
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
A stereodivergent multicomponent approach for the synthesis of C–N atropisomeric peptide analogues†
Natalie J.
Roper
 a,
Aaron D. G.
Campbell
a,
Paul G.
Waddell
a,
Alistair K.
Brown
b,
Kristaps
Ermanis
*c and
Roly J.
Armstrong
*a
a,
Aaron D. G.
Campbell
a,
Paul G.
Waddell
a,
Alistair K.
Brown
b,
Kristaps
Ermanis
*c and
Roly J.
Armstrong
*a
aSchool of Natural and Environmental Sciences, Newcastle University, Newcastle Upon Tyne, NE1 7RU, UK. E-mail: roly.armstrong@newcastle.ac.uk
bBiosciences Institute, Faculty of Medical Sciences, Newcastle University, Newcastle Upon Tyne, NE2 4HH, UK
cSchool of Chemistry, University of Nottingham, University Park, Nottingham, NG7 2RD, UK. E-mail: kristaps.ermanis@nottingham.ac.uk
First published on 16th September 2024
Abstract
A four-component Ugi reaction is described for the stereoselective synthesis of novel C–N atropisomeric peptide analogues. Using this approach, a combination of simple, readily available starting materials (ortho-substituted anilines, aldehydes, carboxylic acids and isocyanides) could be combined to access complex products possessing both central and axial chirality in up to 92% yield and >95![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :5 d.r. Variation of the reaction temperature enabled the development of stereodivergent reactions capable of selectively targeting either diastereoisomer of a desired product from a single set of starting materials with high levels of stereocontrol. Detailed experimental and computational studies have been performed to probe the reaction mechanism and stereochemical outcome of these reactions. Preliminary studies show that novel atropisomeric scaffolds prepared using this method display inhibitory activity against M. tuberculosis with a significant difference in activity observed between different atropisomers.
:5 d.r. Variation of the reaction temperature enabled the development of stereodivergent reactions capable of selectively targeting either diastereoisomer of a desired product from a single set of starting materials with high levels of stereocontrol. Detailed experimental and computational studies have been performed to probe the reaction mechanism and stereochemical outcome of these reactions. Preliminary studies show that novel atropisomeric scaffolds prepared using this method display inhibitory activity against M. tuberculosis with a significant difference in activity observed between different atropisomers.
Introduction
Recent advances in stereoselective synthesis have led to a wealth of processes enabling the synthesis of materials with precisely controlled relative stereochemistry.1 In the majority of cases, diastereoselectivity in such reactions is governed by substrate control in which an existing element of chirality guides the stereochemical outcome of the reaction. However, in the event that a different diastereoisomer is required it can be extremely challenging to override this innate selectivity. Much less common are diastereodivergent reactions capable of selectively converting a common precursor to either diastereoisomer of a desired target.2 For example, elegant methods have been established for diastereodivergent olefination of aldehydes to afford either E- or Z-configured alkenes, along with powerful diastereodivergent reduction processes enabling the synthesis of 1,3-diols (Scheme 1A).3,4 The flexibility of such reactions to selectively and predictably deliver different stereoisomers on demand is hugely valuable, particularly in drug discovery, where subtle configurational changes can dramatically influence biological activity.5 | ||
| Scheme 1 Previous work and strategy for stereodivergent multicomponent synthesis of C–N atropisomeric peptide analogues. | ||
The synthesis of atropisomeric materials is an area that is rapidly gaining traction owing to their emerging applications in materials science, catalysis, and medicine.6 An array of powerful synthetic methods have been reported which enable the construction of scaffolds displaying restricted rotation about C–C, C–N and N–N bonds.6,7 However, the preparation of atropisomeric molecules that contain an additional element of stereogenicity is significantly complicated by the potential for formation of multiple diastereoisomers.8 Comparatively few stereoselective methods are available for the synthesis of such molecules, and only a handful of approaches capable of diastereodivergence have been reported.2b,9,10 In particular, the groups of Miller10a and Sparr10b have independently disclosed elegant stereodivergent syntheses of molecules containing multiple stereogenic C–C axes and Bencivenni10c and Miller10d have reported efficient stereodivergent syntheses of materials combining central chirality with atropisomerism about N–N and C–N bonds respectively (Scheme 1B). Despite these important innovations, selectively accessing scaffolds in which atropisomerism is combined with an additional element of stereochemistry remains a significant challenge.
Given these considerations, we recently questioned whether it might be possible to prepare the first examples of C–N atropisomeric peptide analogues 1 possessing both central and axial chirality (Scheme 1C).11–14 Given the ubiquitous nature of peptides in pharmaceuticals and agrochemicals, the idea of developing C–N atropisomeric analogues would be of significant interest and we proposed that it might be possible to directly access them via a stereoselective Ugi reaction between ortho-substituted anilines, aldehydes, carboxylic acids and isocyanides.15 This approach has the potential to provide access to these novel scaffolds with unparalleled efficiency. However, we recognized that to prove successful, several important challenges would need to be overcome, namely: (i) identification of mild conditions capable of engaging extremely hindered aniline reagents whilst avoiding competing Passerini side reactions; (ii) simultaneously achieving high levels of stereoselectivity; (iii) discovering a complementary method to access the alternative atropisomer of the peptidic products. Here we describe our results in this area and how we were ultimately able to develop efficient stereodivergent multicomponent reactions that allow a single set of starting materials to be converted to either syn- or anti-configured atropisomeric peptide analogues with excellent efficiency, broad scope, and near complete stereocontrol.
