
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Formation of a low-symmetry Pd8 molecular barrel employing a hetero donor tetradentate ligand, and its use in the binding and extraction of C70†
Dharmraj
Prajapati
a,
Jack K.
Clegg
 b and
Partha Sarathi
Mukherjee
*a
b and
Partha Sarathi
Mukherjee
*a
aDepartment of Inorganic and Physical Chemistry, Indian Institute of Science, Bangalore-560012, India. E-mail: psm@iisc.ac.in
bSchool of Chemistry and Molecular Biosciences, The University of Queensland, St. Lucia, Queensland 4072, Australia
First published on 26th June 2024
Abstract
The majority of reported metallo-supramolecules are highly symmetric homoleptic assemblies of MxLy type, with a few reports on assemblies that are obtained using multicomponent self-assembly or using ambidentate ligands. Herein, we report the use of an unsymmetrical tetratopic ligand (Lun) containing pyridyl and imidazole donor sites in combination with a cis-protected Pd(II) acceptor for the formation of a low-symmetry M8Lun4 molecular barrel (UNMB). Four potential orientational isomeric (HHHH, HHHT, HHTT, and HTHT) molecular barrels can be anticipated for the M8Lun4 type metallo-assemblies. However, the formation of an orientational isomer (HHTT) of the barrel was suggested from single-crystal X-ray diffraction and 1H NMR analysis of UNMB. Two large open apertures at terminals and the hydrophobic confined space surrounded by four aromatic panels of Lun make UNMB a potential host for bigger guests. UNMB encapsulates fullerenes C70 and C60 favoured by non-covalent interactions between the fullerenes and aromatic panels of the ligand molecules. Experimental and theoretical studies revealed that UNMB has the ability to bind C70 more strongly than its lower analogue C60. The stronger affinity of UNMB towards C70 was exploited to separate C70 from an equimolar mixture of C70 and C60. Moreover, C70 can be extracted from the C70⊂UNMB complex by toluene, and therefore, UNMB can be reused as a recyclable separating agent for C70 extraction.
Introduction
Nature has used various types of non-covalent interactions to control the preciseness of self-assembly processes to ensure that the individual components combine in specific ratios and orientations to function effectively.1 For instance, protein-based enzymes convene into homo- or hetero-polymeric quaternary structures to execute a variety of biological functions.2 The active sites of these enzymes are enclosed by low symmetrical and chiral cavities containing a combination of different chemical functionalities, such as catalytic sites, recognition sites, and conformational switches.3 Over the past few decades, chemists across the globe in different fields have tried to imitate these principles by applying them to synthetic systems.4 In this regard, a wide range of supramolecular architectures, such as self-assembled coordination cages,5 molecular capsules,6 and molecular barrels7 that demonstrate a vast range of important applications, have been developed.8 Nonetheless, the majority of such self-assembled molecules reported to date are highly symmetric and homoleptic complexes of MxLy type, which were prepared by incorporating a selected metal node (M) and only one type of symmetric ligand (L).5–8 Recently, there has been a push towards unfolding lower symmetry systems intending to get upgraded functionally. In addition to heading towards supramolecular assemblies with reduced symmetry,9 such as heteroleptic cage systems10 and hetero-polymetallic cage systems,11 there has also been a rise in interest in developing self-assembled systems containing bis-monodentate ligands of two different donor groups.12 However, unsymmetrical multitopic ligands (number of donor sites > 2) with distinct binding sites remain largely ignored because of the possibility of the formation of a mixture of different isomers. An isomeric mixture of the complexes might be formed due to the random relative orientational arrangement of the ligands around the metal centres. Thus, selecting an unsymmetrical ligand with distinct donor sites for the synthesis of self-assembled coordination complexes of lower symmetry is quite challenging.Fullerene is one of the stable allotropes of carbon and has a vast range of captivating properties, such as conducting, magnetic, antioxidant, and electronic, due to its unusual symmetry and extended conjugated electronic features.13 Owing to these properties, fullerenes find applications in several fields, for instance in materials science,14 in superconducting materials,15 as electroactive materials in solar cells,16 and for biological applications.17 However, these applications largely depend on their purity and solubility. Unrefined carbon soot contains a mixture of fullerenes of different carbon numbers and amorphous forms of carbon and other allotropes such as carbon nanotubes.18 The popular purification techniques to separate fullerenes from carbon soot are recrystallization, controlled sublimation, and extraction with organic solvents.19 In recent times, chromatographic methods have been used predominantly for the isolation and purification of fullerenes.20 Although efficient columns are available for the isolation of fullerene using HPLC techniques, all of these purification methods need large amounts of solvents and can induce irreversible adsorption, and decomposition of fullerene within the column.20 Moreover, these techniques are often expensive, tedious, energy and time-consuming, and in some instances, it is difficult to get a particular fullerene with high selectivity. Therefore, inventing techniques for the purification of fullerenes is a challenging and highly desirable task in materials chemistry. Over the past few decades, selective separation of fullerenes by encapsulation within soluble supramolecular receptors has attracted the research community's attention21 because this method offers potential selectivity via selective host–guest complexation without any special equipment. Moreover, the encapsulation of fullerene enhances its solubility. Therefore, it is highly appealing to devise a suitable molecular host that has a better binding affinity for one fullerene over the other, leading to their separation from a mixture.
Herein, we report the formation of an unsymmetrical molecular barrel (UNMB) of M8Lun4 type by coordination-driven self-assembly of an unsymmetrical tetratopic donor 4′-(3,5-di(1H-imidazole-1-yl)phenyl)-4,2′:6′,4′′-terpyridine (Lun) containing pyridine and imidazole donor sites with cis-[(tmeda)Pd(ONO2)2] as an acceptor in DMSO (tmeda = N,N,N′,N′-tetramethyl-ethane-1,2-diamine) (Scheme 1). The orientational isomeric product (UNMB) was characterized by 1H NMR, 2D DOSY NMR, and ESI-MS analysis. Furthermore, the molecular structure was unambiguously established by single-crystal X-ray diffraction analysis. UNMB features a rhombohedral hydrophobic cavity fenced by extended π-conjugated aromatic rings of the four ligand units (Lun) along with two large open windows. Above-mentioned features of UNMB assist the encapsulation of C70 and C60 inside its hydrophobic cavity. ESI-MS analysis of C70⊂UNMB and C60⊂UNMB revealed the formation of 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 host–guest complexes (Scheme 1). Furthermore, association constant values (Ka), DFT studies, and competitive guest encapsulation studies suggested that UNMB has better binding affinity towards C70 over C60, which enables the recyclable separation of C70 from an equimolar mixture of C60 and C70.
