
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Unusual quadruple bonds featuring collective interaction-type σ bonds between first octal-row atoms in the alkaline-earth compounds Ae OLi2 (Ae = Be–Ba)†‡
OLi2 (Ae = Be–Ba)†‡

Li-Juan
Cui§
a,
Yu-Qian
Liu§
a,
Sudip
Pan
 *a,
Zhong-Hua
Cui
*ab and
Gernot
Frenking
*cd
*a,
Zhong-Hua
Cui
*ab and
Gernot
Frenking
*cd
aInstitute of Atomic and Molecular Physics, Jilin University, Changchun 130023, China. E-mail: zcui@jlu.edu.cn; sudip@jlu.edu.cn
bKey Laboratory of Physics and Technology for Advanced Batteries (Ministry of Education), Jilin University, Changchun 130023, China
cPhilipps-Universität Marburg, Hans-Meerwein-Strasse 4, D-35043 Marburg, Germany
dInstitute of Advanced Synthesis, School of Chemistry and Molecular Engineering, Nanjing Tech University, Nanjing 211816, China. E-mail: frenking@chemie.uni-marburg.de
First published on 31st July 2024
Abstract
Quantum chemical calculations are reported for the complexes of alkaline earth metals AeOLi2 (Ae = Be–Ba) at the BP86-D3(BJ)/def2-QZVPP and CCSD(T)/def2-QZVPPQZVPP levels. The nature of the Ae–OLi2 bond has been analyzed with a variety of methods. The AeOLi2 molecules exhibit an unprecedented σ donor bond Ae→OLi2 where the (n)s2 lone-pair electrons of the Ae atom are donated to vacant O–Li2 antibonding orbitals having the largest coefficient at lithium. This is a covalent bond where the accumulation of the associated electronic charge is located at two positions above and below the Ae–OLi2 axis. The bifurcated component of orbital interactions is structurally related to the recently proposed collective bonding model, but exhibits a completely different type of bonding. The most stable isomer of AeOLi2 has a C2v geometry and a singlet (1A1) electronic ground state. The bond dissociation energy (BDE) of the Ae–OLi2 bonds exhibits a zig-zag trend from BeOLi2 to BaOLi2, with BeOLi2 having the largest BDE (De = 73.0 kcal mol−1) and MgOLi2 possessing the lowest BDE (De = 42.3 kcal mol−1) at the CCSD(T) level. The calculation of the atomic partial charges by the Hirshfeld and Voronoi methods suggests that Be and Mg carry small negative charges in the lighter molecules whereas the heavier atoms Ca–Ba have small positive charges. In contrast, the NBO and QTAIM methods give positive charges for all Ae atoms that are larger for Ca–Ba than that calculated by the Hirshfeld and Voronoi approaches. The molecules AeOLi2 have large dipole moments where the negative end is at the Ae atom with the polarity Ae→OLi2. The largest dipole moments are predicted for the lighter species BeOLi2 and MgOLi2 and the smallest value is calculated for BaOLi2. The calculation of the vibrational spectra shows a significant red-shift toward lower wave numbers for the Ae–OLi2 stretching mode in comparison to diatomic AeO. Besides the Ae→OLi2 σ-donor bonds there are also three dative bonds due to Ae←OLi2 backdonation which consist of one σ bond and two π bonds. The appearance of strong Ae→OLi2 σ donation leads to quadruple bonds Ae OLi2 in all systems AeOLi2, even for the lightest species with Ae = Be, Mg. The valence orbitals of Ca, Sr, and Ba, which are involved in the dative interactions, are the (n)s and (n−1)d AOs whereas Be and Mg use their (n)s and (n)p AOs. The EDA-NOCV results are supported by the AdNDP calculations which give four 2c–2e bonding orbitals. Three bonding orbitals have occupation numbers ∼2. One σ orbital has smaller occupation numbers between 1.32 and 1.73 due to the delocalization to the lithium atoms. The analysis of the electronic structure with the ELF method suggests multicenter bonds with mainly trisynaptic and tetrasynaptic basins, which also support the results of the EDA-NOCV calculations.
OLi2 in all systems AeOLi2, even for the lightest species with Ae = Be, Mg. The valence orbitals of Ca, Sr, and Ba, which are involved in the dative interactions, are the (n)s and (n−1)d AOs whereas Be and Mg use their (n)s and (n)p AOs. The EDA-NOCV results are supported by the AdNDP calculations which give four 2c–2e bonding orbitals. Three bonding orbitals have occupation numbers ∼2. One σ orbital has smaller occupation numbers between 1.32 and 1.73 due to the delocalization to the lithium atoms. The analysis of the electronic structure with the ELF method suggests multicenter bonds with mainly trisynaptic and tetrasynaptic basins, which also support the results of the EDA-NOCV calculations.
1. Introduction
Multiple bonds between atoms constitute a fundamental cornerstone of chemistry.1 While double and triple bonds are commonly encountered in molecules, compounds featuring bond orders greater than three were until recently only known for chemical bonds between transition metals and possibly actinides, because they use their valence d and f orbitals for covalent bonding.2–5 A previous suggestion that C2 possesses a quadruple bond started a vigorous controversy in the literature with many arguments both in favor6–9 and against.10–15 The controversy was finally solved by an experimental study using high-resolution photoelectron imaging spectrometry, which showed that dicarbon has a strong degenerate π bond but negligible σ bonding due to near cancellation of bonding and antibonding σ-orbital interactions like in Be2.16Quadruple bonding of main-group atoms between boron and various transition metals (TMs) was lately reported in joint experimental and theoretical studies of molecules where the bond multiplicity of TM–B bonds can vary from single to quadruple bonding.17 In particular, diatomic RhB− was experimentally detected and the transition to neutral RhB was studied by photoelectron spectroscopy.18 The analysis of the spectra and quantum chemical calculations suggests that the neutral molecule in its 1Σ+ electronic ground state possesses a quadruple bond Rh![[quadruple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e003.gif) B which consists of two σ and two π bonds. Dative quadruple bonds between beryllium and various d10 transition metals were recently reported by Parameswaran.19
B which consists of two σ and two π bonds. Dative quadruple bonds between beryllium and various d10 transition metals were recently reported by Parameswaran.19
The finding that only one atom of a chemical bond A–B needs to provide valence d orbitals in order to achieve a quadruple bond led us to investigate the chemical bonds of heavy alkaline earth (Ae) atoms, Ca, Sr, and Ba with first octal-row species, because previous studies showed that these Ae atoms utilize their (n−1)d orbitals for covalent bonding like transition metals.20,21 We found that the anions AeB− and AeF−, and neutral AeC, where Ae = Ca, Sr, Ba, have indeed two σ and two π orbitals.22–24 The degenerate π orbitals in AeB− and isoelectronic AeC, which have only six valence electrons and a triplet (3Σ−) ground state, have singly occupied π orbitals.22 In contrast, the anions AeF− of the heavy Ae atoms, which have ten valence electrons, possess two doubly occupied σ and two π bonding orbitals, and thus they have a genuine quadruple bond with four strong dative components Ae F− whereas the lighter species with Ae = Be, Mg have triple bonds Ae
F− whereas the lighter species with Ae = Be, Mg have triple bonds Ae F−.23,24
F−.23,24
In order to find neutral molecules Ae–X, which have genuine quadruple bonds, we calculated the molecules AeOLi2, which are valence isoelectronic with the anions AeF−. The calculated results yielded an unexpected finding, which is presented here. We report about quantum chemical calculations of the geometries, vibrational frequencies, and bond dissociation energies of the title compounds, and we present and discuss the results of a thorough analysis of the bonding situation using a variety of methods. The ligand species OLi2 was experimentally reported before,25 but none of the calculated adducts AeOLi2 is experimentally known so far. The computed Ae–OLi2 bond strength suggests that they can be synthesized at least in the gas phase or in low-temperature matrices. The theoretically predicted vibrational spectra are helpful to identify the molecules AeOLi2 experimentally.
2. Computational details
The exploration of potential energy surfaces (PESs) of the AeOLi2 (Ae = alkaline-earth atoms) systems was undertaken utilizing the CALYPSO (Crystal structure AnaLYsis by Particle Swarm Optimization) code.26 Initial structures for both singlet and triplet spin states were considered at the BP86-D3(BJ)/def2-SVP level,27–30 and subsequently refined to enhance the accuracy of geometrical and frequency predictions at the BP86-D3(BJ)/def2-QZVPP level.30 To attain more precise geometries and relative energies, optimizations followed by the frequency calculations at the CCSD(T)/def2-QZVPP31,32 level, where all electrons are correlated (full core), were conducted on the low-lying minimum energy geometries determined at the BP86-D3(BJ)/def2-QZVPP level. The reliability of the mono-determinantal methodologies employed in this study was affirmed through the attainment of small T1 diagnostic (within 0.02) values from the converged CCSD wavefunction. All of these computational investigations were executed employing the Gaussian 16 package.33Chemical bonding analyses were conducted using the quantum theory of atoms in molecules (QTAIM),34 the adaptive natural density partitioning (AdNDP) analysis35 and the electron localization function (ELF),36 which are implemented in the Multiwfn code.37 The natural partial charges were evaluated using various methods, viz., the natural bond orbital (NBO),38 QTAIM,34 Hirshfeld,39 and Voronoi40 approaches. NBO charge was computed using the NBO7 program,41,42 while Hirshfeld charges were calculated using the Gaussian 16 program. For Voronoi charges, the ADF 2020 software was used.43 We also calculated the bond orders using the Wiberg method44 as well as the Mayer approach.45
To gain deeper insights into the nature of chemical interactions, energy decomposition analysis (EDA)46 in conjunction with the natural orbital for chemical valence theory (NOCV)47 which leads to the combined EDA-NOCV method48 was carried out. This comprehensive analysis was performed at the unrestricted (U)BP86-D3(BJ)/TZ2P level49 where scalar-relativistic effects are considered with the ZORA method50–52 utilizing the ADF 2020 package.53,54 In this analysis, the intrinsic interaction energy (ΔEint) between two fragments is dissected into four distinct energy components, as follows:
| ΔEint = ΔEelstat + ΔEPauli + ΔEorb + ΔEdisp | (1) |
The electrostatic ΔEelstat term represents the quasiclassical electrostatic interaction between the unperturbed charge distributions of the prepared fragments. The Pauli repulsion, ΔEPauli accounts for the energy change during the transformation from the superposition of unperturbed electron densities of the individual fragments into a wavefunction that explicitly adheres to the Pauli principle, achieved through the necessary antisymmetrization and wavefunction renormalization. The orbital term ΔEorb results from the mixing of the orbitals, which causes a charge transfer between the isolated fragments and a polarization within the fragments. The dispersion contribution (ΔEdisp), facilitated by the D3(BJ) method, elucidates the dispersion forces influencing the overall interaction between the fragments.
