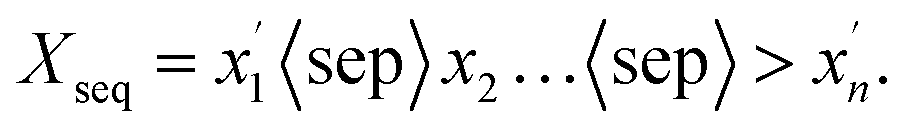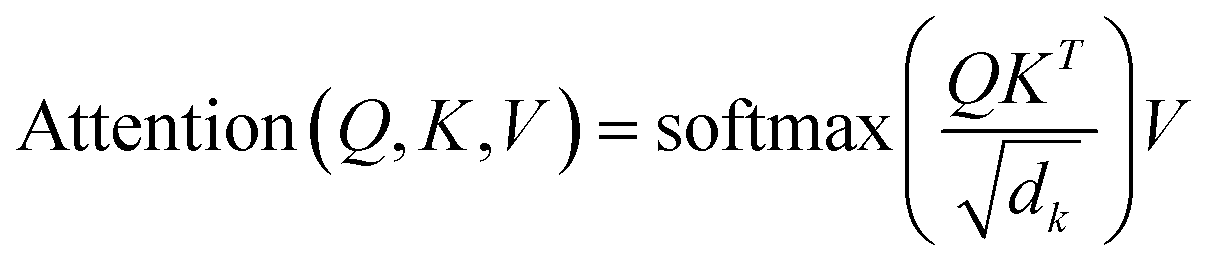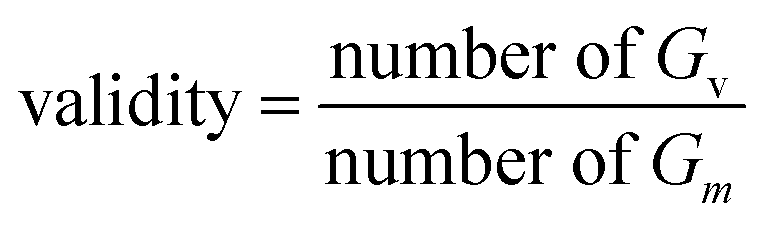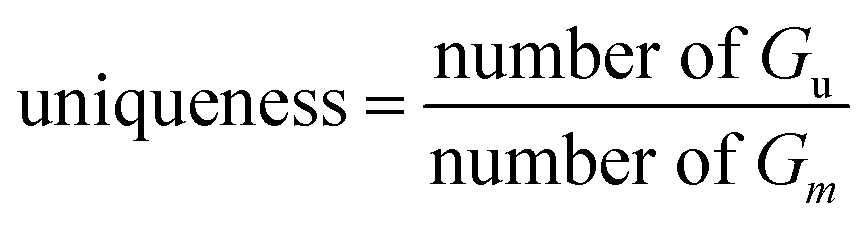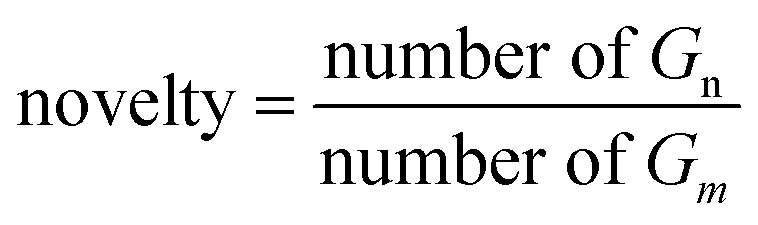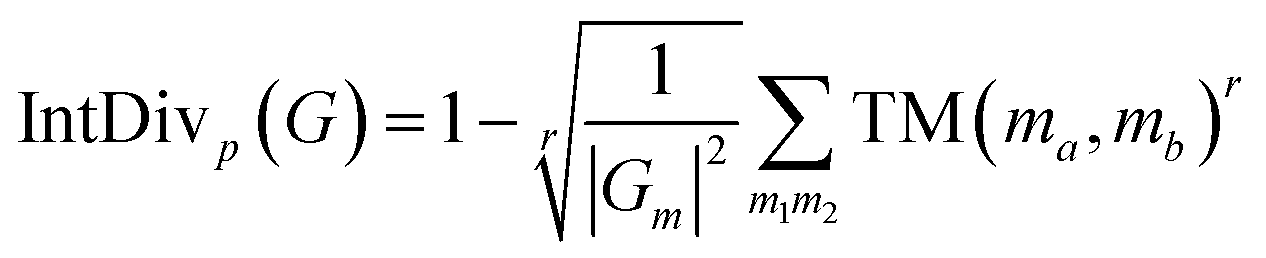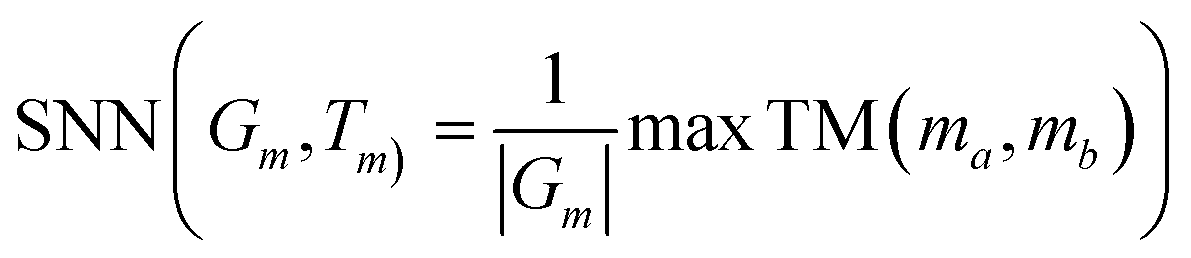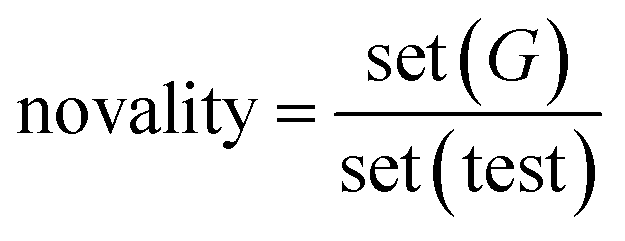DOI:
10.1039/D4SC03744H
(Edge Article)
Chem. Sci., 2024,
15, 13727-13740
Unlocking comprehensive molecular design across all scenarios with large language model and unordered chemical language†
Received
7th June 2024
, Accepted 28th July 2024
First published on 29th July 2024
Abstract
Molecular generation stands at the forefront of AI-driven technologies, playing a crucial role in accelerating the development of small molecule drugs. The intricate nature of practical drug discovery necessitates the development of a versatile molecular generation framework that can tackle diverse drug design challenges. However, existing methodologies often struggle to encompass all aspects of small molecule drug design, particularly those rooted in language models, especially in tasks like linker design, due to the autoregressive nature of large language model-based approaches. To empower a language model for a wider range of molecular design tasks, we introduce an unordered simplified molecular-input line-entry system based on fragments (FU-SMILES). Building upon this foundation, we propose FragGPT, a universal fragment-based molecular generation model. Initially pretrained on extensive molecular datasets, FragGPT utilizes FU-SMILES to facilitate efficient generation across various practical applications, such as de novo molecule design, linker design, R-group exploration, scaffold hopping, and side chain optimization. Furthermore, we integrate conditional generation and reinforcement learning (RL) methodologies to ensure that the generated molecules possess multiple desired biological and physicochemical properties. Experimental results across diverse scenarios validate FragGPT's superiority in generating molecules with enhanced properties and novel structures, outperforming existing state-of-the-art models. Moreover, its robust drug design capability is further corroborated through real-world drug design cases.