:1 host–guest complexes (Scheme 1). Furthermore, association constant values (Ka), DFT studies, and competitive guest encapsulation studies suggested that UNMB has better binding affinity towards C70 over C60, which enables the recyclable separation of C70 from an equimolar mixture of C60 and C70.
 | ||
| Scheme 1 Schematic presentation of the synthesis of unsymmetrical molecular barrel UNMB, and its selective encapsulation of C70. | ||
Results and discussion
Synthesis and characterization of UNMB
The new tetratopic unsymmetrical ligand Lun containing two imidazole and two pyridyl donor sites was synthesized by standard Ullman coupling of 4′-(3,5-dibromophenyl)-4,2′:6′,4′′-terpyridine with imidazole (Scheme S1†).20 Ligand Lun was characterized by 1H and 13C NMR spectroscopy (Fig. S2–S5†). A mixture of ligand Lun (1 equivalent) and metal acceptor cis-[(tmeda)Pd(ONO2)2] (M) (2 equivalents) in DMSO was heated for 12 h at 60 °C under stirring. Afterward, the resulting clear solution was precipitated by treating it with an excess of ethyl acetate. The precipitate was centrifuged and dried under vacuum to get a white powder of UNMB in 98% yield. The obtained white powder of UNMB was analysed by NMR and ESI-MS spectroscopy. Due to the unsymmetrical nature of the tetratopic donor Lun, upon self-assembly with 90° acceptor M, it may produce different orientational isomers of the most common compositions, such as M6Lun3 or M8Lun4. The 1H NMR spectrum of UNMB in D2O displayed a set of eight peaks in the aromatic region and three signals each for ethylene and methyl protons of the acceptor unit in the aliphatic region (2.6–3.3 ppm) (Fig. 1, S6 and S7†). The integral ratio (1:2:1) of the proton signals of ethylene and methyl groups in the aliphatic region probably hints at either the formation of the product that has three different kinds of (tmeda)Pd(II) units in a 1:2:1 ratio (Fig. S8†) or an equilibrium mixture. Moreover, a significant downfield shift was observed in the signals of pyridyl α-protons (Ha) and imidazole protons (Hf) with Δδ = 0.33 and Δδ = 1.16 ppm, respectively, which indicates the formation of ligand-to-metal dative bonds (Fig. 1 and S6†). Additionally, the appearance of a single diffusion band in the 2D DOSY (diffusion order spectroscopy) NMR spectrum indicated the formation of a single self-assembled molecular architecture (Fig. 1c and S8†). Moreover, all the proton signals of UNMB were assigned by a thorough investigation of 1H–1H COSY NMR, which confirmed that all signals are originated from the ligand Lun (Fig. S9†). Thus, altogether, the NMR spectral data provided preliminary information in support of the formation of a self-assembled architecture, but due to the absence of the required splitting patterns of the proton signals, it could not suggest the arrangement of the donors in the final assembly.
 | ||
| Fig. 1 Stacked partial 1H NMR spectra of (a) UNMB in D2O, (b) ligand Lun in CDCl3, and (c) diffusion-ordered 1H NMR of UNMB in D2O. | ||
The molecular composition of the self-assembled molecular architecture in solution was ascertained by electrospray ionization mass spectroscopy (ESI-MS). For this, an aqueous solution of UNMB was reacted with an excess of KPF6 at room temperature overnight. The ESI-MS spectrum of the PF6− analogue was recorded in acetonitrile. The ESI-MS spectrum showed the presence of several noticeable peaks and respective isotopic distribution patterns corresponding to the charge fragments at m/z = 1810.4826 for [M8Lun4(PF6)13]3+, 1321.6334 for [M8Lun4(PF6)12]4+, 1028.3242 for [M8Lun4(PF6)11]5+, 832.7648 for [M8Lun4(PF6)10]6+, and 693.0971 for [M8Lun4(PF6)9]7+, which are well matched with the respective calculated isotopic distribution patterns of the above mentioned charged fragments (Fig. 2, S11 and S12†). Thus, ESI-MS investigation suggested the formation of a molecular architecture with the composition of M8Lun4.
 | ||
| Fig. 2 ESI-MS spectrum of the PF6− analogue of UNMB in acetonitrile. (Inset) Experimental isotopic distribution pattern of the [M8Lun4(PF6)10]6+ fragment. | ||
Due to the different orientations of four molecules of non-symmetric ligand Lun, several orientational isomeric barrels like HHHH, HHHT, HHTT and HTHT (Scheme 1) are possible. In the case of the most symmetric HHHH (having C4V symmetry) isomer, there will be two kinds of palladium centres in a 1:1 ratio. One set of Pd ions will be coordinated to the pyridyl units and another set will be connected with the imidazole units; hence, in this case, the 1H NMR pattern will presumably be simpler.7e,f However, in the case of the HTHT (having D2d symmetry) isomer, there will be only one kind of Pd(II) centre; hence, each pyridyl and imidazole unit will face an identical electronic environment, and therefore, again, a much simple NMR pattern is expected. The aliphatic proton peaks' integrations in a ratio of 1:2:1 hint at the formation of either the HHTT (having C2v symmetry) or HHHT (having Cs symmetry) isomer. Only these two isomers have three different kinds of (tmeda)Pd(II) units. 1H NMR data in combination with the ESI-MS result indicated the formation of one of these isomers.
A computational study was performed to determine the comparative stabilities of these isomeric barrels, depicted in Scheme 1. Initially, the computational optimization for all the isomers was performed by the PM6 semiempirical method in their ground state. Next, single point energy calculations were carried out by employing the DFT method (B3LYP/LanL2DZ, 6-31G). From the DFT calculations, it can be observed that orientational isomer HHHH is found to be the energetically most favoured isomer, while isomer HTHT is the least favourable (Fig. 3). However, the energy difference among the HHHH, HHHT and HHTT isomers was found to be small (<20 kcal mol−1) (Table S2†). Moreover, the energy difference between the HHTT and HHHT isomers (one of which is indicated to be formed by 1H NMR analysis) is almost negligible. Thus, the theoretical study did not give enough information to predict the isomer formed.
 | ||
| Fig. 3 Energy level diagram with the DFT [B3LYP/LanL2DZ, 6-31G] geometry optimized structures in the gas phase for the four possible linkage isomers of the [Pd8(Lun)4]16+ complex. | ||
Although 1H NMR and ESI-MS analyses gave a preliminary idea about the formation of a M8Lun4 barrel, such studies could not predict the actual isomer formed. Therefore, to get precise information about the isomer formed, a single-crystal X-ray diffraction study was necessary. To do this, suitable single crystals were grown by slow diffusion of acetone vapour into an aqueous solution of UNMB at room temperature. The diffraction study was carried out with a synchrotron beam line.22 The single crystal data unequivocally revealed the formation of the HHTT isomer (Fig. 4).
