DOI:
10.1039/D4SC02462A
(Edge Article)
Chem. Sci., 2024,
15, 12488-12495
A cobalt-modified covalent organic framework enables highly efficient degradation of 2,4-dichlorophenol in high concentrations through peroxymonosulfate activation†
Received
15th April 2024
, Accepted 2nd July 2024
First published on 2nd July 2024
Abstract
The development of covalent organic frameworks (COFs) which can rapidly degrade high concentrations of 2,4-dichlorophenol is of great significance for its practical application. In this work, we report a cobalt-doped two-dimensional (2D) COF (JLNU-307-Co) for the ultra-efficient degradation of high concentration 2,4-dichlorophenol (2,4-DCP) by activating peroxymonosulfate (PMS). The JLNU-307-Co/PMS system takes only 3 min to degrade 100% of 50 mg L−1 2,4-DCP and shows excellent catalytic stability in real water. The superoxide radical (O2˙−) and singlet oxygen (1O2) play a major role in the system through capture experiments and electron spin resonance (ESR) tests. Compared to most previously reported catalysts, JLNU-307-Co/PMS showed the highest efficiency to date in degrading 2,4-DCP. This work not only demonstrates the potential of COFs as a catalyst for water environmental treatment, but also provides unprecedented insights into the degradation of organic pollutants.
Introduction
2,4-DCP is a typical chlorophenol compound in wastewater from the production of pesticides, disinfectants, preservatives, etc.1,2 The compound not only pollutes the environment, but also endangers human health by acting as a precursor to synthetic carcinogens, and can have serious adverse effects on the human nervous and respiratory systems even at trace concentrations.3 The presence of 2,4-DCP in surface and groundwater worldwide is frequently observed due to its non-volatile nature and resistance to microbial degradation.4,5 The development of an environmentally friendly and efficient degradation technology for 2,4-dichlorophenol in water is therefore of utmost urgency. The persulfate-based advanced oxidation process (SR-AOPs) has garnered significant attention due to its robust oxidizing capacity and exceptional degradation efficiency.6
Peroxymonosulfate (PMS), being a crucial oxidizer in SR-AOPs, possesses the advantages of excellent stability, cost-effectiveness, and a wide range of pH applications.7,8 The PMS compound is susceptible to single-electron reduction, leading to the generation of active free radicals such as SO4˙−, ˙OH, and O2˙− upon peroxide bond cleavage.9–11 The simultaneous oxidation of PMS generates 1O2, representing a non-radical pathway for pollutant degradation.12–14 The activation methods proposed so far encompass a range of techniques, including thermal treatment, ultraviolet irradiation, alkaline conditions, metal ion incorporation, and utilization of activated carbon.15–17 It is noteworthy that cobalt ions exert a significant influence on the activation of PMS. The catalytic properties of cobalt-based functional materials in PMS activation have been extensively demonstrated by numerous studies.18,19
Covalent organic frameworks (COFs) are a type of porous crystalline material characterized by a well-defined structure and large surface area.20 Through the incorporation of specific unit structures, COFs can offer an increased number of active sites for the doping of the transition metal cobalt.21–24 The design of the pore structure can effectively restrict the growth of transition metals within the skeleton, prevent excessive aggregation of metal ions, enhance the dispersion of metal ions, and significantly enhance the catalytic performance of the Co-COF.23,24 The presence of cobalt ions plays a regulatory role in the electronic structure of the COF, thus forming a highly efficient active site and showing excellent performance in activating PMS. Moreover, the periodic stacking nanosheet structure of the 2D COF provides an extensively conjugated π-electron system, thereby enhancing the efficiency of electron mass transfer during oxidation.25 Therefore, the composite COF nanomaterials with transition metals exhibit remarkable advantages in terms of stability, abundant active sites, high dispersion of transition metals, and exceptional catalytic performance in the process of water purification.25–27
In this study, we utilized 2,4,6-tri(4-aldehyde phenyl)-1,3,5-triazine (TFPT) and 2,2′-bipyridine-5,5′-diamine (Bpy) as ligands to synthesize an imine-linked donor–acceptor (D–A). The obtained material was denoted as JLNU-307 (JLNU = Jilin Normal University), which effectively enhances electron transport by acting as a cobalt-supported carrier. The ultra-efficient degradation of JLNU-307-Co to 2,4-DCP with high concentration was obtained by introducing cobalt into JLNU-307 at room temperature. Various experimental conditions and inorganic anions were investigated to determine their effects on the reaction. Furthermore, excellent performance of JLNU-307-Co/PMS was observed when applied to real water samples. Through free radical capture experiments and ESR tests, the ROS species generated by the JLNU-307-Co system were analyzed, while high performance liquid chromatography-mass spectrometry (HPLC-MS) was employed to identify intermediates produced during the reaction process. Based on these findings, a proposed removal mechanism and degradation pathway for 2,4-DCP were established.
Results and discussion
Structure characterization
As shown in Scheme 1, the condensation of 2,4,6-tri(4-aldehyde phenyl)-1,3,5-triazine (TFPT) and 2,2′-bipyridine-5,5′-diamine (Bpy) through solvothermal reactions resulted in JLNU-307. Cobalt was introduced by using a bipyridine unit to obtain JLNU-307-Co. The crystallinity and stacking patterns of the prepared COFs were confirmed by powder X-ray diffraction (PXRD) (Fig. S1†). Geometric energy minimization using Materials Studio28 based on an overlapping two-dimensional hcb network shows that JLNU-307 crystallizes in the P6 (no. 174) space group (Table S1†). The lattice model was simulated and the most likely structure of the COF in AA and AB stacking modes was obtained (Fig. S3 and S4†). The single-cell parameters of JLNU-307 were determined (a = b = 44.3729 Å, c = 3.5122 Å, α = β = 90°, and γ = 120°). The PXRD pattern was refined using the Pawley pattern matching technique. The peaks observed at 2θ values of 2.30°, 3.98°, 4.60°, and 6.09° in the PXRD pattern of JLNU-307 corresponded to the crystal faces (010), (110), (020), and (120), respectively (Fig. 1a). The obtained cell parameters showed excellent agreement with the observed values and had high coincidence coefficients (Rωp = 2.79% and Rp = 1.96%). By comparing the PXRD patterns of JLNU-307 and JLNU-307-Co, it can be observed that there is a slight decrease in the peak height at 2.30° due to stirring force caused by JLNU-307-Co. However, the original COF skeleton remains unchanged. The observed transition from an orange hue to brown signifies the successful incorporation of Co through doping (Fig. S2†). JLNU-307 exhibited structural integrity even after being immersed in various organic solvents for three days (Fig. S5†). Fourier transform infrared spectra (FT-IR) have confirmed the presence of imine bonds in the COF, as shown in Fig. 1b. It can be observed that JLNU-307 and JLNU-307-Co exhibit C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N stretching patterns at 1620 cm−1 and 1597 cm−1, respectively, while the amino band (3213–3435 cm−1) and aldehyde band (1673–1699 cm−1) present in Bpy and TFPT are no longer detected.29 Additionally, JLNU-307 and JLNU-307-Co exhibit exceptional thermal stability, with thermal decomposition temperatures surpassing 465 °C (Fig. S6–S7†). Furthermore, solid-state 13C cross-polarized magic angle rotation (CP/MAS) NMR spectra provide additional evidence of the presence of imines at 159 ppm for JLNU-307 and 168 ppm for JLNU-307-Co (Fig. 1c and d).
