
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceThe forgotten borole: synthesis, properties and reactivity of a 1-boraindene†
Nele
Wieprecht
ab,
Ivo
Krummenacher
 ab,
Leonie
Wüst
ab,
Maximilian
Michel
ab,
Sonja
Fuchs
ab,
Samuel
Nees
ab,
Marcel
Härterich
ab and
Holger
Braunschweig
*ab
ab,
Leonie
Wüst
ab,
Maximilian
Michel
ab,
Sonja
Fuchs
ab,
Samuel
Nees
ab,
Marcel
Härterich
ab and
Holger
Braunschweig
*ab
aInstitute for Inorganic Chemistry, Julius-Maximilians-Universität Würzburg, Am Hubland, 97074 Würzburg, Germany. E-mail: h.braunschweig@uni-wuerzburg.de
bInstitute for Sustainable Chemistry & Catalysis with Boron, Julius-Maximilians-Universität Würzburg, Am Hubland, 97074 Würzburg, Germany
First published on 8th July 2024
Abstract
The chemistry of unsaturated boron heterocycles, including five-membered boroles, continues to attract substantial interest. Herein, we report the synthesis of 1,2,3-triphenyl-1-boraindene, a benzene-fused borole, and examine its Lewis acidic, electrophilic, and antiaromatic properties relative to non-fused and bis-benzannulated boroles (9-borafluorenes). Reactivity studies with organic azides reveal that the boraindene behaves similarly to other boroles, undergoing ring expansion to a BN-naphthalene through insertion of a nitrogen atom.
Introduction
Boroles, boron heterocycles containing 4π electrons in a five-membered BC4 ring, exhibit fascinating electronic and optical properties as well as unique reactivities, due to their antiaromatic character and three-coordinate boron atom.1 Over a decade of research into boroles has deepened our understanding of the critical relationship between structure and properties, paving the way for diverse applications in synthesis, catalysis, and material science.1Highly reactive by nature, boroles depend on the stabilizing influence of surrounding substituents in order to be isolable.2 Both the presence of aryl groups and ring-fusion strategies, whereby the five-membered borole ring becomes part of a larger conjugated system, have been demonstrated to effectively enhance the kinetic or thermodynamic stability of the antiaromatic unit.1,3–5 Fusing two benzene rings onto the borole core, yielding 9-borafluorenes,3 significantly enhances stability, reflected in both diminished antiaromatic character and reduced Lewis acidity. Dilution of the parent borole's antiaromaticity through ring fusion, a strategy that has resulted in a variety of aromatic–antiaromatic hybrids,4 surprisingly encounters an exception in cases where heteroarene units are fused to boroles. These species, first reported by the group of Yamaguchi in the form of dithiophene-fused boroles,6 deviate from the expected trend by exhibiting even more pronounced antiaromaticity.7 Studies on mixed heteroarene-benzene-fused boroles further support these findings,8 clearly implicating the heteroarene ring in enhancing the antiaromatic and Lewis acidic character of the central borole unit. In addition to electron-rich five-membered heterocycles, fusion with electron-poor pyridine also strengthens the Lewis acidity of boroles, as demonstrated by Marder and coworkers.9
While extensive research has explored the modification of borole properties through ring fusion, our understanding is limited almost entirely to systems where the borole unit sits between two aromatic rings. In contrast, singly fused boroles, like the boraindene scaffold, have been largely neglected in the literature. In 2014, Piers reported a perfluorinated 1-boraindene derivative, bridging the gap between non-fused and doubly fused boroles.10,11 Its reactivity towards dihydrogen revealed some interesting differences between these different classes of borole compounds. Leveraging its slightly lower reactivity towards dihydrogen relative to that of perfluoropentaphenylborole, the perfluorinated 1-boraindene derivative proved valuable as a mediator for the hydrogenation of olefins.10 Kaufmann synthesized one additional derivative, 1-chloro-1-boraindene, using pyrolytic dehydrogenation.12 However, its presence could only be indirectly confirmed through trapping experiments. Beyond these studies, the chemistry of boraindenes remains unexplored.13
In this work, we report the synthesis of 1,2,3-triphenyl-1-boraindene, expanding the class of benzene-fused borole derivatives alongside established families like the 9-borafluorenes. Employing a range of spectroscopic and computational techniques, we explore the properties of the boraindene and compare them to its non-fused and doubly benzene-fused borole analogues, revealing fundamental insights into the effect of single benzene ring fusion. Beyond the exploration of Lewis acid–base adduct formation, this study complements the understanding of the boraindene's reactivity by investigating its reaction with organic azides, leading to the formation of BN-naphthalenes.