 | ||
| Fig. 4 SC-XRD structure of UNMB: (a) space-filling diagram; (b) side view; (c) top view (capped stick model) [color codes: C (light blue), N (blue), Pd (purple)]. Counter anions, hydrogen atoms, and solvent molecules have been omitted for the sake of clarity. | ||
The Pd8 barrel crystallized in the triclinic system with space group P![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) and two molecules were found in the asymmetric unit. The crystal structure of UNMB revealed that there are three types of Pd(tmeda) units present in UNMB. Two Pd centres (labelled as Pd1 and Pd1′) out of eight relate to only imidazole rings and other set of Pd centres (labelled as Pd2 and Pd2′) are coordinated only with pyridine units, whereas remaining four Pd ions (labelled as Pd3, Pd3′, Pd3′′, and Pd3′′′) are each bound to one imidazole unit and one pyridine ring. This mode of coordination bonding between the Pd(tmeda) centres and imidazole/pyridine units of Lun resulted in orientational self-sorting type self-assembly and gave rise to the formation of the HHTT isomer. The presence of three different types of (tmeda)Pd(II) units in the single crystal structure of UNMB thus supports the findings of the 1H NMR data. Moreover, the redissolved crystals gave a very clean 1H NMR spectrum, which exactly matches that of the as synthesized barrel UNMB. This finding further gives primary evidence for the formation of the HHTT isomer in solution. Therefore, single-crystal XRD analysis confirmed the formation of the low symmetry barrel HHTT. The average distance between the Pd(II) ions in opposite corners is ∼21.25 Å for Pd1–Pd2′ and ∼15.65 Å for Pd3–Pd3′′; however, the distance between the adjoining Pd centres is roughly ∼12.67 Å.
and two molecules were found in the asymmetric unit. The crystal structure of UNMB revealed that there are three types of Pd(tmeda) units present in UNMB. Two Pd centres (labelled as Pd1 and Pd1′) out of eight relate to only imidazole rings and other set of Pd centres (labelled as Pd2 and Pd2′) are coordinated only with pyridine units, whereas remaining four Pd ions (labelled as Pd3, Pd3′, Pd3′′, and Pd3′′′) are each bound to one imidazole unit and one pyridine ring. This mode of coordination bonding between the Pd(tmeda) centres and imidazole/pyridine units of Lun resulted in orientational self-sorting type self-assembly and gave rise to the formation of the HHTT isomer. The presence of three different types of (tmeda)Pd(II) units in the single crystal structure of UNMB thus supports the findings of the 1H NMR data. Moreover, the redissolved crystals gave a very clean 1H NMR spectrum, which exactly matches that of the as synthesized barrel UNMB. This finding further gives primary evidence for the formation of the HHTT isomer in solution. Therefore, single-crystal XRD analysis confirmed the formation of the low symmetry barrel HHTT. The average distance between the Pd(II) ions in opposite corners is ∼21.25 Å for Pd1–Pd2′ and ∼15.65 Å for Pd3–Pd3′′; however, the distance between the adjoining Pd centres is roughly ∼12.67 Å.
Fullerene encapsulation studies
Two large open terminal windows and the hydrophobic cavity enclosed by the aromatic rings of four ligands make UNMB a suitable encapsulant for the entrapment of large guest molecules. Therefore, to investigate the guest binding ability of UNMB, we chose large sized insoluble guests C60 and C70. An aqueous solution (0.5 mL) of UNMB was stirred with 2 equivalents of fullerene C60 for 12 h at 55 °C, but the resultant solution showed no colour change. 1H, 13C and ESI-MS spectra showed the spectral data corresponding to only free UNMB. Evidently, there is no interaction between C60 and the host in an aqueous medium. Moreover, stirring C70 and UNMB did not result in any change in the spectral features, indicating no encapsulation of C70 either in an aqueous medium. Stunningly, when two equivalents of solid C60 were added to the acetonitrile solution of UNMB (PF6-analogue) and heated at 55 °C under stirring, the colour of the reaction mixture slowly changed to light violet within 2 h and it got darker after completion of the reaction. The resulting suspension was centrifuged, and the supernatant was examined by ESI-MS, 1H and 13C NMR and electronic absorption spectroscopy. As C60 is insoluble in acetonitrile, the appearance of the characteristic profile in the spectroscopic data suggested the binding of C60 with UNMB in an acetonitrile medium. In contrast to the sharp and simple signals in the 1H NMR spectrum of the free UNMB host, C60⊂UNMB displayed multiple signals suggesting strong enough interaction between the guest C60 and host UNMB (Fig. S13†). Additionally, a single horizontal band in the DOSY NMR spectrum revealed that all the signals belong to a single species (Fig. S14†). Moreover, a sharp and intense signal at 142 ppm was obtained in the 13C NMR spectrum of C60⊂UNMB due to C60, which further confirms the binding of C60 with UNMB (Fig. 5 and S15†). | ||
| Fig. 5 Stacked partial 13C NMR spectra of (a) C70⊂UNMB, (b) UNMB, and (c) C60⊂UNMB recorded in CD3CN at room temperature. | ||
In line with this, the ESI-MS spectrum showed several peaks corresponding to charge fragments associated with host–guest complex C60⊂UNMB. The isotopic distribution pattern corresponded to charge fragments at m/z = 1501.6285 for [C60⊂UNMB(PF6)12]4+, 1172.3069 for [C60⊂UNMB(PF6)11]5+, 952.7668 for [C60⊂UNMB(PF6)10]6+, 795.9456 for [C60⊂UNMB(PF6)9]7+, and 678.3319 for [C60⊂UNMB (12PF6)8]8+ (Fig. 6, S16 and S17†), suggesting the formation of a host–guest adduct with the stoichiometry of 1:1. These isotopic distribution patterns matched well with the simulated isotopic distribution pattern for the respective charge fragments. Furthermore, the ESI-MS spectrum of the C60⊂UNMB adduct showed the presence of peaks for free UNMB in addition to the signals for C60⊂UNMB. This is owing to the decomplexation of the host–guest adduct at the time of ionization.
 | ||
| Fig. 6 (a) Stacked partial ESI-MS spectra of UNMB (red), C60⊂UNMB (violet), and C70⊂UNMB (purple). Experimental isotopic distribution patterns of (b) [C60⊂UNMB(PF6)12]4+, and (c) [C70⊂UNMB(4PF6)12]4+ fragments in ESI-MS. | ||
Similarly, host–guest complexation was performed employing C70 as a guest. A colourless acetonitrile solution of UNMB was treated with two equivalents of solid C70 at 55 °C under stirring, which resulted in a deep purple suspension after 12 h. After removal of the excess guest, the deep purple solution was analysed with various spectroscopic methods. Like C60⊂UNMB, C70⊂UNMB displayed multiple peaks in the 1H NMR spectrum (Fig. S18†); however, the single diffusion band in DOSY NMR spectroscopy revealed that all the signals are associated with a single species (Fig. S19†). Interestingly, five additional signals at 149.55, 146.59, 146.15, 143.87 and 129.16 ppm appeared in the 13C NMR of the C70⊂UNMB complex (Fig. 3 and S20†), contrary to the single extra peak in the case of the C60⊂UNMB adduct. The appearance of these new peaks is due to the presence of chemically different carbon atoms in C70,22 and the presence of these additional resonance peaks strongly suggests the formation of an inclusion complex of UNMB with C70.