Results and discussion
We commenced our study by evaluating a room-temperature multicomponent reaction between an equimolar mixture of 2-tert-butylaniline, benzoic acid, benzaldehyde and tert-butyl isocyanide. Disappointingly, under typical Ugi conditions employing MeOH (0.2 M) as the reaction solvent, only trace conversion to the desired N-arylated amide was observed (Table 1, entry 1). This is likely a consequence of the extremely hindered nature of the aniline reagent. However, tantalizingly the trace amount of anti-1a formed in this reaction appeared to be present as a single diastereoisomer. In an attempt to boost conversion, we explored introducing various additives which have been shown to accelerate other challenging Ugi reactions.16 Brønsted acids as well as scandium and titanium-based Lewis acids all gave comparable or inferior results (Table 1, entries 2–5). However, upon addition of 0.1 equivalent of MgCl2 the desired product was formed in 21% yield and >95:5 d.r. (Table 1, entry 6). Employing MgCl2 as the Lewis acid, we then explored the effect of varying the reaction solvent. Poor results were observed in aprotic solvents such as dichloromethane and acetonitrile (entries 7 and 8), but we were pleased to find that fluorinated alcohol solvents were particularly effective for this transformation, with 71% yield and >95:5 d.r. obtained in 2,2,2-trifluoroethanol (2,2,2-TFE) (Table 1, entries 9 and 10).17 A control experiment performed in the absence of MgCl2 revealed that the Lewis acid additive was no longer beneficial in 2,2,2-TFE, affording the desired product in an improved yield of 82% (Table 1, entry 11). Finally, we found that increasing the concentration to 0.5 M was also beneficial, allowing us to isolate anti-1a in 92% yield as a single diastereoisomer (Table 1, entry 12). We were fortunate to be able to obtain crystals of anti-1a suitable for single crystal X-ray diffraction, which enabled us to unambiguously assign the relative stereochemistry of the major diastereoisomer as anti.
|
|
||||
|---|---|---|---|---|
| Entry | Solvent | Additive (0.1 eq.) | Yield anti-1aa/% | d.r. anti-1ab |
| a Determined by 1H NMR analysis vs. CHCl2CHCl2 as an internal standard; values in parentheses indicate the yield of isolated product. b Diastereoselectivity determined by 1H NMR analysis of the crude reaction mixture. c HCl was added as a 2 M solution in diethyl ether. | ||||
| 1 | MeOH (0.2 M) | — | 8 | >95:5 |
| 2 | MeOH (0.2 M) | p-TSA | <5 | — |
| 3 | MeOH (0.2 M) | HClc | 5 | >95:5 |
| 4 | MeOH (0.2 M) | Sc(OTf)3 | <5 | — |
| 5 | MeOH (0.2 M) | Ti(OiPr)4 | 13 | >95:5 |
| 6 | MeOH (0.2 M) | MgCl2 | 21 | >95:5 |
| 7 | CH2Cl2 (0.2 M) | MgCl2 | <5 | — |
| 8 | MeCN (0.2 M) | MgCl2 | 11 | >95:5 |
| 9 | HFIP (0.2 M) | MgCl2 | 62 | >95:5 |
| 10 | 2,2,2-TFE (0.2 M) | MgCl2 | 71 | >95:5 |
| 11 | 2,2,2-TFE (0.2 M) | — | 82 | >95:5 |
| 12 | 2,2,2-TFE (0.5 M) | — | 90 (92) | >95:5 |

With optimal conditions established to access anti-configured C–N atropisomeric peptide analogues, we set out to establish the generality of the method, initially focusing on the aldehyde partner (Scheme 2). We were pleased to find that a variety of aromatic aldehydes were well tolerated, delivering products anti-1a–l in uniformly high yields and with complete diastereocontrol, including examples containing unprotected phenols (anti-1d), sterically encumbered groups (anti-1i–j), heterocycles (anti-1k) and metallocenes (anti-1l). The chemistry was not restricted to aromatic aldehydes, and a series of vinylic, aliphatic, and saturated heterocyclic products (anti-1m–p) were isolated in excellent yields with >95:5 d.r. We next investigated the scope of the carboxylic acid partner and were pleased to find that a variety of different aromatic, heteroaromatic and aliphatic carboxylic acids reacted smoothly to afford the corresponding amides anti-1q–aa in high yields with complete diastereoselectivity. Notably, N-protected amino acids could also be employed as substrates, leading to dipeptide analogues anti-1ab–1ac in high yields and >95:5 d.r. To highlight the generality of the chemistry, we also investigated the late-stage functionalization of pharmaceutically active carboxylic acids and were delighted to find that atropisomeric peptide analogues anti-1ad–ag derived from isoxepac, bendazac, tolmetin and indomethacin were all obtained in good to excellent yields with complete selectivity in favour of the anti-diastereoisomer. Reactions with other commerically available isocyanides were also successful, enabling the synthesis of N-benzyl and N-cyclohexyl analogues anti-1ah and anti-1ai in yields of 74% and 71% respectively. Moreover, employing commercially available ethyl isocyanoacetate enabled efficient access to atropisomeric di- and tripeptide analogues anti-1aj and anti-1ak, again with complete stereocontrol. A reaction with commercially available (S)-α-methylbenzyl isocyanide afforded anti-1al as a separable mixture of diastereoisomers (53:47 d.r.), thereby providing a means to access enantiopure products (see ESI† for details). Finally, we investigated alternative aniline components bearing two ortho-substituents and were pleased to find that reactions with 2-iodo-4,6-dimethylaniline also proceeded with essentially complete diastereocontrol to afford anti-1am–ao in 70–78% yield. The relative stereochemistry of anti-1am was unambiguously assigned via single crystal X-ray diffraction and confirmed to have the bulky iodine substituent oriented anti to the phenyl side chain. Analogous products anti-1ap and anti-1aq substituted with bromine and chlorine respectively were obtained in good yields, but somewhat lower diastereoselectivity (90:10 d.r. and 77:23 d.r. respectively), which is likely due to the smaller degree of steric differentiation between ortho-substituents. A multicomponent reaction with 2-phenyl-4,6-dimethylaniline delivered anti-1ar in 92% yield as a single diastereoisomer, whose relative configuration was again confirmed by X-ray analysis. We were pleased to discover that substituted naphthylamines were also compatible with our new method, with peptide analogues anti-1as and anti-1at both obtained in good yields and excellent diastereoselectivity. Interestingly, in anti-1as (which lacks a C2-substituent) the naphthyl backbone is orientated anti- to the phenyl group, but in anti-1at the C2-methyl group preferentially adopts the equivalent position. Not all of the anilines we investigated underwent the desired multicomponent reaction. For example, reactions with extremely sterically congested amines such as 2-isopropyl-4-methylpyridin-3-amine and 2-tert-butyl-4,6-diphenylaniline did not afford the desired products anti-1au and anti-1av instead delivering a mixture of unreacted amine, imine and Passerini product (see ESI† for details).