Results and discussion
Synthesis of the 1-boraindene
Using the well-established tin–boron exchange strategy for borole synthesis, we treated 1,1-dimethyl-2,3-diphenyl-1-stannaindene14 with one equivalent of dibromo(phenyl)borane in toluene (Fig. 1). A color change from colorless to red signaled conversion to the boraindene product (1), as evidenced by the observed appearance of a signal at 65.3 ppm in the 11B NMR spectrum.15 Standard workup, which included sublimation, effectively removed the dimethyltin dibromide byproduct, affording 1 as a high-purity red solid in 66% yield. Single crystals of compound 1 suitable for X-ray diffraction analysis were obtained by slow evaporation of a saturated solution in benzene at room temperature. The structure shows a planar boraindene core, with the phenyl substituents twisted out of this plane: the phenyl group at boron is twisted by 32.6°. The pronounced alternation of carbon–carbon single (1.503(4) Å) and double bonds (1.375(3) and 1.415(3) Å) within the boracycle is consistent with electron localization and the antiaromatic nature of the ring.10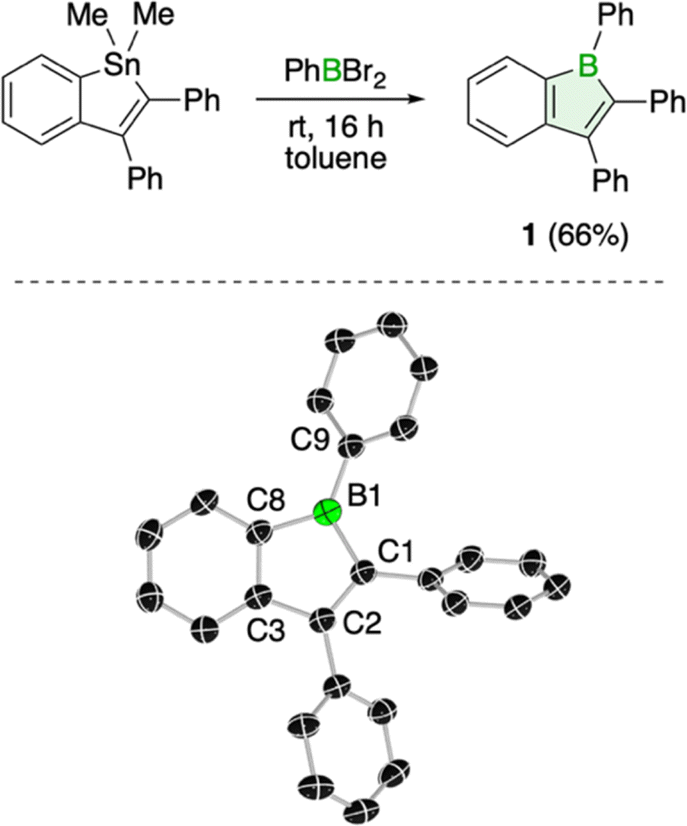 | ||
| Fig. 1 Synthesis and molecular structure of 1 (displacement ellipsoids at 50% probability level and hydrogen atoms removed for clarity). | ||
Occupying an intermediate position between monocyclic boroles and tricyclic 9-borafluorenes, we explored the influence of ring fusion on the properties of compound 1. When comparing the frontier orbital energies of boraindene 1, 9-phenyl-9-borafluorene and pentaphenylborole, a clear trend becomes evident (Fig. 2, see ESI† for computational details). Calculations show a gradual increase in the HOMO–LUMO gap across this series with the number of fused benzene rings, a consequence of both HOMO stabilization and LUMO destabilization. In accordance with this trend, the UV-vis absorption maxima show a hypsochromic shift, with the longest wavelength observed for pentaphenylborole (λ = 560 nm),15 followed by boraindene (λ = 485 nm), and then 9-phenyl-9-borafluorene (λ = 405 nm).16 The trend observed in the frontier orbital energies is further supported by the experimentally determined Lewis acidity and electrophilicity. Based on the Gutmann–Beckett method,17 evaluation by 31P NMR spectroscopy using triethylphosphine oxide (Et3PO) in deuterated benzene revealed an acceptor number (AN) of 76.9 for 1, indicating a slightly higher Lewis acidity than 9-phenyl-9-borafluorene (AN = 73.4).18 This observation is consistent with the general trend of reduced Lewis acidity in boroles with increasing numbers of fused benzene rings (cf. AN = 79.2 for pentaphenylborole).18 A similar trend is seen for the electron deficiency of the boracycle 1. Cyclic voltammetry of 1 in dichloromethane reveals a reduction potential (Epc = −1.75 V) that lies between those of non-fused (pentaphenylborole, Epc = −1.61 V)19 and doubly fused (9-phenyl-9-borafluorene, Epc = −1.87 V) derivatives.20Fig. 2 summarizes these comparisons of various borole structures, collectively confirming the observed trends with increasing benzene ring annulation. More specifically, as the number of fused benzene rings increases, the LUMO energy of the borole unit rises, leading to a decreased Lewis acidity and electron deficiency.
