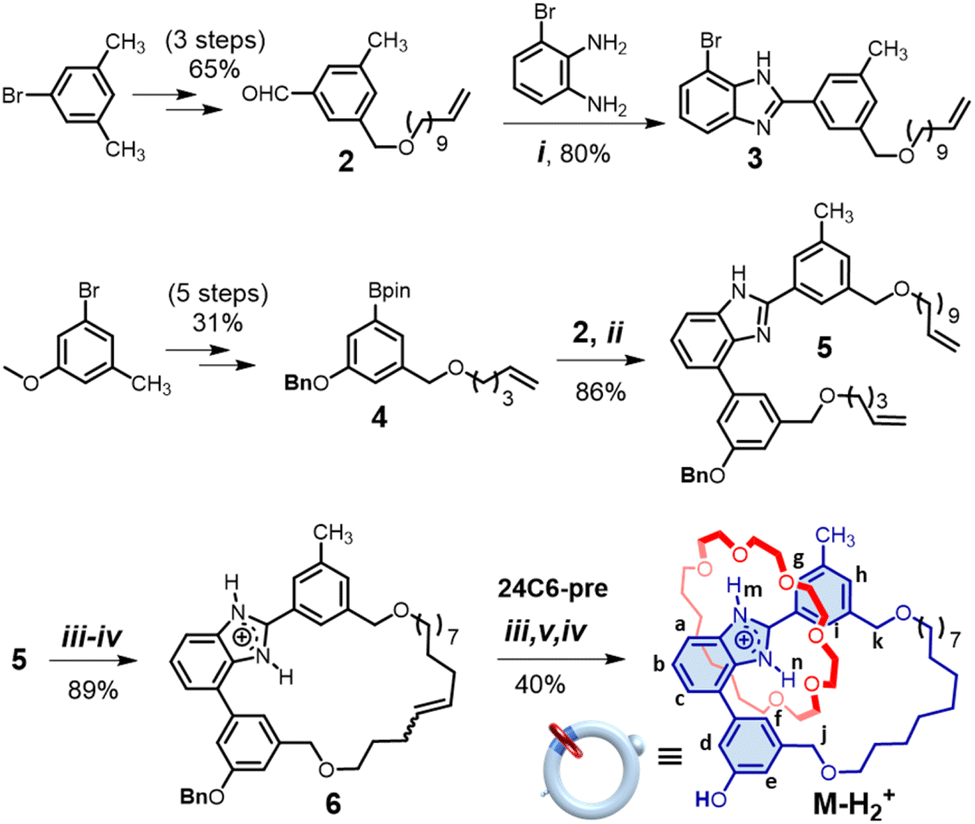 | ||
| Scheme 1 Synthesis of the hydroxyl [2]catenane [M-H2][BF4]. Conditions: (i) ZrCl4, CHCl3, 25 °C, 24 h; (ii) Pd(Ph3P)4, 2 M Na2CO3, THF, 80 °C, 4 h; (iii) Grubbs' catalyst, CH2Cl2, 43 °C, 24 h; (iv) HBF4, 30 min; (v) H2, Pd/C, 25 °C, 4 h. | ||
 | ||
| Fig. 4 Partial 1HNMR (400 MHz, 298 K, CD3CN) spectra of (a) [M-H2]+, (b) [I-BnM-H2]+, and (c) [II-BnM-H2]+. Resonance peak assignment was referenced in Scheme 1. * impurity. | ||
 | ||
| Fig. 5 Crystal structure of M-H. Hydrogen bonds are highlighted in the green dashed lines (i) and (ii). H atoms except OH and NH are omitted for clarity. | ||
The facile removal of the bulkier benzyl group under mild conditions, compared to a methyl group, has prompted us to explore its reinstallation onto [M-H2]+ as the detachable stopper as originally designed (Fig. 1). However, prior to benzylation, it's crucial to assess the stability of [M-H2]+ under basic conditions, as translocation of the 24C6 could occur when the benzimidazolium is deprotonated. Upon adding 10 equivalents of potassium carbonate to [M-H2]+ in dimethyl sulfoxide, the formation of phenolate [M]− was observed, while benzimidazolium was converted to benzimidazole, as verified by 1H NMR analysis (see ESI and Fig. S4†). Furthermore, all the 24C6 wheels were confirmed to reside on the benzimidazole site, indicating no translocation occurs. Gratifyingly, almost identical resonance signals were noticed after heating at 90 °C for 20 h (Fig. S4d†), suggesting a significantly high energy barrier for threading the wheel in phenolate [M]−, which in turn could facilitate the reinstallation of the benzyl stopper. Consequently, implementing benzylation on [M-H2]+ under the verified conditions efficiently afforded the [2]catenane rotary motor [I-BnM-H2]+ as the sole product in an isolated yield of 82% (see ESI† and Fig. 4b). The complete disappearance of the 1H NMR resonance signal for OH, coupled with the emergence of signals from the benzyl group, confirms the successful benzylation of [M-H2]+. Furthermore, similar chemical shifts observed for NHs (13.29 ppm for Hm and 12.84 ppm for Hn) and ethylene protons, compared to [M-H2]+, support the retention of the 24C6 on the benzimidazolium site.