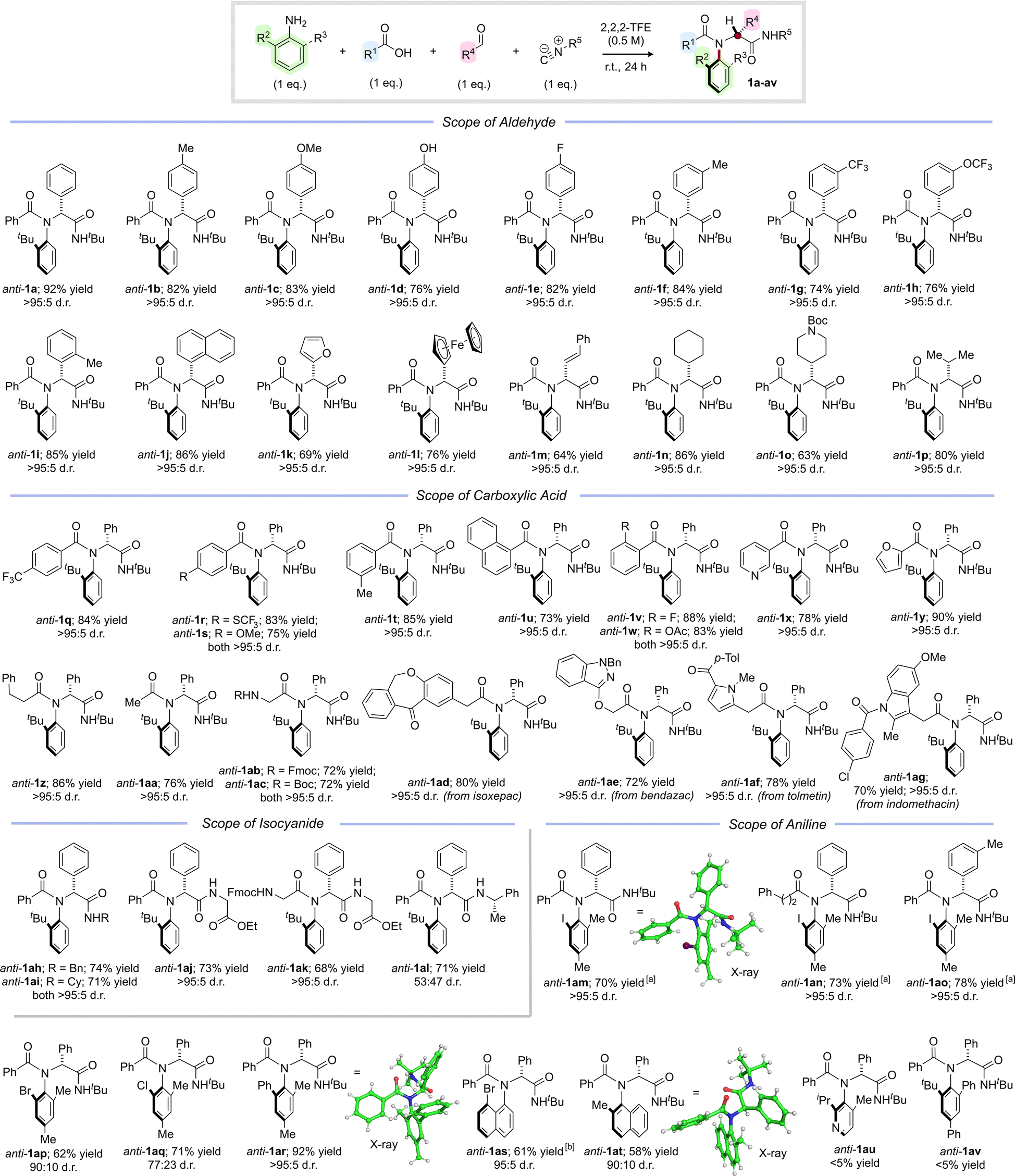 | ||
| Scheme 2 Stereoselective multicomponent synthesis of C–N atropisomeric amides. Reaction conditions: ortho-substituted aniline (1 eq.), aldehyde (1 eq.), carboxylic acid (1 eq.), isocyanide (1 eq.), 2,2,2-trifluoroethanol (0.5 M), r.t., 24 h. Yields refer to isolated material after column chromatography. Diastereoselectivity determined by 1H NMR analysis of the crude reaction mixture. aAn additional portion of isocyanide (1 eq.) was added after 24 h and the reaction was stirred for a further 24 h. bRelative configuration assigned on the basis of a crystal structure of syn-1as (see Scheme 5 and ESI†). | ||
With the generality of the new methodology established, we set out to perform computational modelling with the aim of rationalizing the stereochemical outcome of the transformation (Scheme 3). Although several mechanisms have been proposed for four-component Ugi reactions, there is strong consensus that the final step involves intramolecular acyl transfer (Mumm rearrangement) of an imidate intermediate, and we rationalised that this would be the step which establishes the relative configuration of the atropisomeric products.19 For a representative imidate 2a, geometry minimization was performed at the wB97M-V20/def2-QZVPP/SMD(CF3CH2OH)//PBE0-D321,22/def223-TZVP23/SMD(CF3CH2OH)24 level of theory. We were unable to find any energetically feasible processes involving direct Mumm rearrangement of 2a, so inspired by recent work by Houk, Tan and co-workers, we investigated the potential for an acid mediated rearrangement.16a Protonation of 2a by benzoic acid was calculated to be a mildly endergonic process (ΔG = +6.1 kcal mol−1) to form protonated imidate 3a – this was assumed to be very rapid. This intermediate was observed to undergo facile acyl transfer viaTS-anti (ΔG‡ = 14.0 kcal mol−1) leading to amidinium species anti-1a·BzOH (9.9 kcal mol−1), which was assumed to undergo very rapid deprotonation to form anti-1a (−19.1 kcal mol−1). In line with our experimental observations, formation of the syn-configured product viaTS-syn was calculated to be significantly less favourable (ΔG‡ = 21.2 kcal mol−1). The key difference appears to be that in the favoured anti-transition state, the bulky ortho-tert butyl group can be comfortably accommodated between the two equatorially disposed phenyl groups. Conversely, in the disfavoured syn-transition state, the phenyl substituents are forced closer to an axial arrangement, leading to an unfavourable 1,3-interaction. Notably, in both transition states, benzoic acid was observed to play a key role, forming a stabilizing C–H⋯O interaction with the benzylic hydrogen atom. Interestingly, these computational calculations also predicted that anti-1a produced as the kinetic product of the Ugi reaction is destabilized with respect to syn-1a by 1.4 kcal mol−1. This result raised the interesting possibility of performing a selective thermal atropisomerization of the kinetically formed anti-diastereoisomer to selectively access the thermodynamic syn-atropisomer.