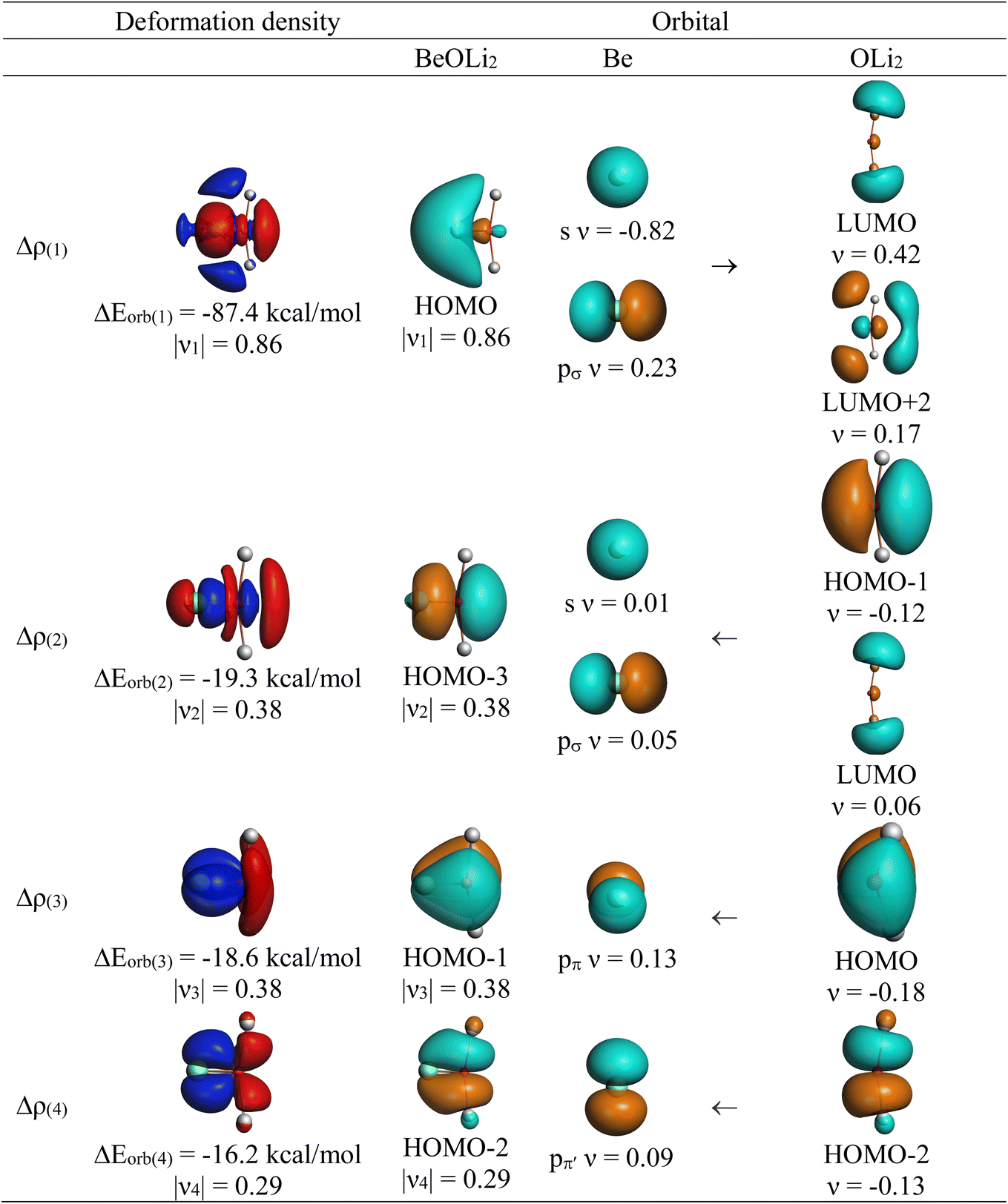
The EDA-NOCV enables the partition of the total ΔEorb into pairwise contributions of the orbital interactions that is very important to get a complete picture of the bonding. The charge deformation Δρk(r), resulting from the mixing of the orbital pairs ψk(r) and ψ−k(r) of the interacting fragments presents the amount and the shape of the charge flow due to the orbital interactions (eqn (2)), and the associated energy term ΔEorb provides the amount of stabilizing orbital energy originating from such an interaction (eqn (3)). Further details about the partitioning are given in the original work.48
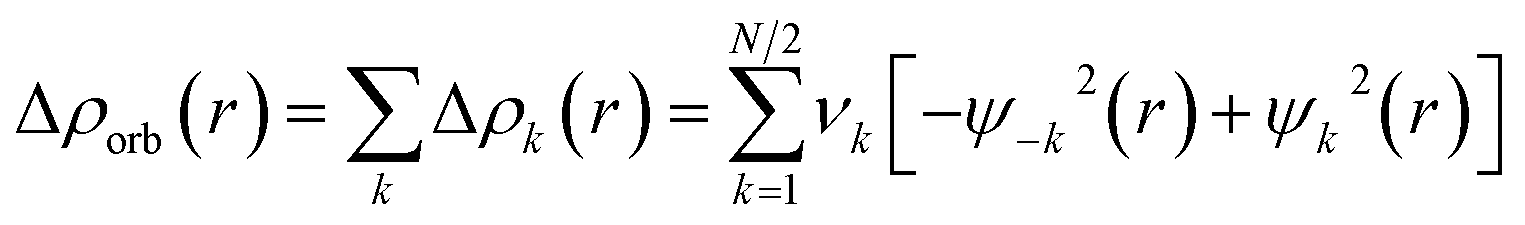 | (2) |
 | (3) |
Several reviews extensively discussed details of the EDA-NOCV method and its application, offering diverse perspectives and viewpoints.55–58
3. Results
Fig. 1 shows the low-lying isomers of the AeOLi2 species which were found on the singlet and triplet potential energy surface (PES) at the BP86-D3(BJ)/def2-QZVPP and CCSD(T)/def2-QZVPP levels. Both methods predict that the C2v structure Ae–OLi2 in the singlet 1A1 state is the global minimum on the PES. The linear singlet isomer (1Σ) with the connectivity Li–Ae–O–Li is energetically higher-lying than the C2v form, but the energy difference for the beryllium species is not very large (4.5 and 5.0 kcal mol−1 at the two levels of theory). The only triplet (3A1) isomer that could be located also has C2v symmetry, which is clearly 9–18 kcal mol−1 higher in energy than the singlet 1A1 species. | ||
| Fig. 1 The relative energies in kcal mol−1 of low-lying minimum energy isomers of AeOLi2 (Ae = Be, Mg, Ca, Sr, Ba) computed at the CCSD(T) and BP86-D3(BJ) (in square brackets) levels with the def2-QZVPP basis set. The green, pink, and gray colors represent the Ae, O, and Li atoms, respectively. #bent structure with Cs symmetry. | ||
Fig. 2 shows the calculated bond lengths and angles of the singlet (1A1) structures of AeOLi2 and the computed bond dissociation energies (BDE) for breaking the Ae–OLi2 bond. The Ae–O bond lengths of the AeOLi2 adducts are clearly longer and the BDEs are smaller than those in diatomic AeO. The free OLi2 ligand is linear but it becomes bent in the AeOLi2 adducts with the bending angle becoming more acute from the lightest compound BeOLi2 to the heaviest adduct BaOLi2. Note that the BDEs for the Ae–OLi2 bond exhibit a zig-zag trend at both levels of theory from BeOLi2, which has the largest BDE, to BaOLi2. Both methods suggest that MgOLi2 has the lowest BDE of the Ae–O bonds.
 | ||
| Fig. 2 Calculated geometrical parameters and bond dissociation energies (De, kcal mol−1) of the lowest-energy structure of AeOLi2 (Ae = Be, Mg, Ca, Sr, Ba) OLi2 and AeO computed at the CCSD(T)/def2-QZVPP level. The BP86-D3(BJ)/def2-QZVPP values are given in parentheses. The bond lengths are given in Å and the bond angles in degrees. | ||
Table 1 gives the calculated vibrational frequencies and IR intensities of the Ae–O stretching mode of the AeOLi2 adduct and free AeO. There is, as expected, a clear shift toward lower wave numbers for the adducts, which may be used as a helpful guide for the experimental studies. The frequency shift in the two methods is very similar. The complete vibrational spectrum of AeOLi2 is given in Table S1 of the ESI.‡
| Bond | BP86-D3(BJ) | CCSD (T) | ||||
|---|---|---|---|---|---|---|
| AeOLi2 | AeO | Shift Δν | AeOLi2 | AeO | Shift Δν | |
| Be–O | 1102.8 (4) | 1468.3 (3) | 360.5 | 1114.8 | 1467.4 | 352.6 |
| Mg–O | 518.2 (0) | 808.6 (16) | 290.4 | 557.3 | 823.8 | 266.5 |
| Ca–O | 506.0 (19) | 770.9 (89) | 264.9 | 487.7 | 699.3 | 211.6 |
| Sr–O | 421.0 (17) | 661.2 (83) | 240.2 | 413.0 | 626.3 | 213.3 |
| Ba–O | 405.6 (32) | 652.0 (143) | 246.4 | 404.0 | 670.4 | 266.4 |
The above results indicate that the BP86 method gives very similar results to the CCSD(T) approach. Thus, we can confidently use BP86 for the bonding analysis of Ae–OLi2 bonds, which is the main topic of this work as discussed below. As a starting point, the atomic partial charges are presented and discussed, which were calculated using four different methods, namely the NBO, QTAIM, Hirshfeld, and Voronoi approaches. The results are shown in Table 2. The data given by the NBO and QTAIM methods differ significantly from those given by the other two approaches, particularly for the oxygen atom. The NBO and QTAIM methods suggest that the oxygen atom has a partial charge of nearly −2e, which in the case of QTAIM is even slightly larger than the value for a full valence shell. It is important to note that the NBO method treats only the (n)s-AOs, but not the (n)p-AOs of the alkali and alkaline earth atoms as true valence orbitals, while the (n)p-AOs are considered as Rydberg AOs, whose contribution is given less weight in the NBO algorithm. Since the covalent bond involves the s/p hybridization of the atoms, this means that the electronic charge of the covalent bonds is excessively assigned to the oxygen atom. The QTAIM method, on the other hand, uses the curvature of the charge distribution as a criterion for the assignment of atomic charges, which is known to lead to highly charged atoms. For example, the QTAIM charges for CO2 at the CCSD(T)/def2-QZVPP level are C (+2.73e) and O (−1.37e) which would mean that CO2 is mainly bonded by electrostatic attraction.59
| Molecules | q O | |||
|---|---|---|---|---|
| NBO | QTAIM | Hirshfeld | Voronoia | |
| BeOLi2 | −1.88 | −2.16 | −0.56 | −0.54 |
| MgOLi2 | −1.92 | −2.02 | −0.66 | −0.69 |
| CaOLi2 | −1.79 | −2.05 | −0.64 | −0.69 |
| SrOLi2 | −1.82 | −2.05 | −0.66 | −0.71 |
| BaOLi2 | −1.80 | −2.08 | −0.64 | −0.69 |
In contrast, the Hirshfeld and Voronoi charges give a much smaller negative charge of a similar magnitude for oxygen in the AeOLi2 molecules between −0.56e and −0.71e. Both methods also suggest that the lighter Ae atoms Be and Mg are weak acceptors for the OLi2 ligand and that the heavy Ae atoms Ca, Sr, and Ba have only small positive charges in the molecules which are markedly smaller than those given by the NBO and QTAIM methods. We think that the Hirshfeld and Voronoi methods present a more faithful indication of the orbital overlap and the partial charges in the molecules. Fig. 3 shows the shape of the five highest-lying occupied orbitals of AeOLi2. It becomes obvious that there is a significant orbital overlap of the oxygen atom with Ae atoms and also with the Li atoms, which agrees with polar covalent bonds. The shape of the orbitals HOMO−1–HOMO−4 also shows that the heavier Ae atoms Ca, Sr, and Ba use their (n−1)d AOs for the covalent bonds.
 | ||
| Fig. 3 Shape of the five highest lying occupied Kohn–Sham MOs of AeOLi2 at BP86-D3(BJ)/def2-QZVPP. | ||
Table 2 gives also the calculated bond order for the Ae–OLi2 bonds using the Wiberg method44 (WBO) and the Mayer partitioning approach (MBO).45 The Wiberg orders are based on the CNDO method which neglects the orbital overlap, whereas the MBO approach explicitly considers the AO overlap. It has been shown that this leads to drastically different values for polar covalent bonds.60–62Table 2 shows that the WBO values are much smaller than the MBO data, which provide a more faithful account of the covalent bonds. However, the MBO values must not be identified with the number of bonded orbitals. Note that polar single bonds have bond orders that may be much smaller than 1 and the MBO values of AeOLi2 between 0.49 and 1.05 do not rule out that there is more than one strongly polar orbital. More sophisticated methods are required to identify the covalent bond multiplicity of a chemical bond.