N stretching patterns at 1620 cm−1 and 1597 cm−1, respectively, while the amino band (3213–3435 cm−1) and aldehyde band (1673–1699 cm−1) present in Bpy and TFPT are no longer detected.29 Additionally, JLNU-307 and JLNU-307-Co exhibit exceptional thermal stability, with thermal decomposition temperatures surpassing 465 °C (Fig. S6–S7†). Furthermore, solid-state 13C cross-polarized magic angle rotation (CP/MAS) NMR spectra provide additional evidence of the presence of imines at 159 ppm for JLNU-307 and 168 ppm for JLNU-307-Co (Fig. 1c and d).
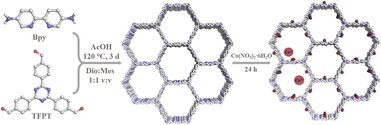 |
| | Scheme 1 The synthesis of JLNU-307 and JLNU-307-Co. | |
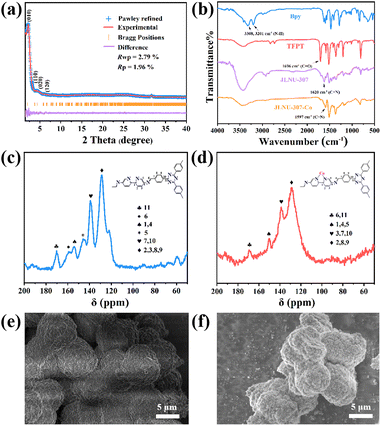 |
| | Fig. 1 (a) PXRD patterns of JLNU-307. (b) FT-IR spectra of JLNU-307 and JLNU-307-Co. Solid-state 13C NMR spectra of (c) JLNU-307 and (d) JLNU-307-Co. SEM images of JLNU-307 (e) and JLNU-307-Co (f). | |
The morphologies of JLNU-307 and JLNU-307-Co were investigated using scanning electron microscopy (SEM). Both exhibit spherical structures with a diameter of approximately 5 μm (Fig. 1e and f). Energy Dispersive Spectrometry (EDS) analysis clearly demonstrates the uniform distribution of atoms (C, N, and Co) within the COF, further confirming successful Co doping (Fig. S8 and S9†). The porosity of both COFs was determined by obtaining the N2 adsorption–desorption isotherm at 77 K (Fig. S10–S15†). The Brunauer–Emmett–Teller (BET) specific surface areas for JLNU-307 and LNU-307-Co were found to be 542.87 and 79.92 m2 g−1, respectively (Fig. S10–S11 and S13–S14†). Based on density functional theory (DFT), the aperture distribution reveals a pore size of 38 Å for JLNU-307 and 29 Å for JLNU-307-Co (Fig. S12 and S15†). Compared to simulated data proposing pore sizes of 51 Å for JLNU-307 and 40 Å for JLNU-307-Co, slight reduction in pore size is observed due to partial migration and accumulation of adjacent layers during solid powder material synthesis under dry conditions.30
The composition and chemical state of the elements were investigated through X-ray photoelectron spectroscopy (XPS) analysis, as shown in Fig. 2. The peaks observed at 284.7 eV and 286.2 eV in the C 1s spectrum in Fig. S17† correspond to the presence of CC and C–N bonds in JLNU-307, respectively, as depicted in Fig. S16–S18.† Additionally, the peak observed at 399.0 eV in the N 1s spectrum in Fig. S18† indicates the existence of a CN bond. In Fig. 2b, the peaks at 284.7 eV and 286.4 eV correspond to the CC and C–N bonds, respectively. In Fig. 2c, the N 1s spectrum exhibits peaks at 399.1 eV and 402.7 eV, which can be attributed to the CN and Co–N bonds, respectively. In the Co 2p spectrum (Fig. 2d), six distinct peaks can be observed, with the Co3+ 2p3/2 and 2p1/2 peaks located at energies of 782.5 and 795.7 eV respectively, while the Co2+ peaks are found at energies of 787.2 and 799.0 eV for the respective 2p3/2 and 2p1/2 states. The presence of cobalt facilitates the transformation of metal valence during PMS activation, thereby enhancing the catalytic degradation performance of PMS. Additionally, satellite peaks were observed at 790.0 and 803.4 eV.12,21,31,32 The hydrophilicity of JLNU-307-Co is significantly enhanced, facilitating improved dispersion of the catalyst in water and enhancing the efficiency of 2,4-DCP degradation in aqueous environments (Fig. S23–S24†). The energy gaps (Eg) of JLNU-307 and JLNU-307-Co are 2.64 eV and 2.47 eV, respectively, as illustrated in Fig. S25–S26.† JLNU-307-Co exhibits a smaller band gap with higher electron transition efficiency.