The ESI-MS spectrum of C70⊂UNMB showed six isotopically well resolved peaks at m/z = 2090.5246, 1531.6380, 1196.3206, 972.7755, 813.0999 and 693.3494 corresponding to charge fragments [C70⊂UNMB(PF6)13]3+, [C70⊂UNMB(PF6)12]4+, [C70⊂UNMB(PF6)11]5+, [C70⊂UNMB(PF6)10]6+, [C70⊂UNMB(PF6)9]7+, [C70⊂UNMB(PF6)8]8+, respectively (Fig. S21 and S22†). The isotopic distribution pattern of these peaks resembled the theoretically simulated patterns, which supports C70⊂UNMB adduct formation in 1:1 host guest stoichiometry. In fact, in comparison to C60⊂UNMB, the ESI-MS data showed mainly peaks corresponding to C70⊂UNMB along with a trace quantity of the free host. This undoubtedly suggests the partial dissociation of C70⊂UNMB over the ionization period and advocates for the stronger binding ability of UNMB for C70 as compared to C60 (Fig. 6a).
In line with this, we employed UV visible absorption spectroscopy to characterize the host–guest complexation. The UV-Vis absorption spectrum of ligand Lun in chloroform showed two absorption bands at λmax 245 and 303 nm, which are due to π–π* transition (Fig. S23†). Meanwhile, the electronic absorption spectrum of UNMB displayed two absorption bands at λmax 232 and 308 nm that can be ascribed to the π–π* transition originating from the Lun units (Fig. 7). The presence of additional broad bands at around λmax = 362 and 520 nm is owing to C60⊂UNMB, and a strong absorption band at λmax = 361 nm along with a broad band around λmax = 473 nm is due to C70⊂UNMB in the respective UV-visible absorption spectrum. This truly speaks for the binding of fullerenes with the host UNMB (Fig. 7).23
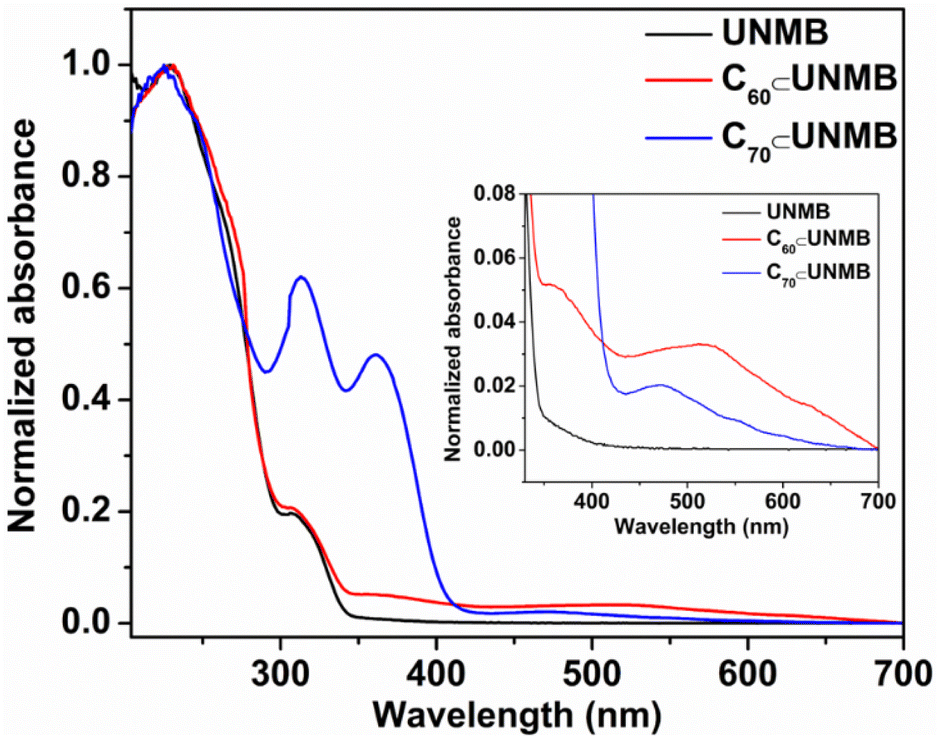 | ||
| Fig. 7 Absorption spectra of UNMB, C60⊂UNMB and C70⊂UNMB at room temperature in acetonitrile (10−5 M solution). Inset: enlarged UV-vis spectra of UNMB, C60⊂UNMB, and C70⊂UNMB showing the absorption band due to encapsulated fullerenes. | ||
After establishing the C60/C70-UNMB host–guest complexation qualitatively using NMR, ESI-MS and UV-vis analysis, the binding constants for their formation were determined by UV-vis titrations. Because of the insolubility of fullerenes in acetonitrile, stock solutions (1 mM) of the fullerenes were prepared in toluene. An acetonitrile solution (10 μM) of UNMB was titrated with the required amount of fullerene in toluene (Fig. S24 and S26†). Changes in absorbance at λmax 306 nm were plotted against the number of equivalents of C60/C70 added, which suggested the formation of a 1:1 inclusion complex in both cases (Fig. S24 and S26†). The Benesi–Hildebrand plots (B–H plots)24 were used to calculate the binding constants, which were roughly found to be 7.15 × 105 M−1 for C70⊂UNMB (Fig. S24 and S25†) and 2.83 × 104 M−1 for C60⊂UNMB (Fig. S26 and S27†), which matches well with the literature reports.18c
Many attempts were made to grow suitable single crystals of fullerene⊂UNMB for SC-XRD diffraction, but they remained unsuccessful. Therefore, to obtain a clear idea about the host–guest interactions between the host UNMB and guest fullerenes, we did energy optimization of these host–guest complexes using the PM6 semiempirical method in the ground state. The optimized structure of the inclusion complexes revealed that fullerenes C60 and C70 fit perfectly within the pocket of host UNMB (Fig. S28†). The distance between the walls of the UNMB and C60 surface is ∼3.3 Å, whereas those of the C70 surface are ∼3.3 and 3.5 Å based on the two orientations of C70. The obtained values lie within the range of the required length for efficient π–π interaction between the host and guest.25 Furthermore, single point energy was computed by employing the DFT method for C70⊂UNMB and C60⊂UNMB. These theoretical studies exhibited that the host–guest complexation of C70⊂UNMB is energetically more stable than that of C60⊂UNMB. Thus, theoretical studies support the stronger affinity of UNMB towards C70 found by the association constant values.