 | ||
| Fig. 2 Comparative analysis of key properties in non-fused (pentaphenylborole), singly benzene-fused (boraindene 1), and doubly benzene-fused borole (9-phenyl-9-borafluorene) systems. Details and references are provided in the main text. | ||
Nucleus-independent chemical shift (NICS) calculations21 reveal a more nuanced relationship between antiaromaticity and structure within this series of three boroles (Fig. 2). NICS calculations indicate NICSzz(−1/+1) values of 26.88/27.90 ppm at the PBE1PBE/6-311+G(d,p)/D3(BJ) level of theory for the borole unit in 1. These values surpass those of 9-phenyl-9-borafluorene (24.62/24.62 ppm), consistent with the expected decline in antiaromaticity as the degree of benzannulation increases.6,7,22 However, pentaphenylborole (24.55/24.88 ppm) deviates from this trend, displaying a slightly lower degree of antiaromatic character than in boraindene 1. The four phenyl groups on the carbon atoms of the borole ring likely influence its π-electron structure. Due to their twisted (propeller-like) arrangement, their ability to donate electrons through resonance will be, however, limited. Interestingly, calculations on 1-phenylborole (PhBC4H4), which lacks the phenyl substituents on the backbone, reveal a more pronounced antiaromatic character (NICSzz(−1/+1) = 28.06/28.05 ppm) than pentaphenylborole and even boraindene 1. In boraindene 1, the two phenyl substituents around the borole ring appear to have a smaller influence on its antiaromaticity, as shown by nearly unchanged NICS values of its hydrogen analogue (NICSzz(−1/+1) = 27.94/27.11 ppm, see ESI† for details).
Due to their pronounced Lewis acidity, boroles are particularly susceptible to adduct formation with Lewis bases, which is manifested by the reported isolation of numerous stable adducts.1,3 In addition to Et3PO, adducts of 1 with THF and the nitrogen bases 4-dimethylaminopyridine (DMAP) and pyridine (Py) were isolated and fully characterized, including structural confirmation by single-crystal X-ray crystallography (Scheme 1, see ESI† for details). The transformations are readily identified by the immediate disappearance of the red boraindene 1 solution and a shift of the 11B NMR signal to lower frequencies, indicating the presence of a four-coordinate boron atom. Despite successful crystallographic characterization, the THF adduct is weakly bound and decomposes under vacuum, slowly releasing THF and reverting to the boraindene 1.