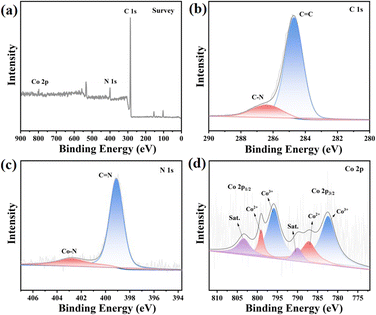 |
| | Fig. 2 XPS spectra (a), high resolution XPS spectra of C 1s (b), N 1s (c) and Co 2p (d) obtained for JLNU-307-Co. | |
The high PMS activation performance of JLNU-307-Co
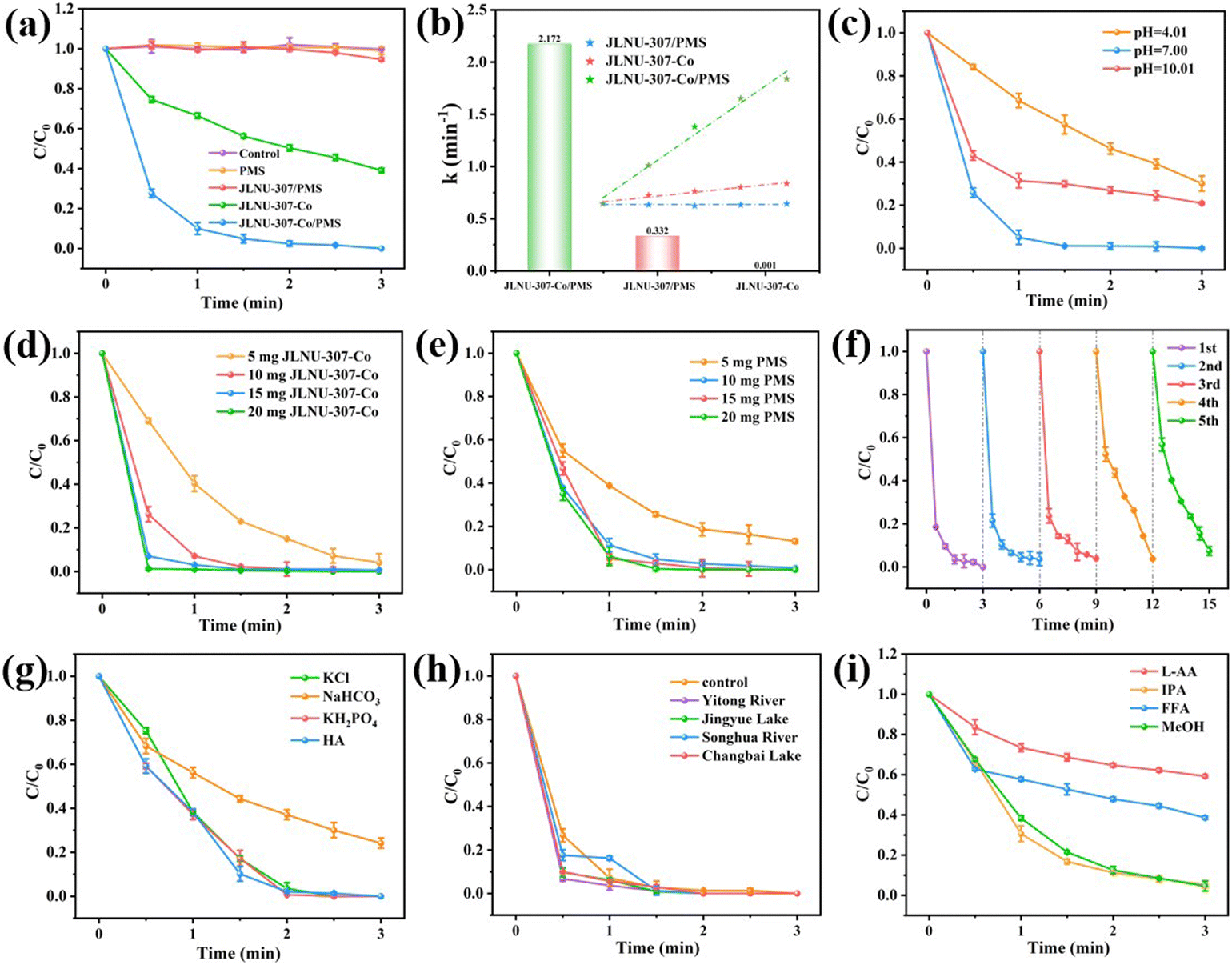
The catalytic activity of the JLNU-307-Co/PMS system was evaluated using 50 mg L−1 of 2,4-DCP as a representative target pollutant in this study. As depicted in Fig. 3a, when solely utilizing JLNU-307-Co as the catalyst, the removal efficiency of 2,4-DCP within 3 minutes reached 59.7%. This can be attributed to the adsorption capacity of JLNU-307-Co for 2,4-DCP due to its large specific surface area. However, adsorption could not achieve the degradation of 2,4-DCP. The presence of PMS alone in the solution did not result in any degradation of 2,4-DCP. However, when JLNU-307-Co was combined with PMS, the degradation rate of 2,4-DCP at a concentration of 50 mg L−1 reached 100% within 3 minutes. This indicates that the catalyst effectively activated PMS to generate free radicals and significantly enhanced the degradation rate of 2,4-DCP. The removal rate of the JLNU-307/PMS system within 3 minutes was found to be negligible at 6.1%. This may be because the PMS in the solution occupied the COF channel, so it did not achieve an obvious adsorption degradation effect. In addition, the experimental findings demonstrate that the highly ordered structure of JLNU-307-Co promotes efficient activation of PMS, thereby enabling ultra-efficient 2,4-DCP removal. The reaction rate constant k = 2.172 min−1 of JLNU-307-Co/PMS was found to be 2172 times higher than that of JLNU-307/PMS (0.001 min−1) and 6.5 times higher than that of JLNU-307-Co (0.332 min−1), as observed in Fig. 3b and S27.† Importantly, compared to most previously reported catalysts (Table S2†), JLNU-307-Co/PMS exhibited the highest efficiency in degrading 2,4-DCP to date.