The directional operation of motor [I-BnM-H2]+ is demonstrated in Fig. 6, illustrating a complete 360° full rotation of the 24C6 wheel along the track loop, consisting of two half-circle rotations corresponding to the pumping and discharging steps, respectively. Initially, by harnessing the benzimidazolium pumping cassette, the 24C6 wheel undergoes a 180° clockwise rotation by converting [I-BnM-H2]+ to its co-conformer [II-BnM-H2]+ with the crown ether wheel positioned on the less-favored aliphatic moiety. Specifically, treatment of [I-BnM-H2]+ with 5.0 equivalents of t-BuOK induces deprotonation, forming the metastable benzimidazolide species [I-BnM]− (Fig. 7a). The electrostatic repulsion between the negative charged benzimidazolide and the electron-rich crown ether generates a thermodynamic driving force, leading to the sliding of the ring onto the alkyl moiety to yield its co-conformer [II-BnM]− (Fig. 6). A τ1/2 of 76 min associated with an energy barrier of 25.8 kcal mol−1 was determined for such a translocation occurring at 338 K (Fig. S5, S6 and Tables S1, S2†). As revealed by 1H NMR (Fig. 7c), approximately 80% of the 24C6 was translocated after heating at 338 K for 3 h, while almost full translocation (>99%) could be achieved in 12 h (Fig. 7d) or in 4 hours by elevating the temperature to 373 K (Fig. S7†). The co-conformational product [II-BnM-H2]+ can be promptly generated by acidification with HBF4, providing a purified product with a 78% yield over two steps (see ESI†). The translocation of 24C6 was confirmed by 1H NMR comparison (Fig. 4c). Protons Hm and Hg exhibit evident upfield chemical shifting (−0.76 and −0.33 ppm, respectively), suggesting the absence of hydrogen bonding. Conversely, noticeable downfield chemical shifting is observed for Hn (+0.27 ppm) Hi (+0.25 ppm), Hh (+0.41 ppm), and Hk (+0.25 ppm), verifying the presence of 24C6 on the alkyl moiety with close proximity to the tolyl moiety. Additionally, 2D nuclear Overhauser enhancement spectroscopy (NOESY) (Fig. S44†) further supported the inferred structure of [II-BnM-H2]+, confirming the successful completion of the half-circle rotation. To power such a pumping process, a chemical energy input of 219.5 kcal mol−1 could be estimated (see ESI†).
 | ||
| Fig. 6 Schematic representation of operating the [2]catenane rotary motor. Conditions: (i) H2, Pd/C, 25 °C; (ii) BnBr, K2CO3, 90 °C. | ||
 | ||
| Fig. 7 Partial 1H NMR (400 MHz, 298 K, DMSO-d6) spectra of [I-BnM]− heated at 338 K for (a) 0 h; (b) 1 h; (c) 3 h; and (d) 12 h * Impurity. | ||
To fulfill the 360° rotation of the motor, [II-BnM-H2]+ was then subjected to the discharging process, that is recovering [II-BnM-H2]+ to [I-BnM-H2]+ with the 24C6 wheel relocated to the benzimidazolium site. As mentioned earlier, removal of the stoppering benzyl group should generate a thermodynamic driving force favoring the benzimidazolium over the alkyl moiety for 24C6. Consequently, implementing palladium-catalyzed hydrogenation on [II-BnM-H2]+ (at 298 K, in 4 h) yielded the hydroxyl [2]catenane [M-H2]+ quantitatively, as evidenced by 1H NMR (Fig. S8†). This implies that clock-wisely translocating the 24C6 wheel along the large loop is much faster than that observed for threading 24C6 onto [1b-H2]+ to form the [2]pseudorotaxane [1b-H2⊂24C6]+. Ultimately, repeating the benzylation of [M-H2]+ completes the discharging process and resets the motor to [I-BnM-H2]+, accomplishing the full circumvolution of the 24C6 wheel in the [2]catenane rotary motor. Correspondingly, an estimated chemical energy input of 195.3 kcal mol−1 is required for achieving the second half-rotation (see ESI†).