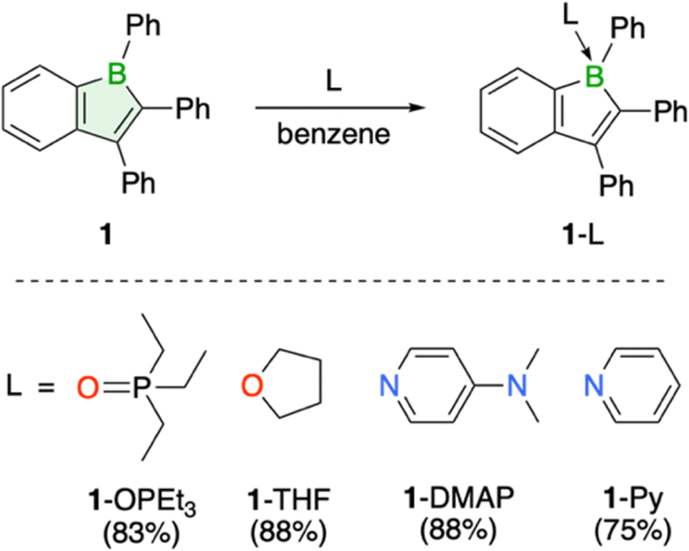 | ||
| Scheme 1 Adducts of compound 1 with triethylphosphine oxide, amines, and THF. | ||
Reactivity with organic azides
Boroles, due to their antiaromatic nature, readily undergo insertion reactions to form ring-expanded products.1d,g A prime example is their reaction with organic azides, leading to the formation of aromatic BN heterocycles through nitrogen atom insertion.1d,g,3,23 To explore the potential of compound 1 to undergo similar ring expansions, its reaction with mesityl and trimethylsilyl azide was studied. Treatment of 1 with mesityl azide at room temperature led to an immediate color change from red to yellow, accompanied by the appearance of a new 11B NMR signal at 38.9 ppm (Scheme 2a).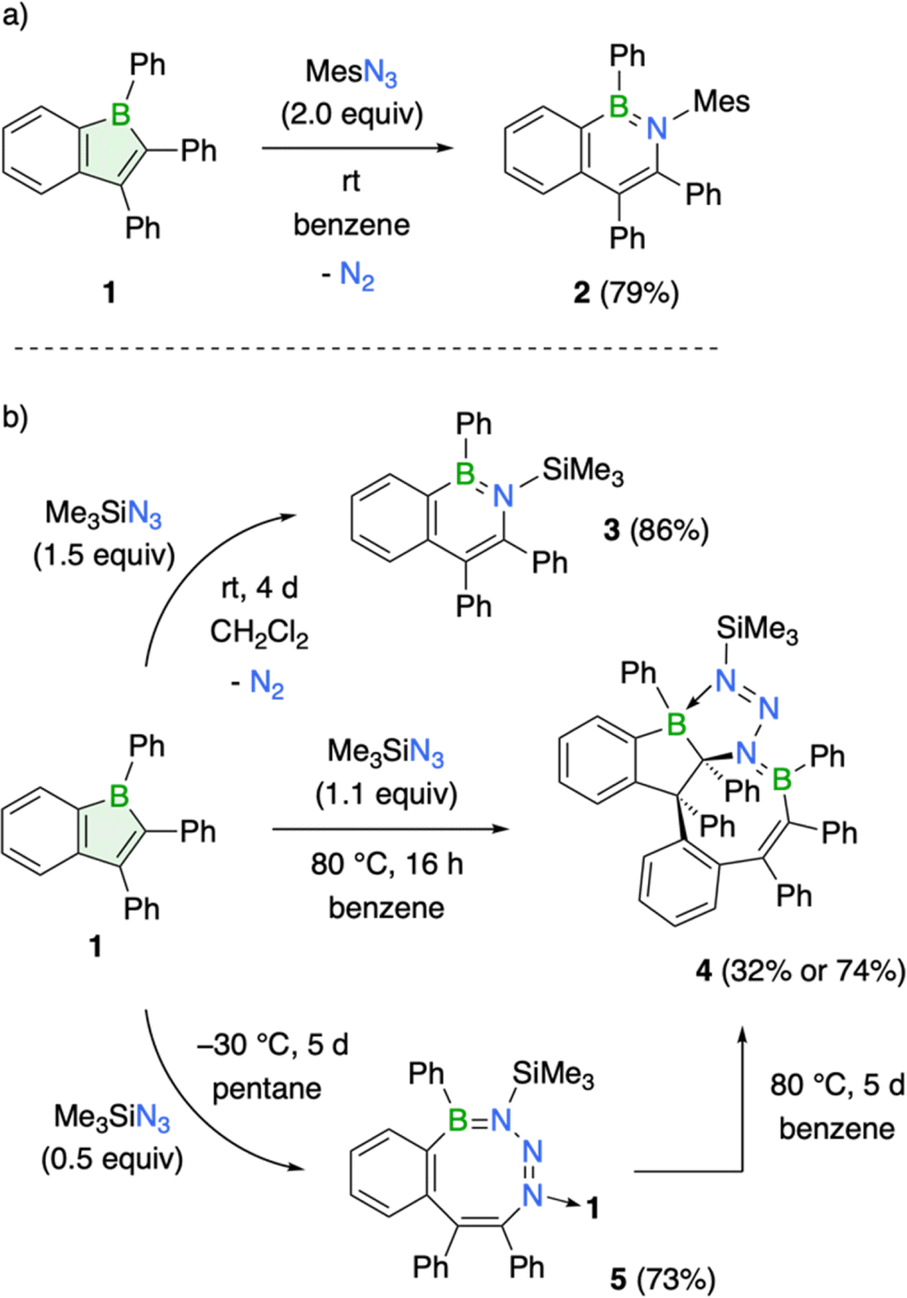 | ||
| Scheme 2 Reactivity of compound 1 with organic azides: (a) mesityl azide and (b) trimethylsilyl azide. | ||
The observed 11B NMR chemical shift aligns well with the range typically reported for aromatic BN heterocycles, suggesting the formation of the corresponding azaborinine ring system.24 The product was isolated as a yellow crystalline solid in 79% yield after standard workup procedure. X-ray diffraction analysis of single crystals grown by slow evaporation of a saturated hexane solution at room temperature confirmed the planar structure of compound 2, a BN analogue of naphthalene (see ESI† for details). Its structural features (e.g. B–N bond: 1.429(2) Å) are consistent with closely related derivatives.25 Notably, the aryl groups on the heterocycle are nearly perpendicular to the ring plane, with the phenyl group on boron showing a more pronounced out-of-plane twist (68.0°) than that of 1. Since Dewar's pioneering work in 1959 on the first BN-naphthalene derivative,26 a variety of synthetic strategies have been developed for these compounds, enabling the generation of a wide range of substitution patterns and isomeric forms.24d Beyond the methods developed by Cui and Wagner, our work presents a new approach for synthesizing the specific BN-naphthalene isomer described herein.25,27
Treatment of 1 with trimethylsilyl azide in dichloromethane afforded the corresponding 2,1-azaboranaphthalene 3, albeit requiring significantly longer reaction times than the reaction with mesityl azide. Stirring 1 with 1.5 equivalents of the azide at room temperature for 4 d resulted in complete conversion of 1 to 3, yielding the BN naphthalene as an off-white solid in a yield of 86% (Scheme 2b). Multinuclear NMR spectroscopy (δ(11B) = 44.8 ppm) and structural analysis (B1–N1 bond: 1.439(2) Å) corroborate with the expected features of 3. It is worth mentioning that the heterocycle in 3 is notably distorted from planarity, likely caused by repulsive steric interactions between the trimethylsilyl group and its neighboring aryl substituents (Fig. 3).