Selective extraction of C70 from a C60/C70 mixture
The stronger binding ability of UNMB for C70 over C60 suggested by ESI-MS and the association constant values is an exciting finding, which gave us the idea to examine the selective binding ability of UNMB for the selective extraction of one fullerene from a mixture of C60/C70. Such extraction is very challenging due to the poor solubility of fullerenes in common solvents. To do this, first, a competitive inclusion experiment was performed. To an acetonitrile solution of UNMB, an equimolar mixture of C60 and C70 (2 equiv. of each) was added, and the mixture was heated at 55 °C for 12 h under stirring. The resulting deep purple supernatant was analyzed by ESI-MS, which exhibited the spectral features of C70⊂UNMB (Fig. 8d). Furthermore, an acetonitrile solution of C70⊂UNMB was treated with 2 equivalents of solid C60 at 55 °C for 12 h. The ESI-MS spectrum of the so-obtained deep purple solution showed the characteristic pattern of C70⊂UNMB (Fig. 8e). Next, we did this experiment in the reverse manner, and 2 equivalents of solid C70 were added to the violet acetonitrile solution of C60⊂UNMB and stirred at 55 °C for 12 h. This again gave a deep purple solution that showed the ESI-MS spectral data of C70⊂UNMB (Fig. 8f). Thus, the C70 introduced into the C60⊂UNMB solution replaces the bound C60, but C60 could not substitute the bound C70 from the cavity of UNMB. Therefore, above experiments also confirm that UNMB has a stronger binding tendency towards C70 than C60. | ||
| Fig. 8 Partial ESI-MS spectra of (a) UNMB, (b) C60⊂UNMB, (c) C70⊂UNMB, and (d) UNMB treated with an equimolar (2 eqv. each) mixture of C60 and C70, after (e) treatment of C70⊂UNMB with C60, and after (f) treatment of C70 with C60⊂UNMB. | ||
To investigate whether UNMB has the overall ability to extract C70 from a C70/C60 mixture, an equimolar mixture of C70/C60 was treated with 1 mL (10 mg) of an acetonitrile solution of UNMB and heated at 55 °C with stirring for 12 h. The centrifuged acetonitrile solution containing predominately C70⊂UNMB was evaporated, and the brown solid thus obtained was treated with 0.5 mL of toluene overnight with stirring at room temperature. The resulting suspension was centrifuged, and the supernatant was investigated by UV-vis analysis. The absorption spectrum of the supernatant in toluene exhibited the absorption profile corresponding to C70 (Fig. S29†). Thus, the UNMB barrel is found to be a potential receptor for the separation of fullerene C70 from its homologue C60 (Scheme 2). This is a very stimulating finding as most of the reported fullerene receptors suffer from strong binding without any selectivity and once the guests are bound, their removal is difficult.
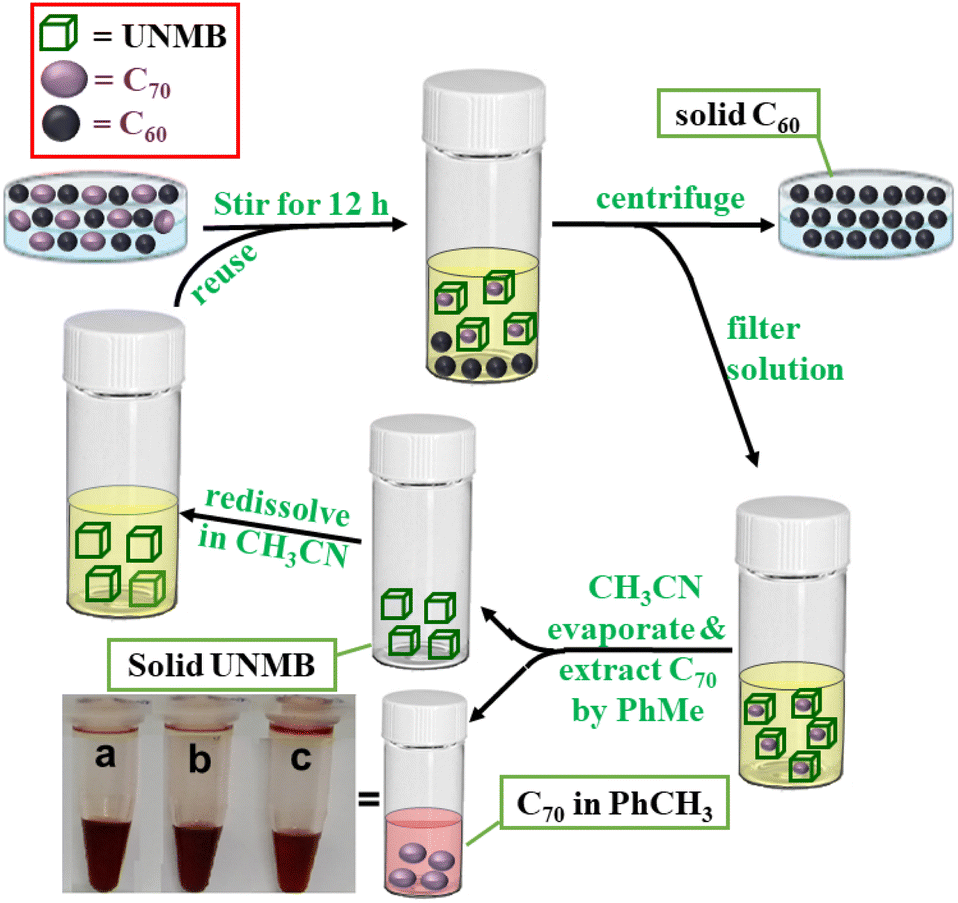 | ||
| Scheme 2 Schematic presentation of the separation of fullerene C70 from a mixture of C60 and C70 by UNMB. (Inset picture shows a, b and c labelled vials containing toluene solutions of extracted C70 after the 1st, 2nd and 3rd cycles, respectively). | ||
The solid residue in the acetonitrile showed an ESI-MS pattern very similar to that of the as-synthesized UNMB, along with the good isotopic distribution patterns of several charge fragments (Fig. S30†). This is a fascinating observation that prompted us to check the reusability of barrel UNMB as a C70 extracting agent. We found that UNMB can be reused for the extraction of C70 with high purity for three cycles.