The charge distribution in a molecule determines the electric dipole moment, which is a vector property that provides useful information about the spatial orientation of the electronic charge. Table 3 shows that the calculated values of the dipole moments of AeOLi2 at the CCSD(T)/def2-QZVPP level are very large between 8.18 D for MgOLi2 and 3.78 D for BaOLi2, where the negative end of the dipole moment lies at the Ae atoms with the polarity Ae→OLi2. The BP86-D3(BJ)/def2-QZVPP values are a bit smaller but the overall trend is the same, except for BeOLi2, which has a slightly higher dipole moment than MgOLi2. A similar situation was reported for the valence isoelectronic anions AeF− which possess large dipole moments with similar magnitude and a polarity Ae→F−.24 This was explained with the formation of a σ lone-pair orbital at atom Ae, whose center is located away from the nucleus of the atom.
| Molecules | μ | |
|---|---|---|
| BP86-D3(BJ) | CCSD(T) | |
| BeOLi2 | 6.37 | 7.89 |
| MgOLi2 | 6.21 | 8.18 |
| CaOLi2 | 4.06 | 6.60 |
| SrOLi2 | 3.54 | 5.80 |
| BaOLi2 | 2.07 | 3.78 |
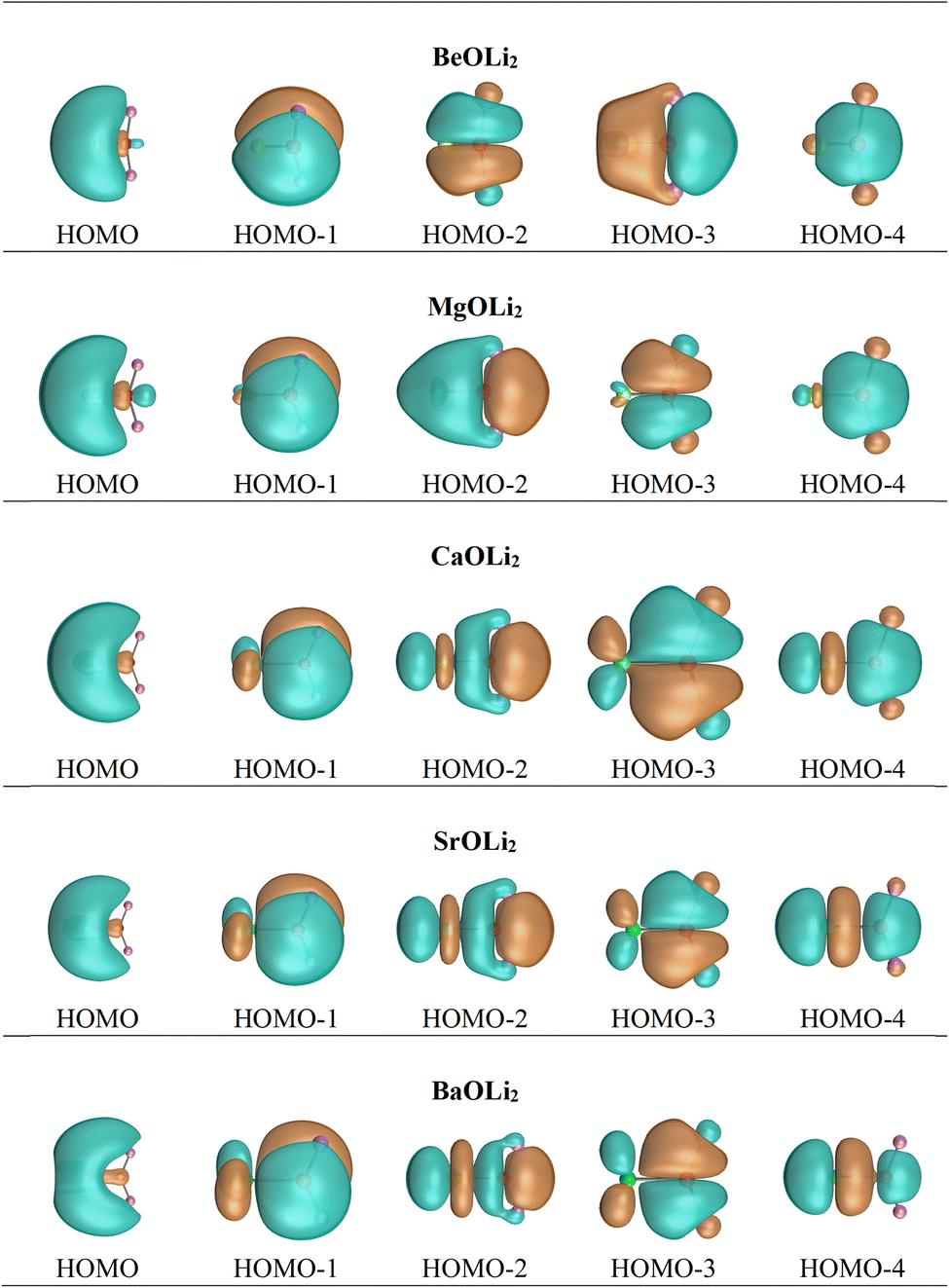
Inspection of the spatial charge distribution in AeOLi2 reveals an apparently similar situation as in AeF−. Fig. 4 shows the Laplacian distribution of electron density, ∇2ρ(r) of AeOLi2 which exhibits a distinct area of charge concentration (red dashed lines) in the σ lone-pair region of BeOLi2 and MgOLi2. It is less visible in the Laplacian distribution of the heavier homologues, because the curvature of the electron-rich species is less pronounced. There are as expected Ae–O and O–F bond paths and bond critical points. Inspection of the occupied orbitals (Fig. 3) shows a very similar shape of the Ae σ lone-pair HOMO which occurs in all AeOLi2 species. A detailed analysis of the individual orbital components of the dipole moments in AeF− showed that the electronic charge of the HOMO has a decisive influence on the overall dipole moment.24 The dipole moments of AeOLi2 show similar characteristics.
 | ||
| Fig. 4 Laplacian distribution ∇2ρ(r) of AeOLi2 (Ae = Be, Mg, Ca, Sr, Ba) at the CCSD(T)/def2-QZVPP level. Red lines indicate the areas of charge concentration (∇2ρ(r) < 0), while blue lines show the areas of charge depletion (∇2ρ(r) > 0). The thick solid lines connecting the atomic nuclei are the bond paths. Blue dots are bond critical points (bcp). The thin lines which cross the bcp show the zero-flux surfaces in the molecular plane that separate the atomic basins. The alkaline earth metal atoms are given at the left. | ||
Very detailed information about the nature of the Ae–OLi2 bonds is available from EDA-NOCV calculations, which has proven to be a very powerful tool for bonding analysis in a variety of main-group compounds, transition-metal complexes as well as lanthanides and actinides.55–58,63–66 The focus of the EDA-NOCV method is the process of bond formation between the chosen fragments, which distinguishes it from most other methods like QTAIM,34 Interacting Quantum Atoms (IQA)67 and ELF36 that analyze the interaction between the atoms in the final molecule after the bond is formed. The choice of the electronic state and the charge of the fragments are crucially important for the results. It has been shown that the size of the orbital term ΔEorb is a very helpful criterion to identify the most suitable fragments for the bonding analysis. The fragments that give the smallest ΔEorb value are the best, as they change the least during bond formation, which proceeds along the path of bond dissociation/bond formation.68–73 We want to point out that the choice of the best fragments does not automatically identify the oxidation state of the atoms, because the oxidation state is related to the hypothetical charge of an atom if all of its bonds to other atoms were fully ionic. This may or may not be the same as the fragments which give the smallest ΔEorb value.
In the case of AeOLi2, we employed several options for neutral charged fragments. It turned out that the neutral species Ae atom and OLi2 in their electronic singlet ground state clearly give the smallest absolute values for ΔEorb and thus, they are used to analyze the bond formation of the Ae–OLi2 bond. The numerical results of EDA-NOCV are shown in Table 4. The EDA results using other fragments are given in Tables S2–S6 in the ESI.†
| Energy | Orbital interaction | Ae (ns2, 1S) + OLi2 (1A1) | ||||
|---|---|---|---|---|---|---|
| BeOLi2 | MgOLi2 | CaOLi2 | SrOLi2 | BaOLi2 | ||
| a The percentage contribution with respect to total attraction is given in parentheses. b The percentage contribution in parentheses is given with respect to total orbital interaction. | ||||||
| ΔEint | −81.0 | −40.2 | −60.9 | −55.2 | −65.0 | |
| ΔEPauli | 253.1 | 130.8 | 137.8 | 131.5 | 155.9 | |
| ΔEdispa | −1.7 | −2.4 | −2.7 | −2.7 | −2.8 | |
| ΔEelstata | −189.0 (56.9%) | −114.2 (67.7%) | −126.9 (64.7%) | −121.4 (66.0%) | −138.7 (63.6%) | |
| ΔEorba | −143.4 (43.1%) | −54.4 (32.3%) | −69.2 (35.3%) | −62.6 (34.0%) | −79.4 (36.4%) | |
| ΔEorb(1)b | Ae→OLi2 σ donation | −87.4 (60.9%) | −35.8 (65.8%) | −34.0 (49.1%) | −28.7 (45.8%) | −29.5 (37.2%) |
| ΔEorb(2)b | Ae←OLi2 σ backdonation | −19.3 (13.5%) | −7.0 (12.9%) | −14.9 (21.5%) | −15.3 (24.4%) | −24.3 (30.6%) |
| ΔEorb(3)b | Ae←OLi2 π backdonation | −18.6 (13.0%) | −5.9 (10.8%) | −10.3 (14.9%) | −9.2 (14.7%) | −12.3 (15.5%) |
| ΔEorb(4)b | Ae←OLi2 π backdonation | −16.2 (11.3%) | −4.9 (9.0%) | −8.6 (12.4%) | −7.8 (12.5%) | −10.3 (13.0%) |
| ΔEorb(rest)b | −1.8 (1.3%) | −0.7 (1.3%) | −1.2 (1.7%) | −1.5 (2.4%) | −2.8 (3.5%) | |
Table 4 shows that the calculated total interaction energies ΔEint of the Ae–OLi2 bonds are only slightly lower than and exhibit the same trend as the bond dissociation energies De (Fig. 2). This is because the two terms differ only in the geometrical deformation/relaxation of the OLi2 fragment, which requires very little energy. The attractive component of ΔEint only has a small contribution from the dispersion interaction ΔEdisp and the major component comes from the electrostatic (Coulomb) attraction ΔEelstat, which provides 57–68% of the total attraction. This is reasonable, because the Ae–OLi2 bonds are very polar and the charge accumulation of the covalent interactions that comes from the interference of the wave functions is shifted toward the nucleus of the more electronegative atom. Note that the shift of the electronic charge from the midpoint of a bond toward one atom does not reduce but rather strengthens the quasiclassical Coulomb attraction, because the distance between the electronic charge in the bonding region and one nucleus becomes smaller. For a very insightful discussion, we refer to the literature.74–76 The electrostatic contribution to the interatomic interaction is sometimes termed as ionic bonding, which is a misleading expression. Ionic bonding occurs between charged fragments with negligible overlap, and is only found in ionic solids and ionic solution. There is no ionic bonding in molecules. The frequent occurrence of “ionic” bonding in molecules stems from the valence bond (VB) approach. The VB method has no explicit expression for a polar bond, which is mathematically described by a mixture of the terms for electron-pair bonding and ionic bonding.77 For details, we refer to a recent publication.78
It is useful to compare the trend of the EDA-NOCV energy components with the bond dissociation energies. Fig. 5 shows that the BDE values and the interaction energies ΔEint between the frozen fragments exhibit the same zig-zag pattern. This means that the geometry relaxation of the OLi2 species has only a negligible effect on the relative bond strength of the Ae–OLi2 bonds. It is interesting to note that both attractive components ΔEorb and ΔEelstat also show the same zig-zag behavior. The bond strengthening/bond weakening may equally well be ascribed to the change in covalent bonding and electrostatic attraction. It is gratifying that the experimentally observed bond dissociation energies correlate well with the energy components of the EDA-NOCV calculations, which emphasizes the relevance of the method.