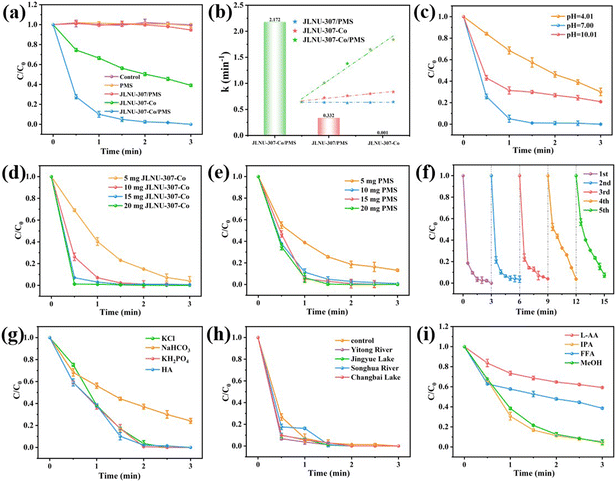 |
| | Fig. 3 (a) Removal efficiency of 2,4-DCP in different reactions within 3 min. (b) The rates of 2,4-DCP degradation in various reaction systems. Effects of (c) pH, (d) catalyst dosage and (e) PMS dosage on 2,4-DCP removal by the JLNU-307-Co/PMS system. (f) The recycling performance of JLNU-307-Co/PMS for 2,4-DCP degradation. (g) Inorganic anion interference experiments. (h) Performance of JLNU-307-Co/PMS in real water bodies. (i) Effects of different quenching agents on degradation of 2,4-DCP, (LAA/IPA/FFA/MeOH = 5 mM). | |
In order to further assess the catalytic performance of JLNU-307-Co/PMS, investigations were conducted on the impact of different pH values, varying catalyst quantities and diverse operating parameters with different amounts of PMS on the catalytic efficiency of JLNU-307-Co/PMS (Fig. 3c–e). The initial pH is a critical factor in the degradation of organic pollutants in SR-AOPs during pH effect studies. Various buffer solutions, including potassium hydrogen phthalate, phosphate, and borax, are utilized to maintain stable reaction system pH levels. As the pH increased from 4.01 to 7.00, the degradation rate of 2,4-DCP increased from 70.0% to 100%. At low pH levels, the excessive presence of protons leads to a depletion of the available SO4˙− and ˙OH ions, resulting in a reduction in the degradation of 2,4-DCP under highly acidic conditions. Conversely, at pH 7.00, the deprotonated form of the 2,4-DCP molecule exhibits higher reactivity compared to its protonated counterpart. Further increasing the initial pH to 10.01 enhances the degradation rate of 2,4-DCP by 79.1%. This can be attributed to the reaction between SO4˙− and ˙OH ions under alkaline conditions leading to the formation of SO42−. Additionally, in a strongly alkaline environment, self-decomposition of PMS also reduces the degradation rate of 2,4-DCP preventing complete removal (Fig. 3c).12,33 With the increase in the amount of catalyst or PMS, from 5 mg to 10 mg, more free radicals can be produced and the removal efficiency of 2,4-DCP can be improved. Further increasing the catalyst or PMS dosage to 10 mg and 20 mg enables complete removal of 2,4-DCP within 3 min (Fig. 3d and e). Therefore, we chose a dose of 10 mg catalyst and 10 mg PMS as ideal conditions for further study. In order to investigate the sustained catalytic capacity of JLNU-307-Co, the JLNU-307-Co/PMS system was used to perform five cycles of 2,4-DCP experiments (Fig. 3f). The final removal rates of 2,4-DCP were 100%, 97.5%, 97.1%, 97.2% and 92.7%, respectively. It can be observed that the JLNU-307-Co/PMS system consistently maintains exceptional efficacy in removing 2,4-DCP even after undergoing five cycles, thereby demonstrating the catalyst's commendable cycling stability. The XPS test was conducted on the catalyst post-circulation (Fig. S19–S22†). The findings demonstrate that cobalt in the catalyst remains stable, existing predominantly as Co2+ and Co3+ ions. The Co3+ 2p3/2 and 2p1/2 peaks are observed at energies of 781.6 and 795.9 eV, respectively, while the Co2+ peaks appear at energies of 786.7 and 797.9 eV. The concentration of exuded Co ions in the reaction solution was determined to be 0.843 mg L−1 using inductively coupled plasma mass spectrometry (ICP-MS).
In the natural water matrix, common anions such as chloride ions (Cl−), carbonate (HCO3−), dihydrogen phosphate (H2PO4−), and humic acid (HA) may exert an influence on the degradation behavior of 2,4-DCP in SR-AOPs.12,34–36 As shown in Fig. 3g, the presence of HCO3− in the system resulted in a significant decrease in the degradation rate of 2,4-DCP. This can be attributed to the decomposition of HCO3− into CO32−, which in turn quenches SO4˙− and ˙OH. The presence of Cl−, KH2PO4, and HA did not exert any influence on the degradation efficiency of the JLNU-307-Co/PMS system. These results demonstrate that the catalyst possesses a robust resistance against ionic interference, further confirming the exceptional stability of the JLNU-307-Co/PMS system. Additionally, as depicted in Fig. 3h, the degradation efficiency of JLNU-307-Co/PMS in actual aquatic environments such as Yitong River (Changchun, China), Jingyue Lake, Songhua River (Songyuan, China) and Changbai Lake is also commendable. The findings demonstrate that JLNU-307-Co/PMS exhibits significant catalytic potential for water treatment.