 | ||
| Fig. 3 Molecular structures of the products from the reaction of 1 with trimethylsilyl azide. Displacement ellipsoids are shown at the 50% probability level (ellipsoids of peripheral phenyl groups and hydrogen atoms removed for clarity). | ||
Switching the solvent to benzene led to the formation of a side product (4), isolable in 32% yield following the completion of the reaction at elevated temperatures (Scheme 2b). Compound 4 is characterized by two different 11B NMR signals (51.1 and 9.7 ppm), suggesting the presence of boron atoms in both three- and four-coordinate environments. Single-crystal X-ray diffraction definitively established its molecular structure. Compound 4 is a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :2 azide-to-borole addition product featuring a polycyclic skeleton with a central eight-membered BNC6 ring (B2–N3 1.442(2) Å) adopting a tub conformation. Two carbon atoms from the BN heterocycle are embedded within a fused boraindane unit. Notably, a nitrogen atom of the diazenyl moiety tethered to the incorporated nitrogen atom coordinates with this boraindane, forming a five-membered BCN3 ring (B1–N1 bond 1.599(2) Å). The structure exhibits a polycyclic architecture reminiscent of those observed from the reaction of a thiophene-benzo-fused borole with organic azides.8b High-resolution mass spectrometry (HRMS) analysis provided further corroboration for the polycyclic structure, as evidenced by the molecular ion peak at m/z = 799.3720.
:2 azide-to-borole addition product featuring a polycyclic skeleton with a central eight-membered BNC6 ring (B2–N3 1.442(2) Å) adopting a tub conformation. Two carbon atoms from the BN heterocycle are embedded within a fused boraindane unit. Notably, a nitrogen atom of the diazenyl moiety tethered to the incorporated nitrogen atom coordinates with this boraindane, forming a five-membered BCN3 ring (B1–N1 bond 1.599(2) Å). The structure exhibits a polycyclic architecture reminiscent of those observed from the reaction of a thiophene-benzo-fused borole with organic azides.8b High-resolution mass spectrometry (HRMS) analysis provided further corroboration for the polycyclic structure, as evidenced by the molecular ion peak at m/z = 799.3720.