 | ||
| Fig. 5 Trend of the bond dissociation energy De and the energy components of the EDA-NOCV calculations ΔEint (total interaction energy of the frozen fragments), ΔEelstat (electrostatic attraction) and ΔEorb (orbital interaction). | ||
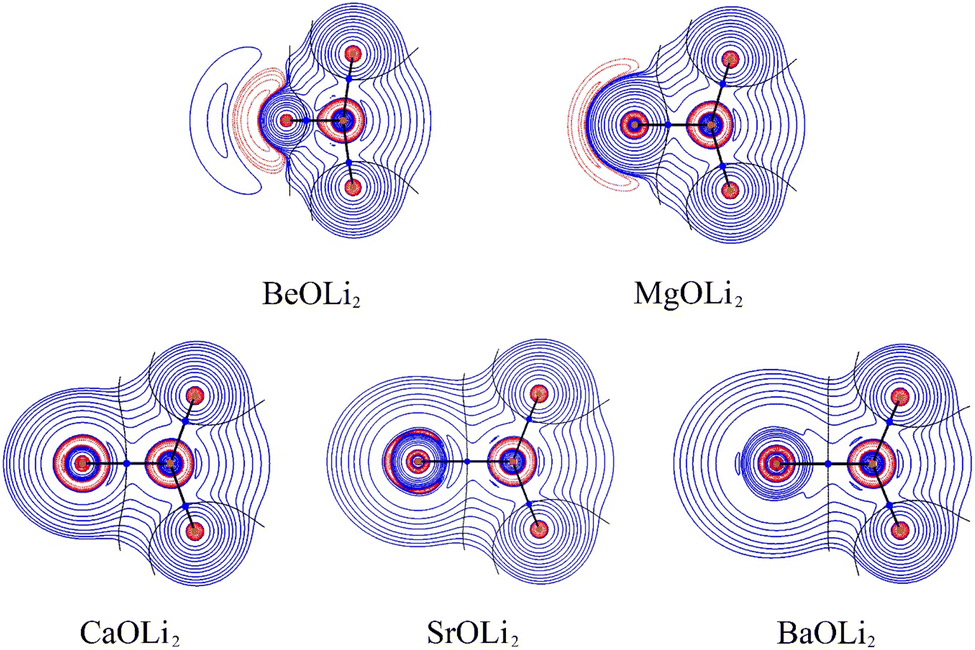
The most important information about Ae–OLi2 comes from the breakdown of the total orbital interaction ΔEorb into pairwise contributions. Table 4 shows that there are four major terms ΔEorb(1)–ΔEorb(4) which provide >96% of the covalent interactions. The other orbital term ΔEorb(rest) comes from the relaxation of the fragment orbitals which are not directly involved in the Ae–OLi2 interactions. The nature of the pairwise orbital terms can be identified by examination of the associated deformation densities Δρ and the connected orbitals. They are shown for BeOLi2 and CaOLi2 in Fig. 6 and 7. The deformation densities and connected orbitals of MgOLi2, SrOLi2 and BaOLi2 are displayed in Fig. S1–S3 of the ESI.†
 | ||
| Fig. 6 Plot of the deformation densities, Δρ(1)–(4) shown as the sum of α and β electronic charges corresponding to ΔEorb(1)–(4) and the related interacting orbitals in the singlet states of BeOLi2 at the BP86-D3(BJ)/TZ2P-ZORA level using Be (2s2, 1S) + OLi2 (1A1) as interacting fragments. The eigenvalues ν indicate the size of the charge flow. The direction of charge flow is red → blue. The isovalue for Δρ(1)–(4) is 0.001 au. | ||
 | ||
| Fig. 7 Plot of the deformation densities, Δρ(1)–(4) shown as the sum of α and β electronic charges corresponding to ΔEorb(1)–(4) and the related interacting orbitals in the singlet states of CaOLi2 at the BP86-D3(BJ)/TZ2P-ZORA level using Ca (4s2, 1S) + OLi2 (1A1) as interacting fragments. The eigenvalues ν indicate the size of the charge flow. The direction of charge flow is red → blue. The isovalue for Δρ(1)–(4) is 0.001 au. | ||
The strongest orbital term ΔEorb(1) comes from Ae→OLi2 σ donation and the other three orbital interactions ΔEorb(2) – ΔEorb(4) are due to Ae←OLi2 backdonation with one σ component and two π components. This is a big difference to the results for the valence isoelectronic anions AeF−, where four components were also found, but all of them originate from the backdonation of Ae←F−.23,24 Unlike the F− ligand, OLi2 has empty orbitals that can act as acceptor orbitals for donation from the occupied orbitals of the Ae atom. This leads to a different bonding situation, particularly in the lighter systems where Ae = Be, Mg. In AeF−, the two σ terms in BeF− and MgF− come from the concomitant σ donation and the polarization (hybridization) of the lone-pair AO of Ae, which are thus two components of the single σ backdonation Ae←F−. The bond multiplicity in BeF− and MgF− is, therefore, a triple bond Ae F−. In contrast, the neutral molecules BeOLi2 and MgOLi2 have genuine quadruple bonds where the occupied orbital of Ae atoms is a donor and the three vacant valence p AOs of Ae are acceptors featuring four dative bonds systems Ae
F−. In contrast, the neutral molecules BeOLi2 and MgOLi2 have genuine quadruple bonds where the occupied orbital of Ae atoms is a donor and the three vacant valence p AOs of Ae are acceptors featuring four dative bonds systems Ae OLi2. The lighter systems with Ae = Be, Mg possess genuine quadruple bonds, because the valence orbitals of the metal atoms built strong dative interactions with the OLi2 fragment.
OLi2. The lighter systems with Ae = Be, Mg possess genuine quadruple bonds, because the valence orbitals of the metal atoms built strong dative interactions with the OLi2 fragment.
Quadruple bonds Ae F− were found for the anions with Ae = Ca, Sr, Ba because the heavier alkaline earth metals utilize their (n−1)d AOs for covalent bonding.23,24 Inspection of Fig. 7, S2 and S3† shows that the orbital interactions in the heavier molecules CaOLi2–BaOLi2 also involve the (n−1)d AOs of the metals as acceptor orbitals for the Ae←OLi2 σ and π backdonation ΔEorb(2)–ΔEorb(4) but the strongest orbital term ΔEorb(1) comes from Ae→OLi2 σ donation where the occupied (n)s AO of the atom Ae acts as a donor orbital. The heavier systems AeOLi2 where Ae = Ca, Sr, Ba have quadruple bonds Ae
F− were found for the anions with Ae = Ca, Sr, Ba because the heavier alkaline earth metals utilize their (n−1)d AOs for covalent bonding.23,24 Inspection of Fig. 7, S2 and S3† shows that the orbital interactions in the heavier molecules CaOLi2–BaOLi2 also involve the (n−1)d AOs of the metals as acceptor orbitals for the Ae←OLi2 σ and π backdonation ΔEorb(2)–ΔEorb(4) but the strongest orbital term ΔEorb(1) comes from Ae→OLi2 σ donation where the occupied (n)s AO of the atom Ae acts as a donor orbital. The heavier systems AeOLi2 where Ae = Ca, Sr, Ba have quadruple bonds Ae OLi2 like the lighter systems with Ae = Be, Mg.
OLi2 like the lighter systems with Ae = Be, Mg.
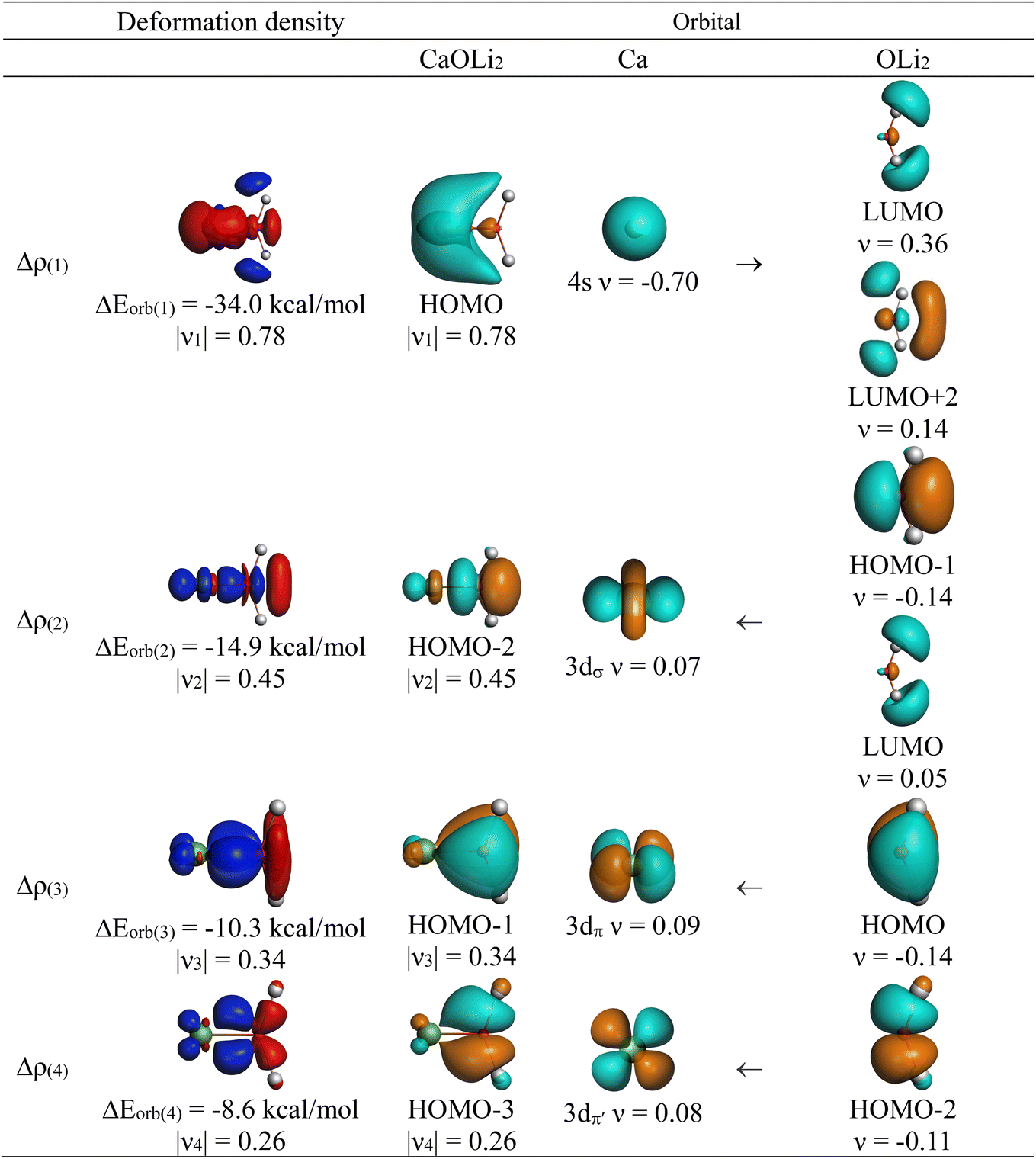
The strongest orbital interaction term ΔEorb(1), which comes from Ae→OLi2 σ donation that does not occur in AeF−, shall be analyzed in more detail. Fig. 6, 7 and S1–S3† show that the acceptor orbitals LUMO and LUMO+2 of OLi2 are antibonding O–Li orbitals where the largest coefficients are at the lithium atoms. Inspection of the areas of charge depletion and charge accumulation Δρ(1) shows that the area along the Ae–O axis undergoes charge depletion while the area of charge accumulation is actually found along the two Ae–Li bond axes. This holds for all molecules AeOLi2 (Ae = Be–Ba). This means that the strongest orbital interaction ΔEorb(1) of AeOLi2 is rather an Ae–Li bond to both lithium atoms than an Ae–O bond. This is a very unusual situation that resembles the collective interactions recently proposed by Pendas and Foroutan-Nejad et al. as ‘exotic bonds’ in organometallic compounds.79 The relevance of collective interactions was disputed by Bickelhaupt, Sola and coworkers80 but it was supported by further work.81–83 A comparison of the nature of the collective interactions proposed by the authors with the bonding in AeOLi2 shows that there is only a structural similarity whereas the nature of the chemical bonds is very different from each other. This is illustrated in Fig. 8a, where the interatomic interactions between a metal atom M, an electron-poor atom E and electron-rich atoms X are shown. The figure is adapted from ref. 83. The authors suggest that there is electrostatic repulsion between atoms M and E (red dotted line) and the attraction comes from electrostatic attraction between M and atoms X. In contrast, the interaction between Ae and O atoms in AeOLi2 (Fig. 8b) comes from triple Ae OLi2 dative bonding enhanced by electrostatic attraction. The feature of collective interaction comes from the unusual σ-backdonation Ae→OLi2 which is a covalent bond with two components that are directed toward the Li atoms. This type of bond is clearly different from the collective interactions proposed by Foroutan-Nejad and co-workers.79,81–83
OLi2 dative bonding enhanced by electrostatic attraction. The feature of collective interaction comes from the unusual σ-backdonation Ae→OLi2 which is a covalent bond with two components that are directed toward the Li atoms. This type of bond is clearly different from the collective interactions proposed by Foroutan-Nejad and co-workers.79,81–83
 | ||
Fig. 8 Schematic representation of (a) the proposed nature of the bonding in collective interactions where the red dashed line indicates Coulomb repulsion and the green dashed line indicates electrostatic attraction; (b) covalent bonding in AeOLi2 which consists of three (σ and 2π) dative interactions Ae OLi2 and one bifurcated σ donation Ae→OLi2. OLi2 and one bifurcated σ donation Ae→OLi2. | ||
It is noteworthy that the charge accumulation Δρ(1) in the Ae–Li bonding region shown by the deformation densities does not lead to separate Ae–Li bond paths in the QTAIM analysis (Fig. 4). The Laplacian distribution ∇2ρ(r) gives the curvature of the density distribution and the absence of a bond path between two atoms does not prove that there is no covalent interaction. This has been shown previously.84 The strength of QTAIM analysis is that it provides detailed information about the overall electronic structure of a molecule after bond formation is complete, which can also be obtained from experiments. The weakness is that it gives no insight into the process of bond formation itself. There is also no direct information about the bond multiplicity. QTAIM also overlooks attractive interactions between atoms that are not strong enough to establish a critical bonding point.