The reactive oxygen species in the JLNU-307-Co/PMS system
As shown in Fig. 3i and S28,† in order to explore more deeply the process of degradation of 2,4-DCP, the sacrificial agents L-ascorbate (L-AA), isopropyl alcohol (IPA), furfuryl alcohol (FFA)/triphenylamine (TPA) and methanol (MeOH) were used to trap superoxide free radicals (O2˙−), hydroxyl free radicals (˙OH), singlet oxygen (1O2) and sulfate free radicals (SO4˙−), respectively, and the active substances in the system were preliminarily determined. In the IPA and MeOH system, the removal rates of 2,4-DCP were 96.9% and 95.9%, respectively. In the SR-AOP system using JLNU-307-Co as an activator for PMS, ˙OH and SO4˙− exhibit a minor inhibitory effect. When L-AA and FFA/TPA were introduced, the removal rates of 2,4-DCP decreased to 40.8% and 60.8%/64.9%, respectively, indicating that O2˙− and 1O2 exhibited a pronounced inhibitory effect on the degradation process and served as the primary active species in the system.21
In order to further validate the aforementioned experimental results, an electron spin resonance (ESR) test was conducted to provide robust evidence for the presence of reactive oxygen species generated during the reaction. As depicted in Fig. 4a–c, the vibrational signals of DMPO-O2˙−, TEMP-1O2, and DMPO-˙OH/SO4˙− in the JLNU-307/PMS system exhibit low intensity. The characteristic signal of JLNU-307-Co/PMS is distinctly identifiable in contrast. Furthermore, the vibration signal exhibits significant enhancement in DMPO-O2˙− and TEMP-1O2, which aligns with the findings from the capture experiment. The JLNU-307-Co/PMS system has been confirmed to possess a heightened capacity for generating reactive oxygen species. In summary, the identified active substances may be generated through a cascading reaction during PMS activation,1,2,21,37 as shown by the following equations:
| | | Co2+ + HSO5− → Co3+ + SO4˙− + HO− | (1) |
| | | Co3+ + HSO5− → Co2+ + SO5˙− + H+ | (2) |
| | | SO4˙− + HO−/H2O → SO42− + ˙OH | (3) |
| | | HSO5− + ˙OH → HO2˙ + HSO4− | (4) |
| | | HSO5− + SO52− → SO42− + HSO4− + 1O2 | (5) |
| | | ˙OH + O2˙− + H+ → H2O + 1O2 | (7) |
| | | 1O2/˙O2−/˙OH/SO4˙− + 2,4-DCP → SO42−/H2O + oxidation products | (8) |
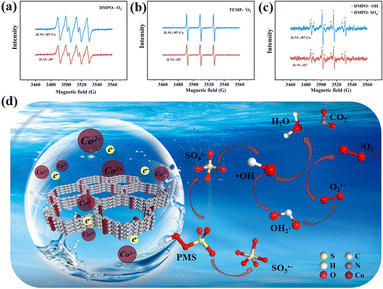 |
| | Fig. 4 ESR spectra of (a) DMPO-O2˙−, (b) TEMP-1O2 and (c) DMPO-˙OH/SO4˙− generated by JLNU-307/PMS and JLNU-307-Co/PMS. (d) The mechanism of PMS activation by conduction electrons. | |
DFT calculations of 2,4-DCP and possible degradation routes
To further elucidate the potential reaction regions and active sites of 2,4-DCP, we conducted density functional theory (DFT) calculations on the Fukui function and electron cloud distribution (ESP), as shown in Fig. 5. The ESP calculation in Fig. 5b vividly illustrates the density of electron clouds on the surface of 2,4-DCP molecules. Notably, a significant concentration of electrons is observed at Cl10 and Cl11, which serve as primary locations for electrochemical reactions. The locations of 2,4-DCP with high values of f+, f−, f0 and charge density difference (CDD) in the presence of free radicals were observed to predict the most probable reactive sites.38Fig. 5c shows the isosurface of electron density of 2,4-DCP molecules plotted by using Fukui indices, and the Fukui index values f+, f−, f0 and CDD corresponding to Fig. 5d–g. The f+, f− and f0 values of atoms are commonly believed to be directly proportional to the susceptibility towards attacks by free radicals, electrophilic substances and nucleophilic substances, respectively. Moreover, a higher value indicates a greater reactivity at the corresponding sites. The CDD value is employed to comprehensively evaluate the reaction potential of atoms when exposed to active substances. A more negative CDD value signifies an enhanced electrophilicity potential at the site.12,21,39,40 According to the calculation of the Fukui function, C3, C6, O10 and Cl13 exhibit high values of CDD, indicating strong electrophilic activities. In summary, these atomic locations are susceptible to attack by electrophiles and are theoretically considered as the most reactive sites.
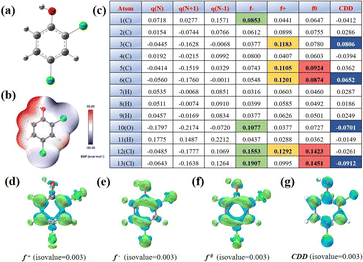 |
| | Fig. 5 (a) Optimized molecular structure and (b) ESP of 2,4-DCP. The isosurface of (d) f+, (e) f−, (f) f0 and (g) CDD of 2,4-DCP. (c) Natural population analysis (NPA) charges and calculated Fukui index results of 2,4-DCP. | |
The degradation pathway of 2,4-DCP in the JLNU-307-Co/PMS system was determined by HPLC-MS analysis. Two degradation pathways in Fig. 6a can be derived. Cl13 with the highest CDD value is vulnerable to attacks by SO4˙− and ˙OH to generate P2/P3 (m/z = 131). Due to the high CDD value of O10 (−0.0701), P2/P3 catalyzes the formation of P4 (m/z = 218) in the presence of O2˙−. Subsequently, P4 undergoes degradation to yield P5 (m/z = 171), which is ultimately converted into smaller, less toxic molecules.40 In addition, there is another path in the system. The Cl12 or Cl13 chlorine atoms in the benzene ring are replaced by free radicals with the help of O2˙− and 1O2 to form P6/P7 (m/z = 157). The generation of 1,2/1,4-phenyldiol from P8/P9 (m/z = 141) is facilitated. In the presence of ˙OH alone, Cl12 or Cl13 can readily undergo chlorine atom removal to acquire hydroxyl groups. Subsequently, P8/P9 reacts with ˙OH to yield isomeric forms of P10/P11 (m/z = 111). During the oxidation of O2˙−, P10/P11 undergoes ring opening and fragmentation into small molecular acids such as oxalic acid, acetic acid and formic acid before ultimately mineralizing pollutants into CO2 and H2O.41–43
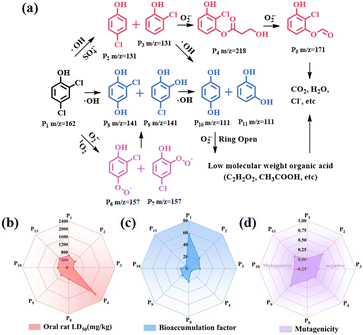 |
| | Fig. 6 (a) Degradation routes of 2,4-DCP by JLNU-307-Co activated PMS, Toxicity evaluation of photodegradation intermediates via the TEST, (b) oral rat LD50, (c) bioaccumulation factor and (d) mutagenicity. | |
The toxicity assessment software tool (TEST) was used to assess the toxicity of degradation intermediates, including oral rat LD50, bioaccumulation factor and mutagenicity (Fig. 6b–d). As shown in Fig. 6b, oral rat LD50 values of the final intermediates P4, P10 and P11 all increased after degradation, indicating that they effectively reduced the original toxicity. The bioaccumulation factor of these intermediates consistently exhibits low values as shown in Fig. 6c. As shown in Fig. 6d, all intermediates demonstrate negative mutagenicity. Based on this analysis, it can be inferred that 2,4-DCP can ultimately degrade into substances with low or non-toxic properties.