The slow conversion rate of 1 to BN-naphthalene 3 enabled the isolation of a kinetic product with an eight-membered BN3C4 ring system, resembling a structure reported by Martin and coworkers.23c Since a second equivalent of borole coordinates to the eight-membered ring, employing half an equivalent of the azide in pentane solution and allowing the reaction mixture to crystallize at −30 °C efficiently afforded product 5 in 73% yield (Scheme 2b). 11B NMR spectroscopy reveals two distinct boron environments at 49.2 and 4.7 ppm, with the lower-frequency signal corresponding to the boron atom of the coordinated boraindene unit. In the solid-state structure, the eight-membered ring adopts a twist-tub conformation similar to that described by Martin.23c The endocyclic (1.453(3) Å) and exocyclic (1.632(3) Å) boron–nitrogen bond lengths align well with the data presented for the earlier example (Fig. 3). Although complete NMR characterization of 5 was achieved, we observed the appearance of new resonances over time, suggesting gradual and selective conversion to the polycyclic product 4. To promote its generation, we heated compound 5 to 80 °C. After 5 d at this temperature, the kinetically favored product 5 is fully consumed, and the thermodynamic product 4 could be isolated as a white solid in 74% yield (Scheme 2b).
Overall, the reactivity profile of boraindene 1 aligns with that of both doubly fused (9-borafluorenes) and non-fused borole derivatives.1,3 All three react with organic azides via formal nitrene insertion, leading to the formation of corresponding BN heterocycles: 9,10-azaboraphenanthrenes for 9-borafluorenes,1g,3,18 2,1-azaboranaphthalenes for boraindenes, and 1,2-azaborinines for boroles.1d,g,23 The observed reactivity divergence of boraindene 1, forming an eight-membered BN3C4 ring23c and a polycyclic product,8b mirrors findings with other borole derivatives, suggesting similar reaction mechanisms for these transformations.
Conclusions
In summary, we have prepared an aryl-substituted boraindene derivative via tin–boron exchange. It exhibits Lewis acidity and electrophilicity properties intermediate between those of monocyclic and double-benzannulated derivatives (9-borafluorene), thereby strengthening the established trend that these properties weaken with increasing numbers of fused benzene rings. NICS calculations reveal a remarkably strong antiaromatic character in 1,2,3-triphenyl-1-boraindene, surpassing even that of the isolable non-fused borole derivative 1,2,3,4,5-pentaphenylborole. Our study further reveals that its reactivity with organic azides mirrors the diverse reaction pathways observed for other borole derivatives, leading to ring-expansion reactions that produce BN-aromatic hydrocarbons, eight-membered rings, and polycyclic products. Notably, boraindene ring expansion using organic azides provides a novel approach to the synthesis of the 2,1-azaboranaphthalene scaffold, one of several possible BN-naphthalene structures.Data availability
The data that support the findings of this study are available in the ESI† of this article.Author contributions
H. B. conceived and designed the research program, N. W. designed studies and performed the research, with support from S. F. L. W. performed the DFT calculations. M. M., S. N., and M. H. conducted the crystallographic studies. I. K. performed the electrochemical studies and wrote the manuscript with contributions from all authors.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We thank the Julius-Maximilians-Universität Würzburg and the Deutsche Forschungsgemeinschaft (DFG grant 466754611) for financial support of this work. We are grateful to Dr Rian D. Dewhurst for insightful discussions and feedback on the manuscript.Notes and references
- For general borole reviews, see: (a) A. Steffen, R. M. Ward, W. D. Jones and T. B. Marder, Coord. Chem. Rev., 2010, 254, 1950–1976 CrossRef CAS; (b) H. Braunschweig and T. Kupfer, Chem. Commun., 2011, 47, 10903–10914 RSC; (c) H. Braunschweig, I. Krummenacher and J. Wahler, Adv. Organomet. Chem., 2013, 61, 1–53 CrossRef CAS; (d) J. H. Barnard, S. Yruegas, K. Huang and C. D. Martin, Chem. Commun., 2016, 52, 9985–9991 RSC; (e) B. Su and R. Kinjo, Synthesis, 2017, 49, 2985–3034 CrossRef CAS; (f) A. Wakamiya, in Main Group Strategies towards Functional Hybrid Materials, ed. T. Baumgartner and F. Jäkle, John Wiley & Sons Ltd, Hoboken (New Jersey), 2018, pp. 1–26 Search PubMed; (g) Y. Su and R. Kinjo, Chem. Soc. Rev., 2019, 48, 3613–3659 RSC; (h) C. Hong, J. Baltazar and J. D. Tovar, Eur. J. Org Chem., 2022, e202101343 CrossRef CAS.