The EDA-NOCV analysis reveals that the unusual σ bond with two components (collective interactions) occurs as the strongest component in the molecules AeOLi2, which leads to quadruple bonding in all compounds, even in BeOLi2 and MgOLi2. In contrast to ΔEorb(1), the deformation densities Δρ(2)–Δρ(4) of the other three orbital terms ΔEorb(2)–ΔEorb(4) show that the charge accumulation (blue area) is along the Ae–O bond axis which indicates the formation of one σ and two π bonds. The Ae OLi2 quadruple bond has two σ and two π bonds where one σ bond is a collective bond between the Ae atom and Li2.
OLi2 quadruple bond has two σ and two π bonds where one σ bond is a collective bond between the Ae atom and Li2.
It is also interesting to analyze the two π backdonations ΔEorb(3) and ΔEorb(4) in more detail. Fig. 6 and 7 show that ΔEorb(3) comes from the out-of-plane π⊥ backdonation Ae←OLi2, which is a bit stronger than the in-plane π‖ backdonation ΔEorb(4). The former donation comes from the π lone-pair of the oxygen atom while the latter is an OLi2 bonding orbital with very small contributions of the Li atoms. But the donor orbitals of OLi2 in the orbital interactions of ΔEorb(2)–ΔEorb(4) are not purely oxygen-based orbitals but also have some lithium valence AOs. The appearance of a vacant orbital of OLi2 in the Ae←OLi2 backdonation is due to the polarization of the occupied orbitals along the orbital interaction. The polarization of the fragment orbitals should be considered as part of the stabilizing orbital interactions, since it is caused by the bond formation.
The deformation densities associated with the orbital interactions ΔEorb(1)–ΔEorb(4) explain nicely why there is still a σ lone-pair type area of charge accumulation at the Ae atoms although the valence electrons of the (n)s AO are engaged in the Ae→OLi2 donation that gives ΔEorb(1). There is a concomitant charge donation in the opposite direction Ae←OLi2 due to the orbital interactions ΔEorb(2)–ΔEorb(4).
The EDA-NOCV results demonstrate that there is a significant difference between the bond formation of F− and valence isoelectronic OLi2 with Ae atoms in AeF− and AeOLi2. The fluorine anion F− may only be a donor whereas OLi2 may also be an electron acceptor, because it has vacant O–Li valence orbitals. The anion F− donates electronic charge from three electron pairs (one σ and two π orbitals) to the three vacant valence orbitals of Be and Mg and it donates four electron pairs (two σ and two π orbitals) to four vacant valence orbitals of Ca, Sr, and Ba, which have an sd-valence space. OLi2 also donates three electron pairs (one σ and two π orbitals) to three vacant valence orbitals of all Ae atoms Be–Ba, but it forms a fourth dative bond through backdonation from the (n)s electron pair of the Ae atom to the vacant O–Li orbitals.
To ensure that the surprising quadruple binding in all systems AeOLi2 is not an artifact of the EDA-NOCV method, but is also suggested by other methods, a further analysis using adaptive natural density partitioning (AdNDP)35 was performed. The AdNDP method is a fundamentally different approach than the EDA-NOCV method and it is particularly well suited for assigning chemical bonding in the present case, where the contribution of the O–Li vacant orbitals to the chemical bonds is addressed. The AdNDP method was developed by Boldyrev and coworkers to give information about chemical bonding in delocalized systems.35
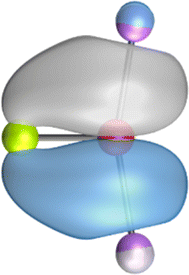
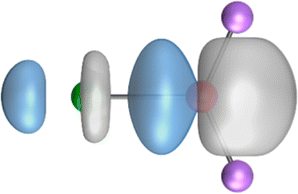
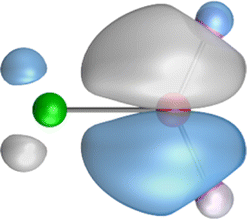
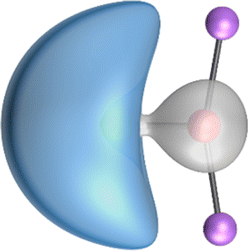
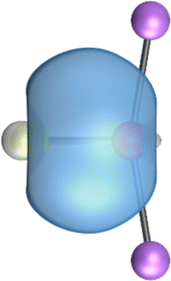
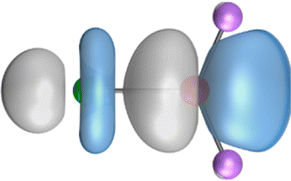
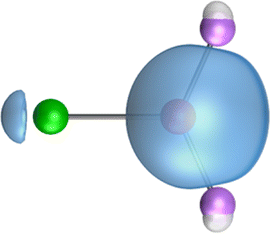
The search of 2c–2e orbitals involving the Ae–O moiety gave five orbitals with high occupation numbers (ONs) consisting of a lone pair at the oxygen atom, two Ae–O σ bonding orbitals and two π bonding orbitals. The numerical results are shown in Table 5 along with the shape of the orbitals for BeOLi2 and BaOLi2. The orbitals of the other species are similar and are shown in Fig. S4 of the ESI.† The four 2c–2e Ae–O bonding orbitals correlate nicely with the four interactions found in the EDA-NOCV analysis. The σ2 orbital and the two π orbitals with a very high ON close to 2 are related to the three orbital terms ΔEorb(2)–(4) of the EDA-NOCV analysis whereas the σ1 orbital with a lower ON (1.32–1.73) is related to the Ae→OLi2 backdonation ΔEorb(1). The latter orbital is more delocalized than the others, which becomes obvious due to the ON number for 4c–2e bonds (Table 5), but it is clearly identified as an Ae–OLi2 bonding orbital. The lower ON of the σ1 orbital is related to the orbital interaction ΔEorb(1) where charge is donated to the LUMO and LUMO+2 of OLi2 (Fig. 6 and 7) which are mainly localized at Li.
| Molecules | σ1 | σ2 | π1 | π2 | Lone pair |
|---|---|---|---|---|---|
| BeOLi2 | 1.67 (2.00) | 1.99 (2.00) | 1.99 (2.00) | 1.98 (2.00) | 2.00 (2.00) |
| MgOLi2 | 1.73 (2.00) | 1.99 (2.00) | 1.98 (2.00) | 1.97 (2.00) | 2.00 (2.00) |
| CaOLi2 | 1.52 (2.00) | 1.98 (2.00) | 1.99 (2.00) | 1.98 (2.00) | 2.00 (2.00) |
| SrOLi2 | 1.50 (2.00) | 1.98 (2.00) | 1.99 (2.00) | 1.98 (2.00) | 2.00 (2.00) |
| BaOLi2 | 1.32 (2.00) | 1.98 (2.00) | 1.99 (2.00) | 1.98 (2.00) | 2.00 (2.00) |
| σ1 | σ2 | π1 | π2 | Lone pair |
|---|---|---|---|---|
| 2c-2e Orbitals | ||||
| BeOLi2 | ||||

|

|

|
|

|
| BaOLi2 | ||||

|
|

|
|

|
| 4c-2e Orbitals | ||||
| BeOLi2 | ||||
|

|

|

|
|
| BaOLi2 | ||||

|
|

|

|
|
It is instructive to compare the 2c–2e AdNDP orbitals with the 4c–2e orbitals, which are also shown in Table 5. The shape of the orbitals σ2, π1, and π2 and the lone-pair are very similar since the 2c–2e orbitals have ON numbers of ∼2. The shape of the 4c–2e orbital σ1 clearly differs from that of the 2c–2e orbital σ1, particularly for BaOLi2, where the delocalization toward lithium becomes obvious. But the shape of the orbitals does not reveal that the covalent interaction takes place mainly between Ae and Li. The deformation densities ρ1 associated with the orbital interaction ΔEorb(1), which are shown in Fig. 6, 7, S1–S3 give more direct evidence for the appearance of collective interactions due to the covalent bonding between the Ae atom and Li. But the AdNDP results support the conclusion of the EDA-NOCV analysis that all systems AeOLi2 have an Ae–O quadruple bond.