 | ||
| Scheme 3 Computational modeling of the reaction pathway. Calculations were carried out at the wB97M-V/def2-QZVPP/SMD(CF3CH2OH)//PBE0-D3/def2-TZVP/SMD(CF3CH2OH) level, with free energy corrections applied for 298.15 K using a qRRHO approximation.18 All free energies are reported in kcal mol−1. | ||
To test this hypothesis experimentally, we investigated the thermal isomerization of anti-1a in heptane at 110 °C (Scheme 4A). In line with our computational predictions, we were delighted to observe an efficient switch in relative configuration to afford syn-1a as the major diastereoisomer (83:17 d.r.). Our computational calculations also suggested that the syn-diastereoisomer is significantly more polar than the anti-isomer (calculated dipole moments for syn-1a and anti-1a are 3.51 a.u. and 2.41 a.u. respectively) and accordingly, we questioned whether the thermodynamic preference for the syn-diastereomer could be exacerbated by a more polar solvent. To test this theory, we explored heating anti-1a in a variety of different solvents and were pleased to find that more polar solvents indeed proved beneficial, with toluene, chlorobenzene and dibutyl ether affording improved selectivity. The best results were obtained in polar protic or aprotic solvents, with 2,2,2-TFE, DMSO and DMF all delivering syn-1a with excellent diastereoselectivity (up to 92:8 d.r.). To probe the kinetics of this thermal atropisomerization process we studied the isomerization of both syn- and anti-1a in d6-DMSO at 110 °C (Scheme 4B).251H NMR spectra were recorded at regular intervals, revealing smooth conversion to a thermodynamic mixture of diastereomers within 2 hours. From the data for anti-1a we calculated free energy barriers for atropisomerization of 28.2 kcal mol−1 (ΔG‡f) and 30.0 kcal mol−1 (ΔG‡b) at 110 °C (see ESI† for details). This corresponds to a half-life of around 1 year at room temperature, confirming these molecules to be configurationally stable atropisomers. To verify that isomerization proceeds by thermal atropisomerization and rule out a pathway involving enolization of the α-stereogenic centre, we performed an isotopic labelling experiment where anti-1a was heated in d3-trifluoroethanol (Scheme 4C). Under these conditions syn-1a was obtained in 88% yield and 91:9 d.r. with no deuterium incorporation at the α-stereogenic centre, strongly supporting a mechanism involving atropisomerization. These thermal isomerization experiments also led us to consider whether it might be possible to augment our anti-selective multicomponent reactions (Scheme 2) with four component couplings performed at elevated temperature to directly access syn-configured peptide analogues. To test this hypothesis, an equimolar mixture of 2-tert-butylaniline, benzoic acid, benzaldehyde and tert-butyl isocyanide in 2,2,2-TFE was stirred overnight in 2,2,2-TFE at room temperature and then heated at 110 °C for 24 h (Scheme 4D). Under these conditions, we were delighted to isolate the desired product syn-1a in an excellent yield of 95% with very high levels of stereocontrol (92:8 d.r.). Excitingly, this result indicates that simply increasing the reaction temperature results in near complete stereodivergence, allowing the alternative stereoisomer of C–N atropisomeric peptide analogues to be selectively assembled from the same set of starting materials.
 | ||
| Scheme 4 Selective atropisomerization studies. (A) anti-1a (0.045 mmol), solvent, 110 °C, 48 h. Diastereomeric ratios determined by 1H NMR analysis of the crude reaction mixture. (B) Isomerization was carried out in an NMR tube and analyzed by variable temperature 1H-NMR spectroscopy (500 MHz, 110 °C) starting with either syn-1a or anti-1a (∼10 mg), d6-DMSO (0.5 mL). (C) Reaction conditions: anti-1a (0.090 mmol), CF3CD2OD (1 mL), 110 °C, 48 h. The extent of deuterium labelling was determined by 1H NMR spectroscopy (see ESI† for details). (D) Reaction conditions: ortho-substituted aniline (1 eq.), aldehyde (1 eq.), carboxylic acid (1 eq.), isocyanide (1 eq.), 2,2,2-trifluoroethanol (0.5 M), r.t. 16 h then 110 °C, 24 h. Yield refers to isolated material after column chromatography. Diastereoselectivity determined by 1H NMR analysis of the crude reaction mixture. | ||
We next explored the scope of this syn-selective multicomponent reaction, initially focusing on the aldehyde component (Scheme 5 – in each case, the result of the corresponding anti-selective reaction from Scheme 2 is reproduced alongside for clarity). We were delighted to find that substituted aldehydes participated efficiently in the thermal four-component reaction, delivering peptide analogues syn-1f and syn-1h in excellent yields and with near complete stereocontrol. Likewise, the acid component could be successfully varied, and reactions of heterocyclic and aliphatic carboxylic acids furnished syn-1x–z, all in excellent yields and diastereoselectivities. Interestingly, for syn-1z, more forcing thermal conditions were required to achieve high levels of stereocontrol and detailed analysis revealed substantially higher free energy barriers for atropisomerization in this case of 31.5 kcal mol−1 (ΔG‡f) and 33.3 kcal mol−1 (ΔG‡b) (see ESI† for details). This is in line with our previous work on other C–N atropisomeric scaffolds where we observed that C–N atropisomeric amides bearing aliphatic acyl substituents undergo atropisomerization significantly more slowly than aromatic analogues.26 The isocyanide component could also readily be varied, with isomeric cyclohexyl substituted amide syn-1ai isolated in excellent yield and stereoselectivity (94:6 d.r.). Other ortho-substituted anilines could also be employed under thermal conditions, leading to syn-1am and syn-1as in good to excellent yields with very high levels of diastereocontrol. The structural and stereochemical assignment of syn-1as was unambiguously confirmed by single crystal X-ray analysis.