Conclusions
In summary, we present a compact and highly efficient [2]catenane rotary motor powered by chemical fuels. This motor accomplishes a full 360° unidirectional rotation through precisely controlled steps, employing a benzimidazolium-crown ether pumping cassette. The process involves distinct pumping and discharging phases: pumping drives a 180° rotation of the 24C6 wheel, while discharging resets the motor to its original state. Detailed experimental protocols confirm the motor's precision and efficiency, laying the foundation for further advances in molecular machines and nanotechnology.Experimental
All reagents purchased from commercial suppliers were directly used without further purification unless otherwise noted. Solvents were either used as purchased or degassed and dried under a Vigor VSGS-5 Solvent Purification System. Detailed synthetic procedures are listed in the ESI (Schemes S1–S3†). The compounds were characterized by 1H NMR, 13C NMR, and 2D NMR spectroscopies (Fig. S9–S44†). NMR spectroscopy, host–guest complexation experiments and kinetics experiments (Fig. S2–S3 and S5†) were performed on a JEOL 400 YH instrument. X-ray diffraction analysis was performed either on a Rigaku SuperNova, Dual, AtlasS2 diffractometer using monochromatized Cu Kα radiation or on a BRUKER D8 VENTURE PHOTON III diffractometer using Ga Kα radiation. The crystal data are summarized in the ESI (Tables S3 and S4†).Data availability
The data supporting this article have been included as part of the ESI.†Author contributions
K. Z. supervised the project. A. L. performed all the synthetic experiments. A. L. collected and analyzed the NMR data with assistance from K. Z., Z. D., J. X., X. L. and Q. C. A. L. collected the SCXRD data. S. Z. and J. C. provided the SCXRD data ananlysis. K. Z. wrote the manuscript with input from A. L.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We thank the National Natural Science Foundation of China (22171295), the Fundamental Research Funds for the Central Universities (23xkjc006), the Starry Night Science Fund of Zhejiang University Shanghai Institute for Advanced Study (SN-ZJU-SIAS-006), and the Guangzhou Science and Technology Program (2024A04J6423) for financial support.Notes and references
- M. Schliwa and G. Woehlke, Nature, 2003, 422(6933), 759–765 CrossRef CAS PubMed.
- C. Toyoshima and G. Inesi, Annu. Rev. Biochem., 2004, 73, 269–292 CrossRef CAS PubMed.
- C. von Ballmoos, G. M. Cook and P. Dimroth, Annu. Rev. Biophys., 2008, 37, 43–64 CrossRef CAS PubMed.
- Molecular Machines in Biology: Workshop of the Cell, ed. J. Frank, Cambridge University Press, 2011 Search PubMed.
- G. Ragazzon, M. Baroncini, S. Silvi, M. Venturi and A. Credi, Nat. Nanotechnol., 2015, 10(1), 70–75 CrossRef CAS PubMed.
- G. Ragazzon, M. Baroncini, S. Silvi, M. Venturi and A. Credi, Beilstein J. Nanotechnol., 2015, 6, 2096–2104 CrossRef CAS PubMed.
- D. A. Leigh, J. K. Y. Wong, F. Dehez and F. Zerbetto, Nature, 2003, 424(6945), 174–179 CrossRef CAS PubMed.
- J. V. Hernandez, E. R. Kay and D. A. Leigh, Science, 2004, 306(5701), 1532–1537 CrossRef CAS PubMed.
- H. Li, C. Cheng, P. R. McGonigal, A. C. Fahrenbach, M. Frasconi, W.-G. Liu, Z. Zhu, Y. Zhao, C. Ke, J. Lei, R. M. Young, S. M. Dyar, D. T. Co, Y.-W. Yang, Y. Y. Botros, W. A. Goddard III, M. R. Wasielewski, R. D. Astumian and J. F. Stoddart, J. Am. Chem. Soc., 2013, 135(49), 18609–18620 CrossRef CAS PubMed.
- C. Cheng, P. R. McGonigal, S. T. Schneebeli, H. Li, N. A. Vermeulen, C. Ke and J. F. Stoddart, Nat. Nanotechnol., 2015, 10(6), 547–553 CrossRef CAS PubMed.
- C. Pezzato, M. T. Nguyen, C. Cheng, D. J. Kim, M. T. Otley and J. F. Stoddart, Tetrahedron, 2017, 73(33), 4849–4857 CrossRef CAS.
- Q.-H. Guo, Y. Qiu, X. Kuang, J. Liang, Y. Feng, L. Zhang, Y. Jiao, D. Shen, R. D. Astumian and J. F. Stoddart, J. Am. Chem. Soc., 2020, 142(34), 14443–14449 CrossRef CAS PubMed.