- Unprotected boroles undergo dimerization: (a) P. J. Fagan, E. G. Burns and J. C. Calabrese, J. Am. Chem. Soc., 1988, 110, 2979–2981 CrossRef CAS; (b) P. J. Fagan, W. A. Nugent and J. C. Calabrese, J. Am. Chem. Soc., 1994, 116, 1880–1889 CrossRef CAS; (c) G. E. Herberich and H. Ohst, Chem. Ber., 1985, 118, 4303–4313 CrossRef CAS; (d) H. Braunschweig, C.-W. Chiu, J. Wahler, K. Radacki and T. Kupfer, Chem.–Eur. J., 2010, 16, 12229–12233 CrossRef CAS PubMed; (e) H. Braunschweig, C.-W. Chiu, A. Damme, K. Ferkinghoff, K. Kraft, K. Radacki and J. Wahler, Organometallics, 2011, 30, 3210–3216 CrossRef CAS; (f) Z. Zhang, Z. Wang, M. Haehnel, A. Eichhorn, R. M. Edkins, A. Steffen, A. Krueger, Z. Lin and T. B. Marder, Chem. Commun., 2016, 52, 9707–9710 RSC; (g) Z. Wang, Y. Zhou, K.-H. Lee, W. H. Lam, R. D. Dewhurst, H. Braunschweig, T. B. Marder and Z. Lin, Chem.–Eur. J., 2017, 23, 11587–11597 CrossRef CAS PubMed; (h) X. Su, J. J. Baker and C. D. Martin, Chem. Sci., 2020, 11, 126–131 RSC.
- X. Su, T. A. Bartholome, J. R. Tidwell, A. Pujol, S. Yruegas, J. J. Martinez and C. D. Martin, Chem. Rev., 2021, 121, 4147–4192 CrossRef CAS PubMed.
- J. He, F. Rauch, M. Finze and T. B. Marder, Chem. Sci., 2021, 12, 128–147 RSC.
- Isolable boroles without aryl groups and fused rings are rare: (a) L. Killian and B. Wrackmeyer, J. Organomet. Chem., 1978, 148, 137–146 CrossRef CAS; (b) A. Sebald and B. Wrackmeyer, J. Organomet. Chem., 1986, 307, 157–165 CrossRef CAS; (c) M. Golfmann and C. P. Sindlinger, Eur. J. Inorg. Chem., 2022, e202200359 CrossRef CAS.
- A. Iida and S. Yamaguchi, J. Am. Chem. Soc., 2011, 133, 6952–6955 CrossRef CAS.
- A. Iida, A. Sekioka and S. Yamaguchi, Chem. Sci., 2012, 3, 1461–1466 RSC.
- (a) W. Zhang, D. Yu, Z. Wang, B. Zhang, L. Xu, G. Li, N. Yan, E. Rivard and G. He, Org. Lett., 2019, 21, 109–113 CrossRef CAS; (b) S. Fuchs, A. Jayaraman, I. Krummenacher, L. Haley, M. Baštovanović, M. Fest, K. Radacki, H. Helten and H. Braunschweig, Chem. Sci., 2022, 13, 2932–2938 RSC.
- (a) J. He, F. Rauch, A. Friedrich, J. Krebs, I. Krummenacher, R. Bertermann, J. Nitsch, H. Braunschweig, M. Finze and T. B. Marder, Angew. Chem. Int. Ed., 2021, 60, 4833–4840 CrossRef CAS; (b) J. He, F. Rauch, I. Krummenacher, H. Braunschweig, M. Finze and T. B. Marder, Dalton Trans., 2021, 50, 355–361 RSC.
- A. Y. Houghton, V. A. Karttunen, W. E. Piers and H. M. Tuononen, Chem. Commun., 2014, 50, 1295–1298 RSC.
- A. Y. Houghton, J. Hurmalainen, A. Mansikkamäki, W. E. Piers and H. M. Tuononen, Nat. Chem., 2014, 6, 983–988 CrossRef CAS PubMed.