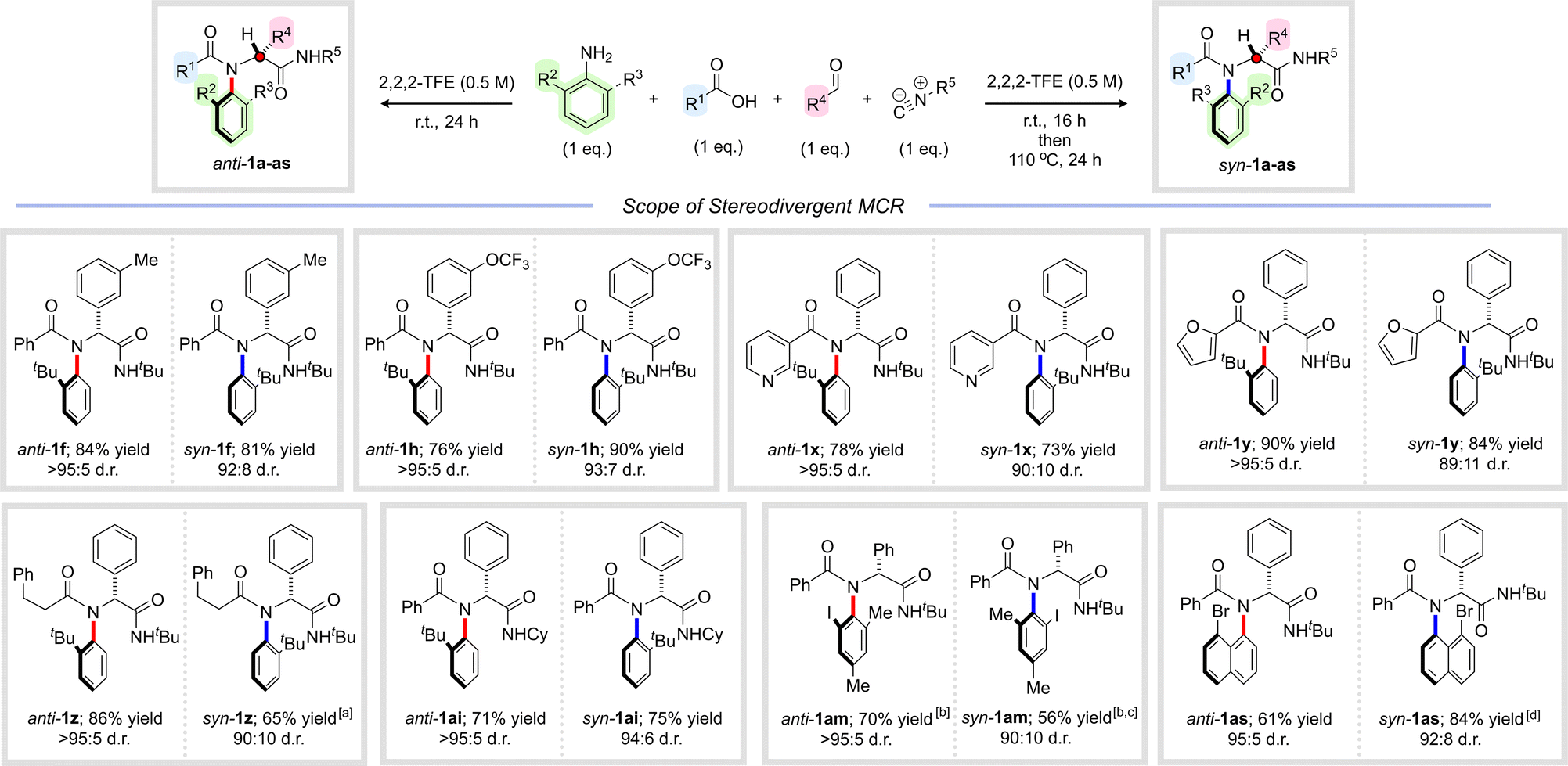 | ||
| Scheme 5 Stereodivergent multicomponent synthesis of C–N atropisomeric amides (results for anti-selective reactions from Scheme 2 are reproduced for clarity). Yields refer to isolated material after column chromatography. Diastereoselectivity determined by 1H NMR analysis of the crude reaction mixture. Reaction conditions for anti-selective reactions are shown in Scheme 2. Reaction conditions for syn-selective reactions: ortho-substituted aniline (1 eq.), aldehyde (1 eq.), carboxylic acid (1 eq.), isocyanide (1 eq.), 2,2,2-trifluoroethanol (0.5 M), r.t., 16 h then 110 °C, 24 h. aHeated to 110 °C in DMSO for 6 days. bAn additional portion of isocyanide (1 eq.) was added after 24 h and the reaction was stirred for a further 24 h at r.t. cHeated to 150 °C in DMSO for 5 hours. dRelative stereochemistry assigned by single crystal X-ray analysis. | ||
Finally, we sought to highlight the potential of our newly developed methodology to prepare medicinally relevant molecules. Inspired by recent elegant reports on the application of scaffolds derived from Ugi multicomponent reactions for the selective inhibition of Mycobacterium tuberculosis (Mtb), we applied our newly developed methodology to prepare both diastereoisomers of atropisomeric peptide analogues syn- and anti-1aw (Scheme 6A).27 The anti-tubercular activity of these stereoisomeric peptide analogues was assessed against a small panel of clinically relevant drug-susceptible and drug-resistant strains of Mtb using the Resazurin Microtiter Assay (REMA) method developed by Palomino and co-workers.28 (Scheme 6B). Excitingly, the syn-configured diastereoisomer displayed a promising level of activity (MIC90 = 31.7 ± 1.5 μM) against the auxotrophic wild-type strain (Mtb mc27902).29 In contrast, the anti-configured diastereoisomer displayed no inhibition up to the maximal concentration of 64 μM employed in the assay. Similar results were observed across a small panel of clinically relevant singly and doubly drug resistant Mtb, with syn-1aw displaying antitubercular activity (MIC90 ≤ 38.1 μM), and the anti-diastereoisomer showing no inhibition (see ESI† for details). These results show significant potential for future development of new highly active and selective antimicrobial agents and underscore the powerful impact atropisomerism can play in the biological activity of small molecules.