DFT calculation
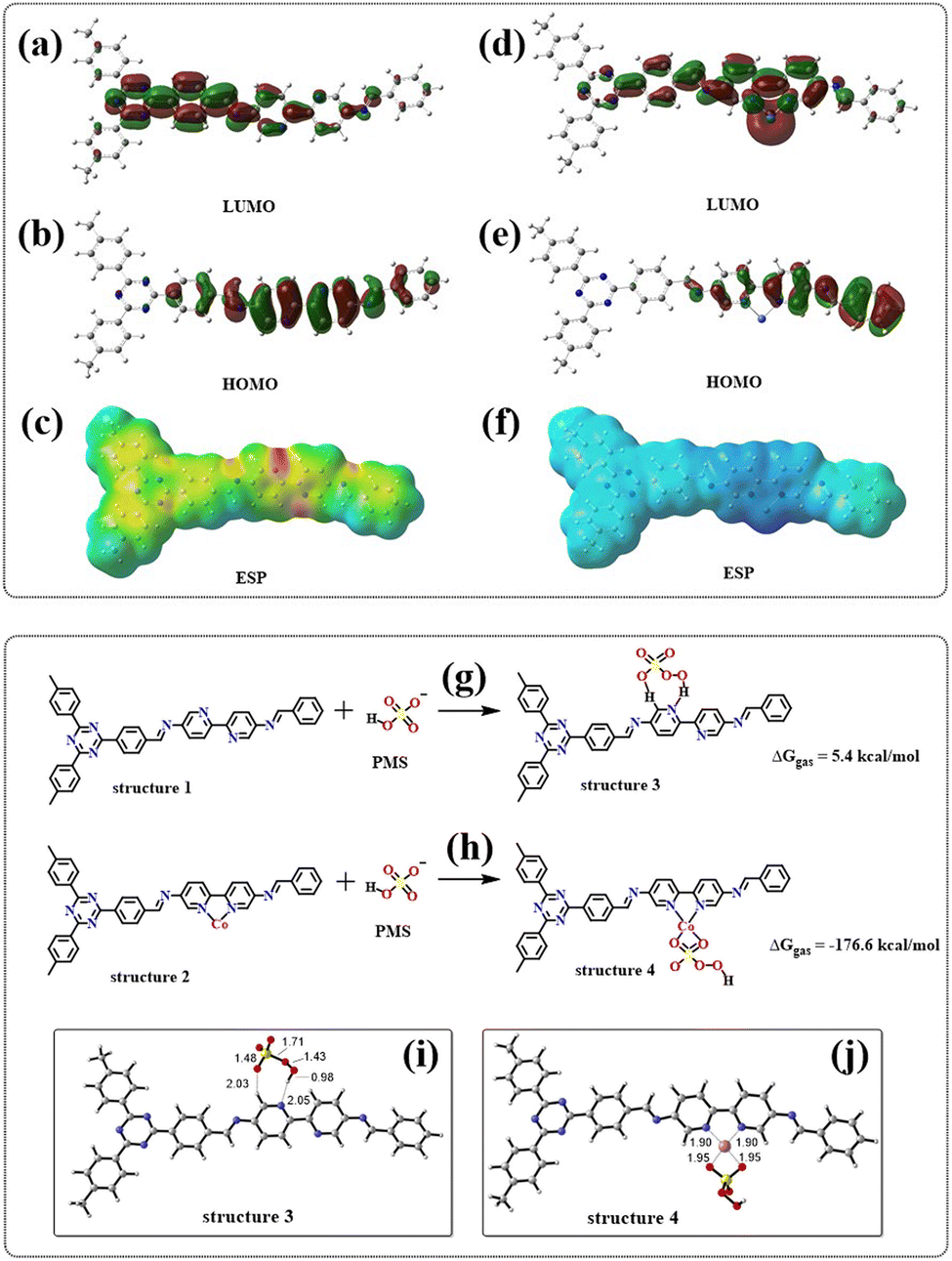
The orbital and surface electrostatic potentials of JLNU-307 and JLNU-307-Co molecules were investigated through DFT theoretical calculations. Simulation results revealed that the LUMO (Fig. 7a) of JLNU-307 is predominantly localized on the TFPT unit, while the HOMO (Fig. 7b) is mainly distributed on the Bpy unit. In contrast, the LUMO for JLNU-307-Co primarily resides on the cobalt atom of the Bpy unit, whereas the HOMO is mainly delocalized over the benzene ring of Bpy, as shown in Fig. 7d and e. The Electrostatic Potential (ESP) in Fig. 7c and f reveals that the distribution state of the electron cloud undergoes changes upon cobalt combination, indicating a predominant concentration of the electron cloud distribution on pyridine N in JLNU-307. The electron density on JLNU-307-Co is predominantly localized near the cobalt atom, providing further evidence that the incorporation of metal Co effectively modulates the electronic structure of COF materials and alters their reactive sites. This redistribution and accumulation of charges at reaction sites significantly enhances charge separation/migration and facilitates redox reactions. Moreover, it has been experimentally confirmed that Co doping within the COF framework can efficiently activate PMS and enhance its catalytic performance.