The suggestion of quadruple bonding Ae OLi2 is based on the energy contributions of the four orbital interactions. This is reasonable because chemical bonding in molecules is foremost an energy occurrence which comes from the interference of the wave functions. A related question concerns the associated charge distribution, which eventually emerges as a result of the chemical bond. Numerous methods have been developed to divide the total electronic charge of a molecule into atomic or electron pair regions, which provide important information about the electronic structure and the bonding situation. The QTAIM method and the Laplacian distribution mentioned above are examples where atomic basins are considered. A more fine-grained method is the ELF (Electron Localization Function) introduced by Becke and Edgecombe in 1990,36 which divides the total electronic charge into basins that can be associated with traditional chemical concepts such as bonding pairs, core electrons, and lone pairs. The concept of synapticity of the basin (or the attractor) was introduced by Savin et al.85,86 According to this approach, a disynaptic bonding basin, V(A,B), represents a covalent A–B bond, whereas monosynaptic basins, V(A), correspond to lone pairs of atom A in the Lewis representation of the valence electrons. The concept was further developed by Silvi for multicenter bonds who introduced the synaptic order.87 There are monosynaptic basins corresponding to electron lone pairs, disynaptic basins corresponding to conventional two-center bonds, trisynaptic basins corresponding to 3c–2e bonds, etc. In our previous work, we used the ELF method for analyzing the chemical bonds in AeF−, which has a maximum synaptic order of two.24 It is interesting to learn about the performance of the ELF method and the synaptic order in the AeOLi2 molecules, because the EDA-NOCV results suggest the occurrence of collective bonds between atoms that are not considered in a conventional Lewis model.
OLi2 is based on the energy contributions of the four orbital interactions. This is reasonable because chemical bonding in molecules is foremost an energy occurrence which comes from the interference of the wave functions. A related question concerns the associated charge distribution, which eventually emerges as a result of the chemical bond. Numerous methods have been developed to divide the total electronic charge of a molecule into atomic or electron pair regions, which provide important information about the electronic structure and the bonding situation. The QTAIM method and the Laplacian distribution mentioned above are examples where atomic basins are considered. A more fine-grained method is the ELF (Electron Localization Function) introduced by Becke and Edgecombe in 1990,36 which divides the total electronic charge into basins that can be associated with traditional chemical concepts such as bonding pairs, core electrons, and lone pairs. The concept of synapticity of the basin (or the attractor) was introduced by Savin et al.85,86 According to this approach, a disynaptic bonding basin, V(A,B), represents a covalent A–B bond, whereas monosynaptic basins, V(A), correspond to lone pairs of atom A in the Lewis representation of the valence electrons. The concept was further developed by Silvi for multicenter bonds who introduced the synaptic order.87 There are monosynaptic basins corresponding to electron lone pairs, disynaptic basins corresponding to conventional two-center bonds, trisynaptic basins corresponding to 3c–2e bonds, etc. In our previous work, we used the ELF method for analyzing the chemical bonds in AeF−, which has a maximum synaptic order of two.24 It is interesting to learn about the performance of the ELF method and the synaptic order in the AeOLi2 molecules, because the EDA-NOCV results suggest the occurrence of collective bonds between atoms that are not considered in a conventional Lewis model.
Fig. 9 shows the ELF results of the five molecules at the CCSD(T)/def2-QZVPP level. For BeOLi2, there are no monosynaptic or disynaptic basins. The calculation gives one trisynaptic basin V(Be,Li,Li) with a population of 2.01e and another trisynaptic basin V(O,Li,Li) with a population of 5.20e. In addition, there is a tetrasynaptic basin V(Be,O,Li,Li) that is populated by 2.56e. The ELF results for MgOLi2 are quite different. There are two separate diatomic basins V(Mg,Li) with a population of 1.93e and there is a diatomic basin V(Mg,O) which is populated by only 1.30e. There are furthermore two trisynaptic basins V(Mg,O,Li) with a total population of 6.52 e. The ELF results clearly show that the transformation of the synaptic basins into conventional Lewis structures without further analysis of the interatomic interactions is not possible for these molecules. The results also show that the lithium atoms are closely involved in the overall covalent bonding, which agrees with the analysis of the strongest ΔEorb(1) term of the EDA-NOCV calculations featuring Ae→OLi2 σ donation between Ae and the Li atoms. We want to point out that the EDA-NOCV method uses the undisturbed electronic structures of Ae and OLi2 for the analysis of the Ae–OLi2 bonds, whereas the ELF approach considers the final electronic structure at the endpoint of bond formation. The two approaches are complimentary, but the EDA-NOCV results are more useful to identify the individual orbital interactions which provide the best Lewis structure for a molecule.
 | ||
| Fig. 9 ELF calculation showing the synaptic basins and the occupation numbers (ONs) of AeOLi2 (Ae = Be–Ba) at CCSD(T)/def2-QZVPP. The contour line diagrams have an isovalue of 0.56e per a.u. | ||
Fig. 9 shows also the ELF basins for the heavier AeOLi2 molecules where Ae = Ca, Sr. Ba. For CaOLi2, there are two separate disynaptic basins V(Ca,Li) with a total population of 1.88e and a tetrasynaptic basin V(Ca,O,Li,Li) that is populated by 7.75e. A similar situation is found for SrOLi2 with two separate diatomic basins V(Sr,Li) with a total population of 1.85e and two separate trisynaptic basins V(Sr,O,Li) that are populated by 7.76e. The finding of two separate trisynaptic basins in the latter molecule instead of a tetrasynaptic basin as in the calcium species is probably a numerical artefact. Somewhat different ELF results are calculated for BaOLi2. Fig. 9 shows that the two separate disynaptic basins V(Ba,Li) are populated by only 1.45e but they are complemented by two monosynaptic basins V(Li) with a total population of only 0.38e. The latter disynaptic and monosynaptic basins of the barium compound correspond to the diatomic basins of the calcium and strontium homologue. There are also two separate trisynaptic basins V(Ba,O,Li) but they are populated by only 6.93e, much less than the two separate trisynaptic basins V(Sr,O,Li) of the strontium molecule.
The ELF results of the heavier systems confirm the picture of the lighter homologues that there is a delocalized bonding interaction in AeOLi2 where the lithium atoms participate to a surprisingly large extent in the covalent bonding of the molecule. This can be explained by the EDA-NOCV results, where the strongest orbital interaction ΔEorb(1) comes from Ae→OLi2 σ donation of the occupied (n)s2 AO into the vacant O–Li antibonding orbitals, which have the largest coefficient at Li. This leads to an unusual covalent Ae–Li2 interaction, which is not considered in the standard Lewis picture of chemical bonding. This is an important component of the quadruple bond between the Ae atoms and the OLi2 ligand which can be considered as an example of the recently introduced collective bonds. The description of the bonding situation with the formula Ae OLi2 comprises direct triple dative backbonding from oxygen to the Ae atom and σ bonding from Ae to Li2 which enhances the overall Ae–OLi2 attraction.
OLi2 comprises direct triple dative backbonding from oxygen to the Ae atom and σ bonding from Ae to Li2 which enhances the overall Ae–OLi2 attraction.
We also carried out ELF calculations of AeOLi2 using the electronic structures at the BP86-D3(BJ)/def2-QZVPP level. The results are very similar to values obtained at the CCSD(T)/def2-QZVPP level. They are shown in Fig. S5 of the ESI.† The atomic basins suggested by the two methods are the same with slightly different occupation numbers except that the BP86-D3(BJ)/def2-QZVPP calculation does not give monosynaptic basins V(Li) for BaOLi2 but a tetrasynaptic basin V(Ba,O,Li,Li) with an occupation of 7.55e.
4. Discussion
The results of the various methods for bond analysis clearly show that the description of the bonding situation in a molecule using standard Lewis formulae, which are a helpful model for describing the atomic structure and connectivity, is only a rough approximation for understanding the electronic structure of a compound. And it becomes clear that a true insight into the nature of interatomic interactions requires the use of multiple methods of charge and energy partitioning, the fundamentals and approximations of which must be known in order to provide meaningful information about the chemical bonds in a molecule. There are a ubiquitous number of publications in which a single method of bonding analysis is used – often without precise knowledge of the basic approximations of the method – and its results are then used as “evidence” for a seemingly authoritative interpretation of the chemical bonds in a molecule. This is particularly dangerous with molecules that have unusual chemical bonds and that differ from reference molecules.The present molecules AeOLi2 (Ae = Be–Ba) and the nature of the Ae–OLi2 bonds are good examples for the above statement. The partial charges calculated using the Hirshfeld and Voronoi approaches indicate an approximate balance between the donation and back-donation between Ae and OLi2, with the back-donation Ae←OLi2 being slightly larger than the donation Ae→OLi2 for the lighter species with Ae = Be, Mg, while the reverse order is predicted for the heavier systems with Ae = Ca–Ba. The NBO and QTAIM methods suggest donation Ae→OLi2 for all systems, but both these methods have methodical deficiencies which make the results of the Hirshfeld and Voronoi approaches more reasonable.
The EDA-NOCV method suggests four distinct pairwise orbital interactions between Ae and OLi2 that clearly establish fourfold bonding between the two fragments. This is supported by the AdNDP approach, which transforms the electronic wavefunction into the most appropriate Lewis structure. Close examination of the four orbital interactions shows that the strongest component comes from Ae→OLi2 σ donation from the (n)s2 electron pair of Ae into vacant OLi2 orbitals, which have the largest coefficient at the Li atoms. Inspection of the associated charge deformation reveals that a covalent bond is present between the Ae atom and the lithium atoms, where the charge accumulation due to the interference of the wavefunction is along the two Ae–Li bond axes. This resembles the recently proposed collective interactions, where a covalent bond is formed between two atoms that are not directly connected when the molecule is sketched with a Lewis structure.
The remaining three orbital interactions come through Ae←OLi2 backdonation from energetically high-lying occupied MOs of OLi2 into vacant AOs of Ae. The latter AOs are the (n)p orbitals of Ae = Be, Mg while for the heavier Ae atoms Ca–Ba, the (n−1)d AOs are the acceptor orbitals. This shows that the heavier elements calcium, strontium and barium bind in molecules like transition metals. The occupied donor orbitals of OLi2 are mainly localized at oxygen with minor contributions at lithium. The four orbital interactions in AeOLi2 suggest some multicenter bonding, which is nicely reflected in the results of the ELF calculations. The ELF calculations of AeOLi2 give mainly trisynaptic and even tetrasynaptic basins for all systems. A proper sketch of the bonding situation in AeOLi2 which accounts for the quadruple bonding with four dative interactions is the formula Ae OLi2.
OLi2.
The results of this work might also stimulate experimental research on the catalytic properties of systems like AeOE2O (Ae = Be, Mg, Ca; E = Li, Na, K) where the bonding situation might be related to our systems.88 We plan to extend our studies in this direction.
5. Conclusion
The results of this work are summarized as follows.• The lowest energy isomer of the AeOLi2 (Ae = Be–Ba) complexes calculated at the BP86-D3(BJ)/def2-QZVPP and CCSD(T)/def2-QZVPP levels has a C2v geometry and a singlet (1A1) electronic ground state. The bond dissociation energy of the Ae–OLi2 bonds exhibit a zig-zag trend at both levels of theory from BeOLi2, which has the largest BDE (De = 73.0 kcal mol−1 at CCSD(T)), to BaOLi2. Both methods suggest that MgOLi2 has the lowest BDE (De = 42.3 kcal mol−1 at CCSD(T)) of the Ae–O bonds.
• The analysis of the chemical bonds with the EDA-NOCV method shows that the strongest component of the covalent interactions comes in all compounds from an unprecedented σ donor bond Ae→OLi2 where the (n)s2 lone-pair electrons of the Ae atom are donated to vacant O–Li2 antibonding orbitals having the largest coefficient at lithium. It is a covalent bond where the accumulation of the associated electronic charge is located at two positions above and below the Ae–OLi2 axis. This bifurcated component of orbital interactions is structurally related to the recently proposed collective bonding model, but exhibits a completely different type of bonding.