- W. Schacht and D. Kaufmann, Angew Chem. Int. Ed. Engl., 1987, 26, 665–666 CrossRef.
- X. Liu, M. Heinz, J. Wang, L. Tan, M. C. Holthausen and Q. Ye, Angew. Chem., Int. Ed., 2023, 62, e202312608 CrossRef CAS PubMed.
- A. Y. Houghton, PhD thesis, University of Calgary, 2014 Search PubMed.
- H. Braunschweig, I. Fernández, G. Frenking and T. Kupfer, Pentaphenylborole exhibits a 11B NMR chemical shift of 65.4 ppm, Angew. Chem., Int. Ed., 2008, 47, 1951–1954 CrossRef CAS PubMed.
- R. Köster and G. Benedikt, Angew. Chem., 1963, 75, 419 CrossRef.
- (a) M. A. Beckett, G. C. Strickland, J. R. Holland and K. Sukumar Varma, Polymer, 1996, 37, 4629–4631 CrossRef CAS; (b) V. Gutmann, Coord. Chem. Rev., 1976, 18, 225–255 CrossRef CAS; (c) P. Erdmann and L. Greb, Angew. Chem. Int. Ed., 2022, 61, e202114550 CrossRef CAS PubMed; (d) A. E. Ashley, T. J. Herrington, G. G. Wildgoose, H. Zaher, A. L. Thompson, N. H. Rees, T. Krämer and D. O'Hare, J. Am. Chem. Soc., 2011, 133, 14727–14740 CrossRef CAS PubMed.
- S. Yruegas, J. J. Martinez and C. D. Martin, Chem. Commun., 2018, 54, 6808–6811 RSC.
- H. Braunschweig and I. Krummenacher, in Organic Redox Systems, ed. T. Nishinaga, John Wiley & Sons Ltd, Hoboken (New Jersey), 2016, pp. 503–519 Search PubMed.
- T. Bischof, X. Guo, I. Krummenacher, L. Beßler, Z. Lin, M. Finze and H. Braunschweig, Chem. Sci., 2022, 13, 7492–7497 RSC.
- (a) P. v. R. Schleyer, C. Maerker, A. Dransfeld, H. Jiao and N. J. R. v. E. Hommes, J. Am. Chem. Soc., 1996, 118, 6317–6318 CrossRef CAS PubMed; (b) A. C. Tsipis, Phys. Chem. Chem. Phys., 2009, 11, 8244–8261 RSC.
- T. B. Tai, V. T. T. Huong and M. T. Nguyen, Chem. Commun., 2013, 49, 11548–11550 RSC.
- For original studies, see: (a) S. Biswas, C. Maichle-Mössmer and H. F. Bettinger, Chem. Commun., 2012, 48, 4564–4566 RSC; (b) H. Braunschweig, C. Hörl, L. Mailänder, K. Radacki and J. Wahler, Chem.–Eur. J., 2014, 20, 9858–9861 CrossRef CAS PubMed; (c) S. A. Couchman, T. K. Thompson, D. J. D. Wilson, J. L. Dutton and C. D. Martin, Chem. Commun., 2014, 50, 11724–11726 RSC.
- (a) M. J. D. Bosdet and W. E. Piers, Can. J. Chem., 2009, 87, 8–29 CrossRef; (b) P. G. Campbell, A. J. V. Marwitz and S.-Y. Liu, Angew. Chem., Int. Ed., 2012, 51, 6074–6092 CrossRef CAS PubMed; (c) G. Bélanger-Chabot, H. Braunschweig and D. K. Roy, Eur. J. Inorg. Chem., 2017, 4353–4368 CrossRef; (d) Z. X. Giustra and S.-Y. Liu, J. Am. Chem. Soc., 2018, 140, 1184–1194 CrossRef CAS PubMed.
- X. Liu, P. Wu, J. Li and C. Cui, J. Org. Chem., 2015, 80, 3737–3744 CrossRef CAS PubMed.
- M. J. S. Dewar and R. Dietz, J. Chem. Soc., 1959, 2728–2730 RSC.
- O. Ouadoudi, T. Kaehler, M. Bolte, H.-W. Lerner and M. Wagner, Chem. Sci., 2021, 12, 5898–5909 RSC.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 2360675, 2360676, 2360677, 2360678, 2360679, 2360680, 2360681, 2360682 and 2360683. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d4sc03817g |
| This journal is © The Royal Society of Chemistry 2024 |