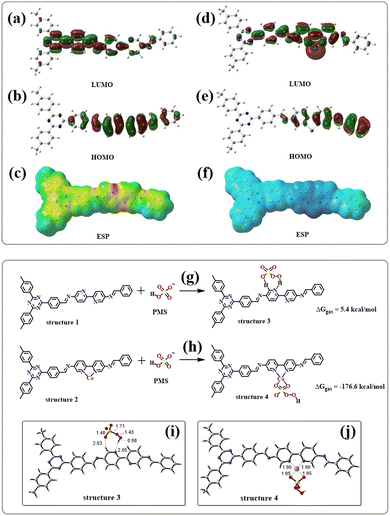 |
| | Fig. 7 (a and b) Optimized LOMO and HUMO of JLNU-307 and (d and e) LOMO and HUMO of JLNU-307-Co; electrostatic potential images of JLNU-307 (c) and JLNU-307-Co (f); adsorption energy of JLNU-307 (g) and JLNU-307-Co (h) with PMS; The PMS adsorption configurations on (i) JLNU-307 and (j) JLNU-307-Co (the bond lengths are in angstroms). | |
Meanwhile, the adsorption energy between the catalyst and PMS was calculated. As we all know, the adsorption energy is positive, indicating that the energy of the adsorbed state exceeds the energy of the non-adsorbed state, which makes the adsorption process unstable and leads to an overall increase in the system energy. In such cases, the adsorption process typically exhibits endothermic characteristics. Hence, a positive value for adsorption energy may suggest instability in PMS/catalyst interactions, potentially resulting in desorption or susceptibility to displacement by other groups. In contrast, a negative adsorption energy indicates an exothermic process in which PMS releases energy when it is transferred to the catalyst surface. Therefore, the greater the negative value of adsorption energy, the stronger the corresponding adhesion effect. As shown in Fig. 7g and h, the adsorption energy of PMS for JLNU-307-Co (ΔGgas = −176.6 kcal mol−1) is much smaller than that of PMS for JLNU-307 (ΔGgas = 5.4 kcal mol−1). Therefore, JLNU-307-Co has strong interaction with PMS. This is also the fundamental reason for the substantial improvement in the degradation performance of JLNU-307-Co. According to the adsorption configuration analysis shown in Fig. 7i and j, in JLNU-307, PMS adsorbs on pyridine N and the adjacent H. It is difficult for this cleavage to promote the generation of the active species required for the system. In JLNU-307-Co, the two O atoms on PMS are adsorbed onto Co, facilitating the breakage of the S–O bond in PMS and subsequently inducing the formation of the primary active species O2˙− and 1O2 within the system.
Conclusions
In summary, a metal-free bipyridine COF (JLNU-307) was synthesized using the solvothermal synthesis method. Subsequently, JLNU-307-Co was obtained as an exceptionally efficient catalyst for the degradation of 2,4-dichlorophenol (2,4-DCP) through post-modified metal. JLNU-307-Co exhibits outstanding degradation activity and remarkable stability. In the absence of any light source, the removal efficiency of 2,4-DCP at a concentration of 50 mg L−1 can reach 100% within a duration of 3 min. The performance of this technology in real water applications is exceptional, surpassing any previous achievements in the degradation of high concentrations of 2,4-DCP. Toxicity analysis revealed that 2,4-DCP was efficiently degraded into low concentrations or non-toxic intermediates. The DFT calculation elucidates the underlying mechanism behind the significant enhancement in the performance of JLNU-307-Co. The charge redistribution and remarkably strong adsorption of JLNU-307-Co and PMS facilitate efficient charge transfer and redox reactions, thereby promoting the generation of key active species O2˙− and 1O2 within the system. Consequently, JLNU-307-Co/PMS showed ultra-efficient catalytic activity for the degradation of 2,4-DCP. The findings of this study offer promising prospects for the practical utilization of COF materials in the degradation of organic pollutants.
Data availability
The data supporting this article have been included as part of the ESI.†
Author contributions
Yunchao Ma: writing-original draft and conceptualization. Yuhang Han: investigation and methodology. Yuxin Yao: validation. Tianyu Zhou: software. Dongshu Sun: data curation. Chunbo Liu: funding acquisition and resources. Guangbo Che: project administration. Bo Hu: writing-review & editing. Valentin Valtchev and Qianrong Fang: supervision and formal analysis.
Conflicts of interest
The authors declare no conflict of interest.
Acknowledgements
This work was supported by the National Natural Science Foundation of China (22205076 and 22001092), the Natural Science Foundation Project of Jilin Province (YDZJ202201ZYTS335, YDZJ202101ZYTS034, 20210203105SF, YDZJ202102CXJD049, and 20230508040RC), and the Project of Development and Reform Commission of Jilin Province (2023C032-5 and 2023C032-2).
Notes and references
- Z. Weng, Y. Lin, S. Guo, X. Zhang, Q. Guo, Y. Luo, X. Ou, J. Ma, Y. Zhou, J. Jiang and B. Han, Angew. Chem., Int. Ed., 2023, 135, e202310934 CrossRef.
- T. Zhang, Y. Chen, Y. Wang, J. L. Roux, Y. Yang and J. P. Croué, Environ. Sci. Technol., 2014, 48, 5868–5875 CrossRef CAS PubMed.
- J. Luo, Y. Wang, D. Cao, K. Xiao, T. Guo and X. Zhao, Chem. Eng. J., 2018, 343, 69–77 CrossRef CAS.
- C. Huang, Y. Wen, J. Ma, D. Dong, Y. Shen, S. Liu, H. Ma and Y. Zhang, Nat. Commun., 2021, 12, 320 CrossRef CAS PubMed.
- Y. Peng, M. Zhao, B. Chen, Z. Zhang, Y. Huang, F. Dai, Z. Lai, X. Cui, C. Tan and H. Zhang, Adv. Mater., 2018, 30, 1705454 CrossRef PubMed.
- T. Ni, H. Zhang, Z. Yang, L. Zhou, L. Pan, C. Li, Z. Yang and D. Liu, J. Colloid Interface Sci., 2022, 625, 466–478 CrossRef CAS PubMed.
- G. Ye, S. Liu, K. Zhao and Z. He, Angew. Chem., Int. Ed., 2023, 62, e202303409 CrossRef CAS PubMed.
- L. Pei, J. Zhong, T. Li, W. Bai, S. Wu, Y. Yuan, Y. Chen, Z. Yu, S. Yan and Z. Zou, J. Mater. Chem. A, 2020, 8, 6795–6803 RSC.
- L. Xie, P. Wang, Y. Li, D. Zhang, D. Shang, W. Zheng, Y. Xia, S. Zhan and W. Hu, Nat. Commun., 2022, 13, 5560 CrossRef CAS PubMed.
- W. Miao, Y. Zhang, H. Li, Z. Zhang, L. Li, Z. Yu and W. Zhang, J. Mater. Chem. A, 2019, 7, 5504–5512 RSC.
- L. Wu, P. Guo, X. Wang, H. Li, X. Zhang, K. Chen and P. Zhou, Chem. Eng. J., 2022, 446, 136759 CrossRef CAS.
- Y. Li, M. Sun, B. Gao, B. Hu, S. Zhou, B. Liu, W. Jiang, C. Liu and G. Che, Sep. Purif. Technol., 2024, 330, 125656 CrossRef CAS.
- C. Zhu, F. Liu, C. Ling, H. Jiang, H. Wu and A. Li, Appl. Catal., B, 2019, 242, 238–248 CrossRef CAS.
- A. A. Ghogare and A. Greer, Chem. Rev., 2016, 116, 9994 CrossRef CAS PubMed.