• There are also three dative bonds due to Ae OLi2 backdonation which consists of one σ bond and two π bonds. The appearance of strong Ae→OLi2 σ donation leads to quadruple bonds Ae
OLi2 backdonation which consists of one σ bond and two π bonds. The appearance of strong Ae→OLi2 σ donation leads to quadruple bonds Ae OLi2 for all AeOLi2 systems, even for the lightest species with Ae = Be, Mg. The valence orbitals of Ca, Sr, and Ba which are involved in the dative interactions are the (n)s and (n−1)d AOs whereas Be and Mg use their (n)s and (n)p AOs.
OLi2 for all AeOLi2 systems, even for the lightest species with Ae = Be, Mg. The valence orbitals of Ca, Sr, and Ba which are involved in the dative interactions are the (n)s and (n−1)d AOs whereas Be and Mg use their (n)s and (n)p AOs.
• The EDA-NOCV results are supported by AdNDP calculations which give four 2c–2e bonding orbitals. Three bonding orbitals have occupation numbers ∼2. One σ orbital has smaller occupation numbers between 1.32 and 1.73 due to the delocalization to the lithium atoms.
• The analysis of the electronic structure with the ELF method suggests multicenter bonds with mainly trisynaptic and tetrasynaptic basins, which also supports the results of the EDA-NOCV calculations.
• The calculation of the atomic partial charges by the Hirshfeld and Voronoi methods suggests that Be and Mg carry small negative charges in the lighter molecules whereas the heavier atoms Ca–Be have small positive charges. In contrast, the NBO and QTAIM methods give positive charges for all Ae atoms that are higher for Ca–Ba than those given by the Hirshfeld and Voronoi approaches.
• The molecules AeOLi2 have large dipole moments where the negative end is at the Ae atom with the polarity Ae→OLi2. The largest dipole moments are predicted for the lighter species BeOLi2 and MgOLi2 and the smallest value is calculated for BaOLi2.
• The calculation of the vibrational spectra shows a significant red-shift toward lower wave numbers for the Ae–OLi2 stretching mode with regard to diatomic AeO.
Data availability
The data supporting this article have been included as part of the ESI.†Author contributions
S. P., Z. C., and G. F. conceived the project, wrote the draft, and finalized it, L. C. performed the calculations. L. C. and Y. L. analyzed the data. All authors took part in the discussions and approved the final version.Conflicts of interest
The authors declare no conflict of interest.Acknowledgements
This work was supported by the National Natural Science Foundation of China (No. 12374255) and by the Fundamental Research Funds for the Central Universities.References
-
G. Frenking and S. Shaik, The Chemical Bond: Chemical Bonding Across the Periodic Table, John Wiley & Sons, 2014 Search PubMed
.
-
F. A. Cotton, C. A. Murillo and R. A. Walton, Multiple bonds between metal atoms, Springer Science & Business Media, 2005 Search PubMed
- G. Frenking and N. Fröhlich, The nature of the bonding in transition-metal compounds, Chem. Rev., 2000, 100, 717 CrossRef CAS
- L. Gagliardi and B. O. Roos, Quantum chemical calculations show that the uranium molecule U2 has a quintuple bond, Nature, 2005, 433, 848 CrossRef CAS PubMed
- G. Frenking and R. Tonner, The six-bond bound, Nature, 2007, 446, 276 CrossRef CAS PubMed
- S. Shaik, D. Danovich, W. Wu, P. Su, H. S. Rzepa and P. C. Hiberty, Quadruple bonding in C2 and analogous eight-valence electron species, Nat. Chem., 2012, 4, 195 CrossRef CAS PubMed
- S. Shaik, H. S. Rzepa and R. Hoffmann, One Molecule, Two Atoms, Three Views, Four Bonds?, Angew. Chem., Int. Ed., 2013, 52, 3020 CrossRef CAS PubMed
- D. Danovich, P. C. Hiberty, W. Wu, H. S. Rzepa and S. Shaik, The Nature of the Fourth Bond in the Ground State of C2: The Quadruple Bond Conundrum, Chem.–Eur. J., 2014, 20, 6220 CrossRef CAS
- S. Shaik, D. Danovich, B. Braida and P. C. Hiberty, The Quadruple Bonding in C2 Reproduces the Properties of the Molecule, Chem.–Eur. J., 2016, 22, 4116 CrossRef CAS PubMed
- M. Hermann and G. Frenking, The Chemical Bond in C2, Chem.–Eur. J., 2016, 22, 4100 CrossRef CAS PubMed
- W. Zou and D. Cremer, C2 in a Box: Determining Its Intrinsic Bond Strength for the X1Σg+ Ground State, Chem.–Eur. J., 2016, 22, 4087 CrossRef CAS PubMed
- M. Piris, X. Lopez and J. M. Ugalde, The Bond Order of C2 from a Strictly N-Representable Natural Orbital Energy Functional Perspective, Chem.–Eur. J., 2016, 22, 4109 CrossRef CAS
- D. L. Cooper, R. Ponec and M. Kohout, New insights from domain-averaged Fermi holes and bond order analysis into the bonding conundrum in C2, Mol. Phys., 2016, 114, 1270 CrossRef CAS
- L. T. Xu and T. H. Dunning Jr, Insights into the Perplexing Nature of the Bonding in C2 from Generalized Valence Bond Calculations, J. Chem. Theory Comput., 2014, 10, 195 CrossRef CAS
- D. W. O. de Sousa and M. A. C. Nascimento, Is There a Quadruple Bond in C2?, J. Chem. Theory Comput., 2016, 12, 2234 CrossRef CAS PubMed
- B. A. Laws, S. T. Gibson, B. R. Lewis and R. W. Field, The dicarbon bonding puzzle viewed with photoelectron imaging, Nat. Commun., 2019, 10, 5199 CrossRef CAS
-
(a) D. M. Merriles, E. Tieu and M. D. Morse, Bond dissociation energies of FeB, CoB, NiB, RuB, RhB, OsB, IrB, and PtB, J. Chem. Phys., 2019, 151, 044302 CrossRef
- L. F. Cheung, T.-T. Chen, G. S. Kocheril, W.-J. Chen, J. Czekner and L.-S. Wang, Observation of Four-Fold Boron–Metal Bonds in RhB(BO–) and RhB, J. Phys. Chem. Lett., 2020, 11, 659 CrossRef CAS
- S. Parambath and P. Parameswaran, Two σ- and two π-dative quadruple bonds between the s-block element and transition metal in [BeM(CO)4; M = Fe−Os], Phys. Chem. Chem. Phys., 2022, 24, 20183 RSC
- I. Fernández, N. Holzmann and G. Frenking, The Valence Orbitals of the Alkaline-Earth Atoms, Chem.–Eur. J., 2020, 26, 14194 CrossRef PubMed
- M. Zhou and G. Frenking, Transition-Metal Chemistry of the Heavier Alkaline Earth Atoms Ca, Sr, and Ba, Acc. Chem. Res., 2021, 54, 3071 CrossRef CAS PubMed
- Y.-q. Liu, M.-h. Wang, B. Yan, L. Li, S. Pan, Z.-h. Cui and G. Frenking, Quest of Quadruple Bonding Between Two Main-Group Atoms in AeB− and AeC (Ae = Ca, Sr, Ba) and the Role of d Orbitals of Heavier Alkaline-Earth Atoms in Covalent Interactions, Chem.–Eur. J., 2023, 29, e202300446 CrossRef CAS PubMed
- R. Liu, L. Qin, Z. Zhang, L. Zhao, F. Sagan, M. Mitoraj and G. Frenking, Genuine quadruple bonds between two main-group atoms. Chemical bonding in AeF− (Ae = Be–Ba) and isoelectronic EF (E = B–Tl) and the particular role of d orbitals in covalent interactions of heavier alkaline-earth atoms, Chem. Sci., 2023, 14, 4872 RSC
- L. Qin, Y. Liu, R. Liu, X. Yang, Z. Cui, L. Zhao, S. Pan, S. Fau and G. Frenking, Analysis of the Unusual Chemical Bonds and Dipole Moments of AeF− (Ae = Be−Ba): A Lesson in Covalent Bonding, Chem.–Eur. J., 2024, e202304136 CrossRef CAS
- D. White, K. S. Seshadri, D. F. Dever, D. E. Mann and M. J. Linevsky, Infrared Spectra and the Structures and Thermodynamics of Gaseous LiO, Li2O, and Li2O2, J. Chem. Phys., 1963, 39, 2463 CrossRef CAS
- J. Lv, Y. C. Wang, L. Zhu and Y. M. Ma, Particle-swarm structure prediction on clusters, J. Chem. Phys., 2012, 137, 084104 CrossRef PubMed
- A. D. Becke, Density-functional exchange-energy approximation with correct asymptotic behavior, Phys. Rev. A: At., Mol., Opt. Phys., 1988, 38, 3098 CrossRef CAS PubMed
- J. P. Perdew, Density-functional approximation for the correlation energy of the inhomogeneous electron gas, Phys. Rev. B: Condens. Matter Mater. Phys., 1986, 33, 8822 CrossRef PubMed
- L. Goerigk and S. Grimme, A thorough benchmark of density functional methods for general main group thermochemistry, kinetics, and noncovalent interactions, Phys. Chem. Chem. Phys., 2011, 13, 6670 RSC
- F. Weigend and R. Ahlrichs, Balanced basis sets of split valence, triple zeta valence and quadruple zeta valence quality for H to Rn: Design and assessment of accuracy, Phys. Chem. Chem. Phys., 2005, 7, 3297 RSC
- G. D. Purvis III and R. J. Bartlett, A full coupled-cluster singles and doubles model: The inclusion of disconnected triples, J. Chem. Phys., 1982, 76, 1910 CrossRef
- J. A. Pople, M. Head-Gordon and K. Raghavachari, Quadratic configuration interaction. A general technique for determining electron correlation energies, J. Chem. Phys., 1987, 87, 5968 CrossRef CAS
-
M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, G. A. Petersson, H. Nakatsuji, X. Li, M. Caricato, A. V. Marenich, J. Bloino, B. G. Janesko, R. Gomperts, B. Mennucci, H. P. Hratchian, J. V. Ortiz, A. F. Izmaylov, J. L. Sonnenberg, M. Williams, F. Ding, F. Lipparini, F. Egidi, J. Goings, B. Peng, A. Petrone, T. Henderson, D. Ranasinghe, V. G. Zakrzewski, J. Gao, N. Rega, G. Zheng, W. Liang, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, K. Throssell, J. A. Montgomery Jr, J. E. Peralta, F. Ogliaro, M. J. Bearpark, J. J. Heyd, E. N. Brothers, K. N. Kudin, V. N. Staroverov, T. A. Keith, R. Kobayashi, J. Normand, K. Raghavachari, A. P. Rendell, J.