Conclusions
In conclusion, we have designed and synthesized an unsymmetrical tetratopic ligand that has two different donor groups (pyridine/imidazole). Its self-assembly with a cis-Pd(II) acceptor in a 1:2 molar ratio yielded a low symmetry tetrafacial molecular barrel M8Lun4 (UNMB). The formation of an isomeric molecular barrel (HHTT) was suggested by NMR studies in solution and by the solid-state single-crystal X-ray structure analysis. UNMB has two large open windows and a large cavity enclosed by aromatic panels from the unsymmetrical ligands. These features make it a suitable host for binding with large guests like C70 and C60 through noncovalent interactions (π–π interaction) between the aromatic panels of the barrel and the fullerenes. Encapsulation of fullerenes resulted in an increase in the solubility of C60/C70 in acetonitrile, which otherwise are insoluble in the absence of UNMB. ESI-MS analysis revealed the formation of 1:1 host–guest inclusion complexes for C70 and C60, which was further supported by UV-vis titration experiments. UV-vis titration experiments and competitive guest uptake experiments corroborated that UNMB has stronger binding affinity towards C70 over its spherical analogue C60, which enabled it to exclusively form a C70⊂UNMB inclusion complex from a mixture of C60 and C70. This preferential binding ability of UNMB for C70 over C60 was employed to separate C70 from a mixture of C60/C70 with high purity. Moreover, the encapsulated fullerene in pure form was extracted using toluene and the recovered UNMB was reused for C70 separation for up to three cycles.
Data availability
All data (NMR, ESI-MS) are provided in the ESI,† and additional data will be available upon request.Author contributions
P. S. M. and D. P. designed the project and devised the experiments. D. P. carried out the experimental work, analysed the data, optimized the structures, and carried out the theoretical calculations. J. K. C. collected and solved the crystallographic data. All the authors contributed to the writing of the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
P. S. M. thanks the SERB (New Delhi, India) for financial support in the form of core research grant, and J. C. Bose Fellowship.References
- S. E. Ahnert, J. A. Marsh, H. Hernández, C. V. Robinson and S. A. Teichmann, Science, 2015, 350, aaa2245 CrossRef PubMed.
- (a) J. E. M. Lewis, A. Tarzia, A. J. P. White and K. E. Jelfs, Chem. Sci., 2020, 11, 677–683 RSC; (b) W. Chen, X. Li, C. Liu, J. He, M. Qi, Y. Sun, B. Shi, H. Sepehrpour, H. Li, W. Tian and P. J. Stang, Proc. Natl. Acad. Sci. U. S. A., 2020, 117, 30942–30948 CrossRef CAS PubMed; (c) Z. Cui, Q. S. Mu, X. Gao and G. X. Jin, J. Am. Chem. Soc., 2023, 145, 725–731 CrossRef CAS PubMed; (d) Y. Hashimoto, Y. Katagiri, Y. Tanaka and M. Yoshizawa, Chem. Sci., 2023, 14, 14211–14216 RSC; (e) H.-Y. Lin, Y.-T. Wang, X. Shi, H.-B. Yang and L. Xu, Chem. Soc. Rev., 2023, 52, 1129–1154 RSC; (f) M. Yuasa, R. Sumida, Y. Tanaka and M. Yoshizawa, Chem.–Eur. J., 2022, 28, e202104101 CrossRef CAS PubMed; (g) Y. Zhu, H. Jiang, W. Wu, X.-Q. Xu, X.-Q. Wang, W.-J. Li, W.-T. Xu, G. Liu, Y. Ke, W. Wang and H.-B. Yang, Nat. Commun., 2023, 14, 5307 CrossRef CAS PubMed.
- (a) J. Liu, T. Luo, Y. Xue, L. Mao, P. J. Stang and M. Wang, Angew. Chem., Int. Ed., 2021, 60, 5429–5435 CrossRef CAS PubMed; (b) S. Pullen, J. Tessarolo and G. H. Clever, Chem. Sci., 2021, 12, 7269–7293 RSC; (c) B. Shi, P. Qin, Y. Chai, W.-J. Qu, L. Shangguan, Q. Lin, Y.-M. Zhang, Y. Sun, F. Huang and P. J. Stang, Inorg. Chem., 2022, 61, 8090–8095 CrossRef CAS PubMed; (d) Y. Wang, Y. Zhang, Z. Zhou, R. T. Vanderlinden, B. Li, B. Song, X. Li, L. Cui, J. Li, X. Jia, J. Fang, C. Li and P. J. Stang, Nat. Commun., 2020, 11, 2727 CrossRef CAS PubMed; (e) M. J. Wiester, P. A. Ulmann and C. A. Mirkin, Angew. Chem., Int. Ed., 2011, 50, 114–137 CrossRef CAS PubMed; (f) H. N. Zhang and G. X. Jin, Angew. Chem., Int. Ed., 2023, 62, e202313605 CrossRef CAS PubMed.
- (a) R. Chakrabarty, P. S. Mukherjee and P. J. Stang, Chem. Rev., 2011, 111, 6810–6918 CrossRef CAS PubMed; (b) Y. Gu, E. A. Alt, H. Wang, X. Li, A. P. Willard and J. A. Johnson, Nature, 2018, 560, 65–69 CrossRef CAS PubMed; (c) J. M. Lehn, Science, 2002, 295, 2400–2403 CrossRef CAS PubMed; (d) S. R. Seidel and P. J. Stang, Acc. Chem. Res., 2002, 35, 972–983 CrossRef CAS PubMed; (e) L. Tian, C. Wang, H. Zhao, F. Sun, H. Dong, K. Feng, P. Wang, G. He and G. Li, J. Am. Chem. Soc., 2021, 143, 8631–8638 CrossRef CAS PubMed; (f) B. Woods, R. D. M. Silva, C. Schmidt, D. Wragg, M. Cavaco, V. Neves, V. F. C. Ferreira, L. Gano, T. S. Morais, F. Mendes, J. D. G. Correia and A. Casini, Bioconjugate Chem., 2021, 32, 1399–1408 CrossRef CAS PubMed; (g) Y. Xue, X. Hang, J. Ding, B. Li, R. Zhu, H. Pang and Q. Xu, Catalysis within coordination cages, Coord. Chem. Rev., 2021, 430, 213656 CrossRef CAS.
- (a) P. Howlader, E. Zangrando and P. S. Mukherjee, J. Am. Chem. Soc., 2020, 142, 9070–9078 CrossRef CAS PubMed; (b) A. Platzek, S. Juber, C. Yurtseven, S. Hasegawa, L. Schneider, C. Drechsler, K. E. Ebbert, R. Rudolf, Q. Q. Yan, J. J. Holstein, L. V. Schäfer and G. H. Clever, Angew. Chem., Int. Ed., 2022, 61, e202209305 CrossRef CAS PubMed; (c) H. P. Ryan, C. J. E. Haynes, A. Smith, A. B. Grommet and J. R. Nitschke, Adv. Mater., 2021, 33, e2004192 CrossRef PubMed; (d) T. R. Schulte, J. J. Holstein and G. H. Clever, Angew. Chem., Int. Ed., 2019, 58, 5562–5566 CrossRef CAS PubMed; (e) D. Zhang, T. K. Ronson and J. R. Nitschke, Functional Capsules via Subcomponent Self-Assembly, Acc. Chem. Res., 2018, 51, 2423–2436 CrossRef CAS PubMed.