- D. B. Miklos, C. Remy, M. Jekel, K. G. Linden, J. E. Drewes and U. Hübner, Water Res., 2018, 139, 118–131 CrossRef CAS PubMed.
- S. Patnaik, D. P. Sahoo and K. Parida, Carbon, 2021, 172, 682–711 CrossRef CAS.
- Y. Li, H. Dong, L. Li, L. Tang, R. Tian, R. Li, J. Chen, Q. Xie, Z. Jin, J. Xiao, S. Xiao and G. Zeng, Water Res., 2021, 192, 116850 CrossRef CAS PubMed.
- H. Che, X. Gao, J. Chen, J. Hou, Y. Ao and P. Wang, Angew. Chem., Int. Ed., 2021, 60, 25546–25550 CrossRef CAS PubMed.
- Z. Wu, Y. Wang, Z. Xiong, Z. Ao, S. Pu, G. Yao and B. Lai, Appl. Catal., B, 2020, 277, 119136 CrossRef CAS.
- Y. Ma, L. Shang, J. Li, Y. Wu, G. Che, C. Liu and Q. Fang, Chem. Eng. J., 2024, 484, 149347 CrossRef CAS.
- Y. Han, M. Tai, Y. Yao, J. Li, Y. Wu, B. Hu, Y. Ma and C. Liu, J. Colloid Interface Sci., 2024, 663, 238–250 CrossRef CAS PubMed.
- L. Ran, Z. Li, B. Ran, J. Cao, Y. Zhao, T. Shao, Y. Song, M. K. H. Leung, L. Sun and J. Hou, J. Am. Chem. Soc., 2022, 144, 17097–17109 CrossRef CAS PubMed.
- B. Han, X. Ding, B. Yu, H. Wu, W. Zhou, W. Liu, C. Wei, B. Chen, D. Qi, H. Wang, K. Wang, Y. Chen, B. Chen and J. Jiang, J. Am. Chem. Soc., 2021, 143, 7104–7113 CrossRef CAS PubMed.
- Y. Yue, P. Cai, K. Xu, H. Li, H. Chen, H. Zhou and N. Huang, J. Am. Chem. Soc., 2021, 143(43), 18052–18060 CrossRef CAS PubMed.
- J. Wang and S. Zhuang, Coord. Chem. Rev., 2019, 400, 213046 CrossRef CAS.
- Y. Lv, C. Zhang, A. He, S. Yang, G. Wu, S. B. Darling and Z. Xu, Adv. Funct. Mater., 2017, 27, 1700251 CrossRef.
- B. Wang, D. He, D. Zhu, Y. Lu, C. Li, X. Li, S. Dong and C. Lyu, Sep. Purif. Technol., 2022, 296, 121350 CrossRef CAS.
-
Materials Studio ver. 7.0, Accelrys Inc., San Diego, CA, 2013 Search PubMed.
- S. Wan, F. Gándara, A. Asano, H. Furukawa, A. Saeki, S. K. Dey, L. Liao, M. W. Ambrogio, Y. Y. Botros, X. Duan, S. Seki, J. F. Stoddart and O. M. Yaghi, Chem. Mater., 2011, 23, 4094–4097 CrossRef CAS.
- Y. Ma, Y. Fu, W. Jiang, Y. Wu, C. Liu, G. Che and Q. Fang, J. Mater. Chem. A, 2022, 10, 10092–10097 RSC.
- H. Dai, W. Zhou, W. Wang and Z. Liu, J. Hazard. Mater., 2022, 426, 127784 CrossRef CAS PubMed.
- X. Zhang, B. Xu, S. Wang, X. Li, C. Wang, B. Liu, F. Han, Y. Xu, P. Yu and Y. Sun, Chem. Eng. J., 2022, 431, 133477 CrossRef CAS.
- P. K. Klu, M. A. Nasir Khan, C. Wang, J. Qi and X. Sun, Environ. Res., 2022, 207, 112148 CrossRef CAS PubMed.
- S. Wang and J. Wang, J. Hazard.
Mater., 2022, 423, 127207 CrossRef CAS PubMed.
- S. Wang and J. Wang, Chem. Eng. J., 2018, 351, 688–696 CrossRef CAS.
- Q. Zhong, Y. Sun, C. Xu, Y. Li, D. Sun, L. Wu, S. Yang, Y. Liu, C. Qi, Z. Xu, H. He, S. Li, Z. Wang and S. Wang, Appl. Catal., B, 2023, 339, 123113 CrossRef CAS.
- Y. Jiang, J. Wang, B. Liu, W. Jiang, T. Zhou, Y. Ma, G. Che and C. Liu, Chem. Eng. J., 2022, 446, 137361 CrossRef CAS.
- M. Ren, S. Sun, Y. Wu, Y. Shi, Z.-j. Wang, H. Cao and Y. Xie, Chemosphere, 2022, 296, 134071 CrossRef CAS PubMed.
- C. Lin, Z. Liu, Y. Zhao, C. Song, F. Meng, B. Song, G. Zuo, Q. Qi, Y. Wang, L. Yu and M. Song, Sep. Purif. Technol., 2023, 313, 123445 CrossRef CAS.
- Y. Wu, J. Chen, H. Che, X. Gao, Y. Ao and P. Wang, Appl. Catal., B, 2022, 307, 121185 CrossRef CAS.
- Y. Chu, X. Zheng and J. Fan, Chem. Eng. J., 2022, 431, 134020 CrossRef CAS.
- M. Zhu, X. Liu, L. Liu, Q. Liu, F. Li and J. Tang, J. Environ. Chem. Eng., 2022, 10, 108803 CrossRef CAS.
- C. Zhang, J. Tang, F. Gao, C. Yu, S. Li, H. Lyu and H. Sun, Chem. Eng. J., 2022, 431, 134063 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Materials and characterization, FT-IR spectra, PXRD patterns, TGA analysis, SEM images, XPS spectra, contact angles, BET plots, the stability test, degradation performance, unit cell parameters, and comparison performance. See DOI: https://doi.org/10.1039/d4sc02462a |
|
| This journal is © The Royal Society of Chemistry 2024 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence *b,
Guangbo
Che
*c,
Bo
Hu
*a,
Valentin
Valtchev
*b,
Guangbo
Che
*c,
Bo
Hu
*a,
Valentin
Valtchev