 | ||
| Scheme 6 Synthesis and inhibitory activity of N-aryl peptide analogues against drug-susceptible and drug-resistant strains of Mycobacterium tuberculosis. a2-tert-Butylaniline (1 eq.), nicotinaldehyde (1 eq.), carboxylic acid (1 eq.), cyclohexylisocyanide (1 eq.), 2,2,2-trifluoroethanol (0.5 M), r.t., 24 h. b2-tert-Butylaniline (1 eq.), nicotinaldehyde (1 eq.), carboxylic acid (1 eq.), cyclohexylisocyanide (1 eq.), 2,2,2-trifluoroethanol (0.5 M), r.t., 16 h then 110 °C, 24 h. cYields and diastereomeric ratios refer to material obtained after column chromatography (d.r. values in parentheses measured from crude 1H NMR spectrum). Additional purification by recrystallization was carried out prior to biological assays. dValues correspond to minimum concentrations required to achieve 90% inhibition (MIC90) as determined by the Resazurin Microtiter Assay (REMA) method against drug susceptible and resistant Mtb. Values of – correspond to no activity observed at 64 μM. See ESI† for details. | ||
Conclusions
C–N atropisomeric amides are rapidly emerging as powerful scaffolds for catalysis and medicinal chemistry but methods for their stereoselective synthesis are underdeveloped, particularly in cases where the presence of additional stereogenic elements leads to the potential for multiple diastereoisomers. We have developed a new multicomponent approach for synthesis of the first examples of C–N atropisomeric peptide analogues possessing both central and axial chirality. These reactions enable simple, readily available building blocks (aldehydes, carboxylic acids, anilines and isocyanides) to be assembled under mild conditions to deliver a wide range of C–N atropisomeric peptide analogues with very high efficiency and complete diastereocontrol. The flexibility, generality and functional group tolerance of the method is particularly noteworthy – extensive variation of all four reaction components was possible including the use of complex pharmaceutically active molecules as reagents. Remarkably, we found that increasing the reaction temperature could enable stereodivergent reactions capable of selectively targeting the other diastereoisomer of C–N atropisomeric products, again with broad scope, excellent yields and near complete stereocontrol. The mechanism of these reactions was interrogated employing detailed experimental and computational studies, shedding light on the kinetic and thermodynamic aspects of stereocontrol. Finally, we employed our new methodology to prepare novel C–N atropisomeric peptide scaffolds that display inhibitory activity against mc27092, a H37Rv BSL-2 arroved model organism, with a significant difference in activity observed between different atropisomers. We anticipate that this chemistry will spur the development of other new C–N atropisomeric scaffolds based upon biologically relevant peptide scaffolds, especially efforts to achieve stereocontrolled synthesis of atropisomers by exploiting thermodynamic versus kinetic control.Data availability
The data that support the findings of this study are available in the ESI† of this article, including detailed experimental procedures, characterization data for new compounds, details of computational methods, details of biological assays. Crystallographic data have been deposited with the Cambridge Crystallographic Data Centre (CCDC 2355697–2355699 and 2382977–2382979).Author contributions
N. J. R., A. D. G. C. and R. J. A. conceived and designed the study. N. J. R. and A. D. G. C. performed the synthetic experiments and analysed data for all compounds. P. G. W. performed single crystal X-ray analysis and analysed the associated data. A. K. B. and N. J. R. carried out biological assays. K. E. performed the computational calculations. R. J. A. and K. E. wrote the manuscript with input from all other authors.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We are grateful for financial support from EPSRC (EP/S022791/1), the Bill and Milica Beck PhD Endowment Fund, Newcastle University, and the Royal Society (RGS\R1\221162). We also thank Vertex Pharmaceuticals for generous provision of equipment. Calculations were performed using the Sulis Tier 2 HPC platform hosted by the Scientific Computing Research Technology Platform at the University of Warwick. Sulis is funded by EPSRC Grant EP/T022108/1 and the HPC Midlands+ consortium. We thank Dr Benjamin Horrocks (Newcastle University) for helpful discussions regarding kinetic analysis, and Dr Corinne Wills and Dr Casey Dixon (Newcastle University) for assistance with NMR experiments. We are grateful to Prof. William R. Jacobs Jr. and Torin Weisbrod from the Albert Einstein College of Medicine for supplying the bacterial strains used in this study.Notes and references
- E. M. Carreira and L. Kvaerno, Classics in Stereoselective Synthesis, Wiley-VCH, Weinheim, 2009 Search PubMed.
- For review articles, see: (a) S. Krautwald and E. M. Carreira, J. Am. Chem. Soc., 2017, 139, 5627 CrossRef CAS; (b) D. Moser, T. A. Schmidt and C. Sparr, JACS Au, 2023, 3, 2612 CrossRef CAS PubMed; (c) M. Bihani and J. C.-G. Zhao, Adv. Synth. Catal., 2017, 359, 534 CrossRef CAS; (d) I. P. Beletskaya, C. Nájera and M. Yus, Chem. Rev., 2018, 118, 5080 CrossRef CAS PubMed; (e) A. Kumari, A. Jain and N. K. Rana, Tetrahedron, 2024, 150, 133754 CrossRef CAS; (f) L. Lin and X. Feng, Chem.–Eur. J., 2017, 23, 6464 CrossRef CAS PubMed; (g) X. Huo, G. Li, X. Wang and W. Zhang, Angew. Chem., Int. Ed., 2022, 61, e202210086 CrossRef CAS PubMed.
- (a) L. Horner, H. Hoffmann and H. G. Wippel, Chem. Ber., 1958, 91, 61 CrossRef CAS; (b) W. S. Wadsworth and W. D. Emmons, J. Am. Chem. Soc., 1961, 83, 1733 CrossRef CAS; (c) W. C. Still and C. Gennari, Tetrahedron Lett., 1983, 24, 4405 CrossRef CAS.
- (a) D. A. Evans, K. T. Chapman and E. M. Carreira, J. Am. Chem. Soc., 1988, 110, 3560 CrossRef CAS; (b) A. K. Saksena and P. Mangiaracina, Tetrahedron Lett., 1983, 24, 273 CrossRef CAS; (c) K. Narasaka and H. C. Pai, Chem. Lett., 1980, 9, 1415 CrossRef; (d) K.-M. Chen, G. E. Hardtmann, K. Prasad, O. Repič and M. J. Shapiro, Tetrahedron Lett., 1987, 28, 155 CrossRef CAS.