- (a) T. R. Li, G. Piccini and K. Tiefenbacher, J. Am. Chem. Soc., 2023, 145, 4294–4303 CrossRef CAS PubMed; (b) I. Némethová, D. Schmid and K. Tiefenbacher, Angew. Chem., Int. Ed., 2023, 62, e202218625 CrossRef PubMed; (c) Y. Tsunoda, M. Takatsuka, R. Sekiya and T. Haino, Angew. Chem., Int. Ed., 2017, 56, 2613–2618 CrossRef CAS PubMed; (d) J. Xie, H. J. Peng, J. Q. Huang, W. T. Xu, X. Chen and Q. Zhang, Angew. Chem., Int. Ed., 2017, 56, 16223–16227 CrossRef CAS PubMed; (e) Y. Yang, X. Jing, Y. Shi, Y. Wu and C. Duan, J. Am. Chem. Soc., 2023, 145, 10136–10148 CrossRef CAS PubMed; (f) X. Fu, X. Lin, X. Ren, H. Cong, C. Liu and J. Huang, Chin. Chem. Lett., 2021, 32, 565–568 CrossRef CAS; (g) Z. Wang, H. F. Su, Y. Z. Tan, S. Schein, S. C. Lin, W. Liu, S. A. Wang, W. G. Wang, C. H. Tung, D. Sun and L. S. Zheng, Proc. Natl. Acad. Sci. U. S. A., 2017, 114, 12132–12137 CrossRef CAS PubMed; (h) B.-L. Han, Z. Wang, R. K. Gupta, L. Feng, S. Wang, M. Kurmoo, Z.-Y. Gao, S. Schein, C.-H. Tung and D. Sun, ACS Nano, 2021, 15, 8733–8741 CrossRef CAS PubMed; (i) H. Han, L. Kan, P. Li, G. Zhang, K. Li, W. Liao, Y. Liu, W. Chen and C. T. Hu, Sci. China: Chem., 2021, 64, 426–431 CrossRef CAS.
- (a) R. Banerjee, D. Chakraborty and P. S. Mukherjee, J. Am. Chem. Soc., 2023, 145, 7692–7711 CrossRef CAS PubMed; (b) P. Bhandari, R. Modak, S. Bhattacharyya, E. Zangrando and P. S. Mukherjee, JACS Au, 2021, 1, 2242–2248 CrossRef CAS PubMed; (c) I. A. Bhat, R. Jain, M. M. Siddiqui, D. K. Saini and P. S. Mukherjee, Inorg. Chem., 2017, 56, 5352–5360 CrossRef CAS PubMed; (d) P. Das, A. Kumar, P. Howlader and P. S. Mukherjee, Chem.–Eur. J., 2017, 23, 12565–12574 CrossRef CAS PubMed; (e) P. Howlader, B. Mondal, P. C. Purba, E. Zangrando and P. S. Mukherjee, J. Am. Chem. Soc., 2018, 140, 7952–7960 CrossRef CAS PubMed; (f) R. Saha, A. Devaraj, S. Bhattacharyya, S. Das, E. Zangrando and P. S. Mukherjee, J. Am. Chem. Soc., 2019, 141, 8638–8645 CrossRef CAS PubMed.
- (a) D. P. Giannopoulos, A. Thuijs, W. Wernsdorfer, M. Pilkington, G. Christou and T. C. Stamatatos, Chem. Commun., 2014, 50, 779–781 RSC; (b) P. Howlader, P. Das, E. Zangrando and P. S. Mukherjee, J. Am. Chem. Soc., 2016, 138, 1668–1676 CrossRef CAS PubMed; (c) Y. Kim, W. Li, S. Shin and M. Lee, Acc. Chem. Res., 2013, 46, 2888–2897 CrossRef CAS PubMed; (d) R. Saha, B. Mondal and P. S. Mukherjee, Chem. Rev., 2022, 122, 12244–12307 CrossRef CAS PubMed.
- (a) J. E. M. Lewis and J. D. Crowley, ChemPlusChem, 2020, 85, 815–827 CrossRef CAS PubMed; (b) J. E. M. Lewis, A. Tarzia, A. J. P. White and K. E. Jelfs, Chem. Sci., 2019, 11, 677–683 RSC; (c) D. Prajapati, P. Bhandari, N. Hickey and P. S. Mukherjee, Inorg. Chem., 2023, 62, 9230–9239 CrossRef CAS PubMed; (d) S. Pullen, J. Tessarolo and G. H. Clever, Chem. Sci., 2021, 12, 7269–7293 RSC; (e) R. G. Siddique, K. S. A. Arachchige, H. A. Al-Fayaad, J. D. Thoburn, J. C. McMurtrie and J. K. Clegg, Angew. Chem., Int. Ed., 2022, 61, e202115555 CrossRef CAS PubMed; (f) D. Prajapati, P. Bhandari, E. Zangrando and P. S. Mukherjee, Chem. Sci., 2024, 15, 3616–3624 RSC; (g) R. G. Siddique, J. J. Whittaker, H. A. Al-Fayaad, J. C. McMurtrie and J. K. Clegg, Dalton Trans., 2023, 52, 13487–13491 RSC.
- (a) D. Bardhan and D. K. Chand, Chem.–Eur. J., 2019, 25, 12241–12269 CrossRef CAS PubMed; (b) W. M. Bloch and G. H. Clever, Chem. Commun., 2017, 53, 8506–8516 RSC; (c) S. Pullen and G. H. Clever, Acc. Chem. Res., 2018, 51, 3052–3064 CrossRef CAS PubMed.
- (a) W.-X. Gao, H.-N. Zhang and G.-X. Jin, Coord. Chem. Rev., 2019, 386, 69–84 CrossRef CAS; (b) M. Hardy and A. Lützen, Chem.–Eur. J., 2020, 26, 13332–13346 CrossRef CAS PubMed; (c) H. Li, Z.-J. Yao, D. Liu and G.-X. Jin, Coord. Chem. Rev., 2015, 293–294, 139–157 CrossRef CAS; (d) L. S. Lisboa, D. Preston, C. J. McAdam, L. J. Wright, C. G. Hartinger and J. D. Crowley, Angew. Chem., Int. Ed., 2022, 61, e202201700 CrossRef CAS PubMed; (e) Z. Zhou, J. Liu, T. W. Rees, H. Wang, X. Li, H. Chao and P. J. Stang, Proc. Natl. Acad. Sci. U. S. A., 2018, 115, 5664–5669 CrossRef CAS PubMed.