C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, J. M. Millam, M. Klene, C. Adamo, R. Cammi, J. W. Ochterski, R. L. Martin, K. Morokuma, O. Farkas, J. B. Foresman and D. J. Fox, Gaussian 16, Revision C.01, Gaussian, Inc., Wallingford CT, 2019 Search PubMed
-
R. F. W. Bader, Atoms in Molecules: A Quantum Theory, Oxford University Press, Oxford, 1990 Search PubMed
- D. Y. Zubarev and A. I. Boldyrev, Developing Paradigms of Chemical Bonding: Adaptive Natural Density Oartitioning, Phys. Chem. Chem. Phys., 2008, 10, 5207 RSC
- A. D. Becke and K. E. Edgecombe, A Simple Measure of Electron Localization in Atomic and Molecular Systems, J. Chem. Phys., 1990, 92, 5397 CrossRef CAS
- T. Lu and F. W. Chen, Multiwfn: A multifunctional wavefunction analyzer, J. Comput. Chem., 2012, 33, 580 CrossRef CAS
-
F. Weinhold and C. R. Landis, Discovering Chemistry With Natural Bond Orbitals, Wiley, New Jersey, 2012 Search PubMed
- F. L. Hirshfeld, Bonded-Atom Fragments for Describing Molecular Charge Densities, Theor. Chim. Acta, 1977, 44, 129 CrossRef CAS
- C. Nieuwland, P. Vermeeren, F. M. Bickelhaupt and C. F. Guerra, Understanding chemistry with the symmetry-decomposed Voronoi deformation density charge analysis, J. Comput. Chem., 2023, 44, 2108 CrossRef CAS
- E. D. Glendening, C. R. Landis and F. Weinhold, NBO 7.0: New Vistas in Localized and Delocalized Chemical Bonding Theory, J. Comput. Chem., 2019, 40, 2234 CrossRef CAS PubMed
-
E. D. Glendening, J. E. Carpenter, J. A. Bohmann, C. M. Morales, P. Karafiloglou, C. R. Landis and F. Weinhold, NBO 7.0, Theoretical Chemistry Institute, University of Wisconsin, Madison, 2018 Search PubMed
- ADF 2020, ADF SCM, Theoretical Chemistry, Vrije Universiteit, Amsterdam, The Netherlands, https://www.scm.com/.
- K. B. Wiberg, Application of the pople-santry-segal CNDO method to the cyclopropylcarbinyl and cyclobutyl cation and to bicyclobutane, Tetrahedron, 1968, 24, 1083 CrossRef CAS
- I. Mayer, Charge, bond order and valence in the AB initio SCF theory, Chem. Phys. Lett., 1983, 97, 270 CrossRef CAS
- T. Ziegler and A. Rauk, On the calculation of bonding energies by the Hartree Fock Slater method, Theor. Chem. Acc., 1977, 46, 1 Search PubMed
-
(a) M. Mitoraj and A. Michalak, Donor-acceptor properties of ligands from the natural orbitals for chemical valence, Organometallics, 2007, 26, 6576 CrossRef CAS
-
(a) A. Michalak, M. Mitoraj and T. Ziegler, Bond orbitals from chemical valence theory, J. Phys. Chem. A, 2008, 112, 1933 CrossRef CAS PubMed
- E. V Lenthe and E. J. Baerends, Optimized slater-type basis sets for the elements 1-118, J. Comput. Chem., 2003, 24, 1142 CrossRef
- C. Chang, M. Pelissier and P. Durand, Regular Two-Component Pauli-Like Effective Hamiltonians in Dirac Theory, Phys. Scr., 1986, 34, 394 CrossRef CAS
- J.-L. Heully, I. Lindgren, E. Lindroth, S. Lundquist and A.-M. Martensson-Pendrill, Diagonalisation of the Dirac Hamiltonian as a basis for a relativistic many-body procedure, J. Phys. B: Atom. Mol. Phys., 1986, 19, 2799 CrossRef CAS
- E. van Lenthe, J. G. Snijders and E. J. Baerends, The zero-order regular approximation for relativistic effects: The effect of spin–orbit coupling in closed shell molecules, J. Chem. Phys., 1996, 105, 6505 CrossRef CAS
- G. te Velde, F. M. Bickelhaupt, E. J. Baerends, C. F. Guerra, S. J. A. Van Gisbergen, J. G. Snijders and T. Ziegler, Chemistry with ADF, J. Comput. Chem., 2001, 22, 931 CrossRef CAS
- ADF 2020, ADF SCM, Theoretical Chemistry, Vrije Universiteit, Amsterdam, The Netherlands, http://www.scm.com.
- L. Zhao, S. Pan and G. Frenking, Energy Decomposition Analysis of the Chemical Bond: Scope and Limitation, Comprehensive Computational Chemistry, 2024, 2, 322 Search PubMed
- F. M. Bickelhaupt, C. F. Guerra, M. Mitoraj, F. Sagan, A. Michalak, S. Pan and G. Frenking, Clarifying notes on the bonding analysis adopted by the energy decomposition analysis, Phys. Chem. Chem. Phys., 2022, 24, 15726 RSC
- L. L. Zhao, M. Hermann, W. H. E. Schwarz and G. Frenking, The Lewis electron-pair bonding model: modern energy decomposition analysis, Nat. Rev. Chem, 2019, 3, 48 CrossRef CAS
- L. Zhao, M. von Hopffgarten, D. M. Andrada and G. Frenking, Energy decomposition analysis, Wiley Interdiscip. Rev.: Comput. Mol. Sci., 2018, 8, e1345 Search PubMed
- S. Pan, and G. Frenking, unpublished.
- A. J Bridgeman, G. Cavigliasso, L. R. Ireland and J. Rothery, The Mayer bond order as a tool in inorganic chemistry, J. Chem. Soc., Dalton Trans., 2001, 14, 2095 RSC
- I. Mayer, Bond order and valence indices: A personal account, J. Comput. Chem., 2007, 28, 204 CrossRef CAS
- L. Zhao, S. Pan and G. Frenking, The nature of the polar covalent bond, J. Chem. Phys., 2022, 157, 034105 CrossRef CAS PubMed
- L. Zhao, S. Pan, N. Holzmann, P. Schwerdtfeger and G. Frenking, Chemical Bonding and Bonding Models of Main-Group Compounds, Chem. Rev., 2019, 119, 8781 CrossRef CAS PubMed
- G. Frenking, I. Fernández, N. Holzmann, S. Pan, I. Krossing and M. F. Zhou, Metal-CO Bonding in Mononuclear Transition Metal Carbonyl Complexes, JACS Au, 2021, 1, 623 CrossRef CAS PubMed
- W. Su, S. Pan, X. Sun, S. Wang, L. Zhao, G. Frenking and C. Zhu, Double Dative Bond Between Divalent Carbon(0) and Uranium, Nat. Commun., 2018, 9, 4997 CrossRef
- J. Jin, S. Pan, X. Jin, S. Lei, L. Zhao, G. Frenking and M. Zhou, Octacarbonyl Anion Complexes of the Late Lanthanides Ln(CO)8− (Ln = Tm, Yb, Lu) and the 32-Electron Rule, Chem.–Eur. J., 2019, 25, 3229 CrossRef CAS PubMed
- J. M. Guevara-Vela, E. Francisco, T. Rocha-Rinza and A. M. Pendás, Interacting Quantum Atoms - A Review, Molecules, 2020, 25, 4028 CrossRef CAS PubMed
- Q. N. Zhang, W. L. Li, C. Q. Xu, M. H. Chen, M. F. Zhou, J. Li, D. M. Andrada and G. Frenking, Formation and Characterization of the Boron Dicarbonyl Complex [B(CO)2]−, Angew. Chem., Int. Ed., 2015, 54, 11078 CrossRef CAS PubMed
- P. Jerabek, H. W. Roesky, G. Bertrand and G. Frenking, Coinage Metals Binding as Main Group Elements: Structure and Bonding of the Carbene Complexes [TM(cAAC)2] and [TM(cAAC)2]+ (TM = Cu, Ag, Au), J. Am. Chem. Soc., 2014, 136, 17123 CrossRef CAS PubMed
- P. Jerabek, P. Schwerdtfeger and G. Frenking, Dative and electron-sharing bonding in transition metal compounds, J. Comput. Chem., 2019, 40, 247 CrossRef CAS PubMed
- D. M. Andrada, J. L. Casals-Sainz, A. M. Pendas and G. Frenking, Dative and Electron-Sharing Bonding in C2F4, Chem.–Eur. J., 2018, 24, 9083 CrossRef CAS PubMed
- S. Pan and G. Frenking, Comment on “Realization of Lewis Basic Sodium Anion in the NaBH3− Cluster”, Angew. Chem., 2020, 132, 8836 CrossRef
- C. Chi, S. Pan, L. Meng, M. Luo, L. Zhao, M. Zhou and G. Frenking, Alkali Metal Covalent Bonding in Nickel Carbonyl Complexes ENi(CO)3−, Angew. Chem., Int. Ed., 2019, 131, 1746 CrossRef
-
M. W. Schmidt, J. Ivanic and K. Ruedenberg, The Physical Origin of the Chemical Bond, in The Chemical Bond. Fundamental Aspects of Chemical Bonding, ed. G. Frenking and S. Shaik, Wiley-VCH, Weinheim, 2014, pp. 1–68 Search PubMed
- W. Kutzelnigg, The Physical Mechanism, of the Chemical Bond, Angew Chem. Int. Ed. Engl., 1973, 12, 546 CrossRef
- F. Rioux, The Covalent Bond Examined Using the Virial Theorem, Chem. Educ., 2003, 8, 1 Search PubMed
-
S. Shaik and P. C. Hiberty, A Chemist's Guide to Valence Bond Theory, John Wiley & Sons, Inc., 2007 Search PubMed
- S. Pan and G. Frenking, A critical look at Linus Pauling's influence on the understanding of chemical bonding, Molecules, 2021, 26, 4695 CrossRef CAS PubMed
- S. Sowlati-Hashijn, V. Sadek, S. A. Sadjadi, M. Karttunen, A. M. Pendas and C. Foroutan-Nejad, Collective interactions among organometallics are exotic bonds hidden on lab shelves, Nat. Commun., 2022, 13, 2069 CrossRef PubMed
- J. Poater, P. Vermeeren, T. A. Hamlin, F. M. Bickelhaupt and M. Sola, On the existence of collective interactions reinforcing the metal-ligand bond in organometallic compounds, Nat. Commun., 2023, 14, 3872 CrossRef CAS PubMed
- V. Sadek, S. Sowlati-Hashijn, S. A. Sadjadi, M. Karttunen, A. M. Pendas and C. Foroutan-Nejad, Reply to: On the existence of collective interactions reinforcing the metal-ligand bond in organometallic compounds, Nat. Commun., 2023, 14, 3873 CrossRef CAS PubMed
- Z. Badri and C. Foroutan-Nejad, Classical versus Collective Interactions in Asymmetric Trigonal Bipyramidal Alkaline Metal-Boron Halide Complexes, Chem.–Eur. J., 2024, e202400156 CrossRef CAS PubMed
- R. Pino-Rios, R. Baez-Frez and C. Foroutan-Nejad, Anti-electrostatic cation⋯π hole and cation⋯lp-hole interactions are stabilized via collective interaction, Chem. Commun., 2024, 60, 400 RSC
- M. Mousavi and G. Frenking, Bonding Analysis of Trimethylenemethane (TMM) Complexes [(CO)3M-TMM] (M= Fe, Ru, Os, Rh+). Absence of Expected Bond Paths, J, Organomet. Chem., 2013, 748, 2 CrossRef CAS
- A. Savin, B. Silvi and F. Colonna, Topological analysis of the electron localization function applied to delocalized bonds, Can. J. Chem., 1996, 74, 1088 CrossRef CAS
- B. Silvi and A. Savin, Classification of chemical bonds based on topological analysis of electron localization functions, Nature, 1994, 371, 683 CrossRef CAS
- B. Silvi, The synaptic order: a key concept to understand multicenter bonding, J. Mol. Struct., 2002, 614, 3 CrossRef CAS
- D. Faron, P. Skurski and I. Anusiewicz, Tracing the acid-base catalytic properties of MON2O mixed oxides (M = Be, Mg, Ca; N = Li, Na, K) by theoretical calculations, J. Mol. Model., 2021, 27, 210 CrossRef CAS
Footnotes |
| † In memoriam Josef Michl. |
| ‡ Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4sc01979b |
| § These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2024 |