- For selected recent examples of stereodivergent reactions, see: (a) S. Doobary, A. J. D. Lacey, S. G. Sweeting, S. B. Coppock, H. P. Caldora, D. L. Poole and A. J. J. Lennox, Nat. Chem., 2024 DOI:10.1038/s41557-024-01561-6; (b) B. R. Brutiu, G. Iannelli, M. Riomet, D. Kaiser and N. Maulide, Nature, 2024, 626, 92 CrossRef CAS; (c) T. Long, C. Zhu, L. Li, L. Shao, S. Zhu, M. Rueping and L. Chu, Nat. Commun., 2023, 14, 55 CrossRef CAS PubMed; (d) S. Manna, S. Paul, W.-Y. Kong, D. Aich, R. Sahoo, D. J. Tantillo and S. Panda, Angew. Chem., Int. Ed., 2023, 62, e202309136 CrossRef CAS PubMed; (e) J. Merad, P. S. Grant, T. Stopka, J. Sabbatani, R. Meyrelles, A. Preinfalk, J. Matyasovsky, B. Maryasin, L. González and N. Maulide, J. Am. Chem. Soc., 2022, 144, 12536 CrossRef CAS; (f) M. E. Fairchild, A. Noble and V. K. Aggarwal, Angew. Chem., Int. Ed., 2022, 61, e202205816 CrossRef CAS; (g) D. Kaldre, I. Klose and N. Maulide, Science, 2018, 361, 664 CrossRef CAS PubMed; (h) S. Kassem, A. T. L. Lee, D. A. Leigh, V. Marcos, L. I. Palmer and S. Pisano, Nature, 2017, 549, 374 CrossRef CAS; (i) R. J. Armstrong, C. García-Ruiz, E. L. Myers and V. K. Aggarwal, Angew. Chem., Int. Ed., 2017, 56, 786 CrossRef CAS; (j) S.-L. Shi, Z. L. Wong and S. L. Buchwald, Nature, 2016, 532, 353 CrossRef CAS PubMed.
- For selected reviews on the applications of atropisomers, see: (a) S. R. LaPlante, P. J. Edwards, L. D. Fader, A. Jakalian and O. Hucke, ChemMedChem, 2011, 6, 505 CrossRef CAS; (b) S. T. Toenjes and J. L. Gustafson, Future Med. Chem., 2018, 10, 409 CrossRef CAS; (c) P. W. Glunz, Bioorg. Med. Chem. Lett., 2018, 28, 53 CrossRef CAS; (d) M. Basilaia, M. H. Chen, J. Secka and J. L. Gustafson, Acc. Chem. Res., 2022, 55, 2904 CrossRef CAS; (e) H.-U. Blaser, Adv. Synth. Catal., 2002, 344, 17 CrossRef CAS.
- For selected review articles on atropselective synthesis, see: (a) J. K. Cheng, S.-H. Xiang, S. Li, L. Ye and B. Tan, Chem. Rev., 2021, 121, 4805 CrossRef CAS PubMed; (b) S.-H. Xiang, W.-Y. Ding, Y.-B. Wang and B. Tan, Nat. Catal., 2024, 7, 483 CrossRef CAS; (c) J. Wencel-Delord, A. Panossian, F. R. Leroux and F. Colobert, Chem. Soc. Rev., 2015, 44, 3418 RSC; (d) J. S. Sweet and P. C. Knipe, Synthesis, 2022, 54, 2119 CrossRef CAS; (e) Y.-J. Wu, G. Liao and B.-F. Shi, Green Synth. Catal., 2022, 3, 117 CrossRef; (f) P. Rodríguez-Salamanca, R. Fernández, V. Hornillos and J. M. Lassaletta, Chem.–Eur. J., 2022, 28, e202104442 CrossRef PubMed; (g) G.-J. Mei, W. L. Koay, C.-Y. Guan and Y. Lu, Chem, 2022, 8, 1855 CrossRef CAS; (h) A. D. G. Campbell and R. J. Armstrong, Synthesis, 2023, 55, 2427 CrossRef CAS; (i) G. Centonze, C. Portolani, P. Righi and G. Bencivenni, Angew. Chem., Int. Ed., 2023, 62, e202303966 CrossRef; (j) E. Kumarasamy, R. Raghunathan, M. P. Sibi and J. Sivaguru, Chem. Rev., 2015, 115, 11239 CrossRef CAS PubMed; (k) X. Xiao, B. Chen, Y.-P. Yao, H.-J. Zhou, X. Wang, N.-Z. Wang and F.-E. Chen, Molecules, 2022, 27, 6583 CrossRef CAS; (l) O. Kitagawa, Acc. Chem. Res., 2021, 54, 719 CrossRef CAS; (m) V. Corti and G. Bertuzzi, Synthesis, 2020, 52, 2450 CrossRef CAS; (n) H.-H. Zhang and F. Shi, Acc. Chem. Res., 2022, 55, 2562 CrossRef CAS PubMed; (o) W. Qin, Y. Liu and H. Yan, Acc. Chem. Res., 2022, 55, 2780 CrossRef CAS PubMed.
- (a) H.-H. Zhang, T.-Z. Li, S.-J. Liu and F. Shi, Angew. Chem., Int. Ed., 2024, 63, e202311053 CrossRef CAS PubMed; (b) X.-F. Bai, Y.-M. Cui, J. Cao and L.-W. Xu, Acc. Chem. Res., 2022, 55, 2545 CrossRef CAS.
- X. Bao, J. Rodriguez and D. Bonne, Angew. Chem., Int. Ed., 2020, 59, 12623 CrossRef CAS PubMed.
- (a) O. M. Beleh, E. Miller, F. D. Toste and S. J. Miller, J. Am. Chem. Soc., 2020, 142, 16461 CrossRef CAS; (b) D. Moser and C. Sparr, Angew. Chem., Int. Ed., 2022, 61, e202202548 CrossRef CAS; (c) C. Portolani, G. Centonze, S. Luciani, A. Pellegrini, P. Righi, A. Mazzanti, A. Ciogli, A. Sorato and G. Bencivenni, Angew. Chem., Int. Ed., 2022, 61, e202209895 CrossRef CAS PubMed; (d) Y. Kwon, A. J. Chinn, B. Kim and S. J. Miller, Angew. Chem., Int. Ed., 2018, 57, 6251 CrossRef CAS PubMed; (e) D. Lotter, A. Castrogiovanni, M. Neuburger and C. Sparr, ACS Cent. Sci., 2018, 4, 656 CrossRef CAS; (f) X. Bao, J. Rodriguez and D. Bonne, Chem. Sci., 2020, 11, 403 RSC; (g) X. Wu, R. M. Witzig, R. Beaud, C. Fischer, D. Häussinger and C. Sparr, Nat. Catal., 2021, 4, 457 CrossRef CAS; (h) T. A. Schmidt, S. Schumann, A. Ostertag and C. Sparr, Angew. Chem., Int. Ed., 2023, 62, e202302084 CrossRef CAS.