- (a) R. J. Li, A. Marcus, F. Fadaei-Tirani and K. Severin, Chem. Commun., 2021, 57, 10023–10026 RSC; (b) R. J. Li, A. Tarzia, V. Posligua, K. E. Jelfs, N. Sanchez, A. Marcus, A. Baksi, G. H. Clever, F. Fadaei-Tirani and K. Severin, Chem. Sci., 2022, 13, 11912–11917 RSC; (c) S. S. Mishra, S. V. K. Kompella, S. Krishnaswamy, S. Balasubramanian and D. K. Chand, Inorg. Chem., 2020, 59, 12884–12894 CrossRef CAS PubMed; (d) D. Ogata and J. Yuasa, Angew. Chem., Int. Ed., 2019, 58, 18424–18428 CrossRef CAS PubMed; (e) D. Preston and J. D. Evans, Angew. Chem., Int. Ed., 2023, 62, e202314378 CrossRef CAS PubMed.
- (a) H. W. Kroto, J. R. Heath, S. C. O'Brien, R. F. Curl and R. E. Smalley, Nature, 1985, 318, 162–163 CrossRef CAS; (b) P. W. Stephens, D. Cox, J. W. Lauher, L. Mihaly, J. B. Wiley, P.-M. Allemand, A. Hirsch, K. Holczer, Q. Li, J. D. Thompson and F. Wudl, Nature, 1992, 355, 331–332 CrossRef CAS; (c) Z. Yao and K. C. Tam, Macromol. Rapid Commun., 2011, 32, 1863–1885 CrossRef CAS PubMed.
- M. Prato, J. Mater. Chem., 1997, 7, 1097–1109 RSC.
- Y. Iwasa, Nature, 2010, 466, 191–192 CrossRef CAS PubMed.
- (a) G. Dennler, M. C. Scharber and C. J. Brabec, Adv. Mater., 2009, 21, 1323–1338 CrossRef CAS; (b) S. K. Park, J. H. Kim and S. Y. Park, Adv. Mater., 2018, 30, 1704759 CrossRef PubMed.
- (a) H. Aoshima, S. Yamana, S. Nakamura and T. Mashino, J. Toxicol. Sci., 2010, 35, 401–409 CrossRef CAS PubMed; (b) H. Kazemzadeh and M. Mozafari, Drug Discovery Today, 2019, 24, 898–905 CrossRef CAS PubMed; (c) G.-F. Liu, M. Filipović, I. Ivanović-Burmazović, F. Beuerle, P. Witte and A. Hirsch, Angew. Chem., Int. Ed., 2008, 47, 3991–3994 CrossRef CAS PubMed.
- (a) C. García-Simón, M. Costas and X. Ribas, Chem. Soc. Rev., 2016, 45, 40–62 RSC; (b) J. Pfeuffer-Rooschüz, S. Heim, A. Prescimone and K. Tiefenbacher, Angew. Chem., Int. Ed., 2022, 61, e202209885 CrossRef PubMed; (c) P. C. Purba, M. Maity, S. Bhattacharyya and P. S. Mukherjee, Angew. Chem., Int. Ed., 2021, 60, 14109–14116 CrossRef CAS PubMed.
- (a) J. Theobald, M. Perrut, J. V. Weber, E. Millon and J. F. Muller, Sep. Sci. Technol., 1995, 30, 2783–2819 CrossRef CAS; (b) C. Yeretzian, J. B. Wiley, K. Holczer, T. M. Su, S. L. Nguyen, R. B. Kaner and R. L. Whetten, J. Phys. Chem., 1993, 97, 10097–10101 CrossRef CAS.
- A. Hirsch and M. Brettreich, Fullerenes: Chemistry and Reactions, Wiley-VCH, Weinheim, Germany, 2005 Search PubMed.
- (a) J. L. Atwood, G. A. Koutsantonis and C. L. Raston, Nature, 1994, 368, 229–231 CrossRef CAS; (b) S. Bera, S. Das, M. Melle-Franco and A. Mateo-Alonso, Angew. Chem., Int. Ed., 2023, 62, e202216540 CrossRef CAS PubMed; (c) W. Meng, B. Breiner, K. Rissanen, J. D. Thoburn, J. K. Clegg and J. R. Nitschke, Angew. Chem., Int. Ed., 2011, 50, 3479–3483 CrossRef CAS PubMed; (d) K. Tashiro and T. Aida, Chem. Soc. Rev., 2007, 36, 189–197 RSC; (e) L. Wang, G. T. Wang, X. Zhao, X. K. Jiang and Z. T. Li, J. Org. Chem., 2011, 76, 3531–3535 CrossRef CAS PubMed; (f) M. Zhang, H. Xu, M. Wang, M. L. Saha, Z. Zhou, X. Yan, H. Wang, X. Li, F. Huang, N. She and P. J. Stang, Inorg. Chem., 2017, 56, 12498–12504 CrossRef CAS PubMed.
- D. Aragão, J. Aishima, H. Cherukuvada, R. Clarken, M. Clift, N. P. Cowieson, D. J. Ericsson, C. L. Gee, S. Macedo, N. Mudie, S. Panjikar, J. R. Price, A. Riboldi-Tunnicliffe, R. Rostan, R. Williamson and T. T. Caradoc-Davies, J. Synchrotron Radiat., 2018, 25, 885–891 CrossRef PubMed.
- G. Orlandi and F. Negri, Photochem. Photobiol. Sci., 2002, 1, 289–308 CrossRef CAS PubMed.
- (a) Y. Shi, K. Cai, H. Xiao, Z. Liu, J. Zhou, D. Shen, Y. Qiu, Q.-H. Guo, C. Stern, M. R. Wasielewski, F. Diederich, W. A. Goddard III and J. F. Stoddart, J. Am. Chem. Soc., 2018, 140, 13835–13842 CrossRef CAS PubMed; (b) P. Thordarson, Chem. Soc. Rev., 2011, 40, 1305–1323 RSC.
- (a) J.-H. Deng, J. Luo, Y.-L. Mao, S. Lai, Y.-N. Gong, D.-C. Zhong and T.-B. Lu, Sci. Adv., 2020, 6, eaax9976 CrossRef CAS PubMed; (b) C. Janiak, J. Chem. Soc., Dalton Trans., 2000, 3885–3896 RSC.
Footnote |
| † Electronic supplementary information (ESI) available: NMR spectra, ESI-MS, binding constant calculations, optimized structures, and experimental details (PDF). CCDC 2300816. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d4sc01332h |
| This journal is © The Royal Society of Chemistry 2024 |