- Y. Qiu, B. Song, C. Pezzato, D. Shen, W. Liu, L. Zhang, Y. Feng, Q.-H. Guo, K. Cai, W. Li, H. Chen, M. T. Nguyen, Y. Shi, C. Cheng, R. D. Astumian, X. Li and J. F. Stoddart, Science, 2020, 368(6496), 1247–1253 CrossRef CAS PubMed.
- J. S. W. Seale, B. Song, Y. Qiu and J. F. Stoddart, J. Am. Chem. Soc., 2022, 144(37), 16898–16904 CrossRef CAS PubMed.
- L. Zhang, Y. Qiu, W.-G. Liu, H. Chen, D. Shen, B. Song, K. Cai, H. Wu, Y. Jiao, Y. Feng, J. S. W. Seale, C. Pezzato, J. Tian, Y. Tan, X.-Y. Chen, Q.-H. Guo, C. L. Stern, D. Philp, R. D. Astumian, W. A. Goddard and J. F. Stoddart, Nature, 2023, 613(7943), 280–286 CrossRef CAS PubMed.
- Z. Meng, J.-F. Xiang and C.-F. Chen, J. Am. Chem. Soc., 2016, 138(17), 5652–5658 CrossRef CAS PubMed.
- M. R. Wilson, J. Solà, A. Carlone, S. M. Goldup, N. Lebrasseur and D. A. Leigh, Nature, 2016, 534(7606), 235–240 CrossRef CAS PubMed.
- S. Erbas-Cakmak, S. D. P. Fielden, U. Karaca, D. A. Leigh, C. T. McTernan, D. J. Tetlow and M. R. Wilson, Science, 2017, 358(6361), 340–343 CrossRef CAS PubMed.
- S. Amano, S. D. P. Fielden and D. A. Leigh, Nature, 2021, 594(7864), 529–534 CrossRef CAS PubMed.
- L. Binks, C. Tian, S. D. P. Fielden, I. J. Vitorica-Yrezabal and D. A. Leigh, J. Am. Chem. Soc., 2022, 144(34), 15838–15844 CrossRef CAS PubMed.
- Y. Feng, M. Ovalle, J. S. W. Seale, C. K. Lee, D. J. Kim, R. D. Astumian and J. F. Stoddart, J. Am. Chem. Soc., 2021, 143(15), 5569–5591 CrossRef CAS PubMed.
- R. D. Astumian, Chem. Sci., 2017, 8(2), 840–845 RSC.
- J. P. Sauvage, Angew Chem. Int. Ed. Engl., 2017, 56(37), 11080–11093 CrossRef CAS PubMed; Q. Chen and K. Zhu, Chem. Soc. Rev., 2024, 53, 5677–5703 RSC.
- V. K. Ahluwalia and R. Aggarwal, Catenanes, Rotaxanes and Knots, in Alicyclic Chemistry, ed. V. K. Ahluwalia and R. Aggarwal, Springer Nature, Cham, Switzerland, 2023, pp. 203–208 Search PubMed.
- S. Erbas-Cakmak, D. A. Leigh, C. T. McTernan and A. L. Nussbaumer, Chem. Rev., 2015, 115(18), 10081–10206 CrossRef CAS PubMed.
- S. Kassem, T. van Leeuwen, A. S. Lubbe, M. R. Wilson, B. L. Feringa and D. A. Leigh, Chem. Soc. Rev., 2017, 46(9), 2592–2621 RSC.
- C. Pezzato, C. Cheng, J. F. Stoddart and R. D. Astumian, Chem. Soc. Rev., 2017, 46(18), 5491–5507 RSC; I. Aprahamian, ACS Cent. Sci., 2020, 6(3), 347–358 CrossRef CAS PubMed.
- A. Li, Z. Tan, Y. Hu, Z. Lu, J. Yuan, X. Li, J. Xie, J. Zhang and K. Zhu, J. Am. Chem. Soc., 2022, 144(5), 2085–2089 CrossRef CAS PubMed.
- N. Noujeim, K. Zhu, V. N. Vukotic and S. J. Loeb, Org. Lett., 2012, 14(10), 2484–2487 CrossRef CAS PubMed.
- H. Xu and K. Zhu, Sci. Sin.: Chim., 2023, 53(12), 2509–2522 CrossRef PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Details of syntheses, 1H NMR spectroscopy experiments and X-ray solutions for structures M-H (CCDC 2349579) and [1b-H2⊂24C6][BF4] (CCDC 2349580). For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d4sc04292a |
| This journal is © The Royal Society of Chemistry 2024 |