- For atropisomeric N-aryl peptoids, see: (a) B. Paul, G. L. Butterfoss, M. G. Boswell, P. D. Renfrew, F. G. Yeung, N. H. Shah, C. Wolf, R. Bonneau and K. Kirshenbaum, J. Am. Chem. Soc., 2011, 133, 10910 CrossRef CAS; (b) B. Paul, G. L. Butterfoss, M. G. Boswell, M. L. Huang, R. Bonneau, C. Wolf and K. Kirshenbaum, Org. Lett., 2012, 14, 926 CrossRef CAS; (c) Y.-J. Wu, P.-P. Xie, G. Zhou, Q.-J. Yao, X. Hong and B.-F. Shi, Chem. Sci., 2021, 12, 9391 RSC; (d) Z.-S. Jia, Y.-J. Wu, Q.-J. Yao, X.-T. Xu, K. Zhang and B.-F. Shi, Org. Lett., 2022, 24, 304 CrossRef CAS PubMed; (e) T.-Y. Jiang, Y.-T. Ke, Y.-J. Wu, Q.-J. Yao and B.-F. Shi, Chem. Commun., 2023, 59, 13518 RSC.
- For macrocyclic peptide atropisomers, see: (a) I. J. Knudson, T. L. Dover, D. A. Dilworth, C. Paloutzian, H. Cho, A. Schepartz and S. J. Miller, ChemRxiv, 2024, preprint, DOI:10.26434/chemrxiv-2024-kvdpq; (b) P. Nanudorn, S. Thiengmag, F. Biermann, P. Erkoc, S. D. Dirnberger, T. N. Phan, R. Fürst, R. Ueoka and E. J. N. Helfrich, Angew. Chem., Int. Ed., 2022, 61, e202208361 CrossRef CAS; (c) M. C. Morejón, A. Laub, B. Westermann, D. G. Rivera and L. A. Wessjohann, Org. Lett., 2016, 18, 4096 CrossRef; (d) P. W. Glunz, L. Mueller, D. L. Cheney, V. Ladziata, Y. Zou, N. R. Wurtz, A. Wei, P. C. Wong, R. R. Wexler and E. S. Priestley, J. Med. Chem., 2016, 59, 4007 CrossRef CAS PubMed.
- For amino acid-derived atropisomeric sulfonamides, see: (a) Z. Li, H. Zhang and S. Yu, Org. Lett., 2019, 21, 4754 CrossRef CAS PubMed; (b) Z. Li, M. Tang, C. Hu and S. Yu, Org. Lett., 2019, 21, 8819 CrossRef CAS.
- For an application of Ugi reactions to construct atropisomeric heterocycles, see: M. Fragkiadakis, M. Thomaidi, T. Stergiannakos, E. Chatziorfanou, M. Gaidatzi, A. Michailidis Barakat, C. Stoumpos and C. Neochoritis, Chem.–Eur. J., 2024, 30, e202401461 CrossRef PubMed.
- I. Ugi, Angew. Chem., Int. Ed., 1962, 1, 8 CrossRef.
- (a) J. Zhang, P. Yu, S.-Y. Li, H. Sun, S.-H. Xiang, J. (Joelle) Wang, K. N. Houk and B. Tan, Science, 2018, 361, eaas8707 CrossRef; (b) B.-B. Sun, K. Liu, Q. Gao, W. Fang, S. Lu, C.-R. Wang, C.-Z. Yao, H.-Q. Cao and J. Yu, Nat. Commun., 2022, 13, 7065 CrossRef CAS; (c) K. M. Bonger, T. Wennekes, D. V. Filippov, G. Lodder, G. A. van der Marel and H. S. Overkleeft, Eur. J. Org Chem., 2008, 3678 CrossRef CAS.
- S. Balalaie, H. Motaghedi, D. Tahmassebi, M. Bararjanian and H. R. Bijanzadeh, Tetrahedron Lett., 2012, 53, 6177 CrossRef CAS.
- S. Grimme, Chem.–Eur. J., 2012, 18, 9955 CrossRef CAS.
- In line with other reports in the literature, we believe the rate limiting step of the reaction is the addition of isocyanide to an imine or iminium intermediate. This is supported by the observation of imine intermediates in multicomponent reactions which are terminated prematurely.
- N. Mardirossian and M. Head-Gordon, J. Chem. Phys., 2016, 144, 214110 CrossRef.
- C. Adamo and V. Barone, J. Chem. Phys., 1999, 110, 6158 CrossRef CAS.
- S. Grimme, J. Antony, S. Ehrlich and H. Krieg, J. Chem. Phys., 2010, 132, 154104 CrossRef.
- (a) F. Weigend and R. Ahlrichs, Phys. Chem. Chem. Phys., 2005, 7, 3297 RSC; (b) F. Weigend, Phys. Chem. Chem. Phys., 2006, 8, 1057 RSC.
- A. V. Marenich, C. J. Cramer and D. G. Truhlar, J. Phys. Chem. B, 2009, 113, 6378 CrossRef CAS.
- J.-P. Heeb, J. Clayden, M. D. Smith and R. J. Armstrong, Nat. Protoc., 2023, 18, 2745 CrossRef CAS.
- A. D. G. Campbell, N. J. Roper, P. G. Waddell, C. Wills, C. M. Dixon, R. M. Denton, K. Ermanis and R. J. Armstrong, Chem. Commun., 2024, 60, 3818 RSC.
- V. B. Makane, V. S. Krishna, E. V. Krishna, M. Shukla, B. Mahizhaveni, S. Misra, S. Chopra, D. Sriram, V. N. A. Dusthackeer and H. B. Rode, Eur. J. Med. Chem., 2019, 164, 665 CrossRef CAS.
- J.-C. Palomino, A. Martin, M. Camacho, H. Guerra, J. Swings and F. Portaels, Antimicrob. Agents Chemother., 2002, 46, 2720 CrossRef CAS PubMed.
- C. Vilchèze, J. Copeland, T. L. Keiser, T. Weisbrod, J. Washington, P. Jain, A. Malek, B. Weinrick and W. R. Jacobs, mBio, 2018, 9 DOI:10.1128/mbio.00938-18.
Footnote |
| † Electronic supplementary information (ESI) available: Detailed experimental procedures and characterization data for new compounds. CCDC 2355697–2355699 and 2382977–2382979. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d4sc04700a |
| This journal is © The Royal Society of Chemistry 2024 |