Introduction
The inherently intricate process of drug development has been expedited by the emergence of artificial intelligence (AI). However, researchers are confronted with a range of design challenges in real-world scenarios, including synthesizing novel active compounds guided by ligand specifications, devising linkers to connect functional groups, and completing structural fragments based on partial molecular data. To address these multifaceted challenges, AI-driven molecular design models have proliferated in recent years.
Recent advancements have yielded noteworthy methodologies for handling individual tasks. Within the realm of de novo molecule generation, Bagal et al. utilized a language model to interpret molecular simplified molecular input line entry system (SMILES)1 character sequences, ultimately leading to the development of MolGPT, a novel framework that leverages the self-attention mechanism with masking.2 Moreover, Juan-Ni et al. introduced a fragment-based approach for de novo molecular design, significantly enhancing both the effectiveness and uniqueness of the synthesized molecules.3 Considering the importance of generating molecules with desired pharmaceutical properties in lead discovery, Wang et al. presented MCMG,4 a novel methodology that facilitates the generation of molecules compliant with multiple constraints. Additionally, other frameworks, such as REINVENT2,5 MIMOSA,6 and Mol-CycleGAN,7 have achieved remarkable results in the generation of molecules with specific property constraints.
In 2020, Imrie et al. introduced the groundbreaking linker design paradigm, DeLinker, rooted in the variational autoencoder (VAE) framework.8,9 This innovative model was designed to amalgamate two fragments or partial structures, thus orchestrating the synthesis of a molecule that embodies both components. Later, Igashov et al. pioneered the development of 3D equivariant molecular features using equivariant networks, subsequently employing a diffusion process for linker synthesis.10 This model enables accurate prediction of the linker's size, facilitating the generation of a diverse array of linkers and exhibiting state-of-the-art (SOTA) performance across various benchmark datasets such as ZINC,11 CASF,12 and GEOM.13 Then, Imrie et al. further refined the DeLinker framework to develop DEVELOP,14 a versatile tool applicable to linker and R-group design,15,16 scaffold hopping,17,18 and PROTAC design,19,20 all demonstrating promising results. More recently, Jin and colleagues introduced FFLOM,21 a model that utilizes molecular graphs to represent molecular fragments. By integrating node and edge flow layers to regulate atom and bond sampling, this model enhances crucial metrics such as traceability and molecular binding affinity for across diverse molecular generation applications.
While the achievements of these molecular design methodologies are undoubtedly noteworthy, it is crucial to recognize the inherent complexity of drug research and development. The current methodologies tend to focus on modeling specific generation contexts, thus limiting their adaptability to the wide range of challenges encountered in drug design. However, in recent years, substantial progress has been achieved in the development of large-scale general natural language models.22–25 These models have demonstrated remarkable efficacy across diverse domains, attributed to their utilization of pretraining and fine-tuning methodologies.26–29
Utilizing textual representation enables comprehensive utilization of the modeling approach offered by pre-trained language models, making SMILES-based pre-trained models more adaptable and effective in molecular generation compared to graph-based methodologies.30,31 Moreover, SMILES-based language models, utilizing architectures like transformer, are capable of handling lengthy molecular sequences.32–35 Studies reveal that these models outperform graph generation models in capturing complex molecular distributions and possess superior generative capabilities.36 Traditional autoregressive language models, typically utilized in SMILES or SELFIES, generate molecules sequentially, atom by atom, from left to right. However, this approach is prone to exposure bias, where the accuracy of subsequent atom generation hinges heavily on the preceding fragment, potentially leading to error accumulation. Additionally, it lacks the capability to handle molecular design tasks such as linker design, which require filling gaps within the molecular structure.
In response to this challenge, our study introduces FU-SMILES, a novel molecular representation that identifies disconnection points among molecular fragments, enabling their seamless integration into whole molecules. Unlike the traditional left-to-right sequential representation, FU-SMILES incorporates fragment details from any part of the molecule into the context. Building upon FU-SMILES, we propose FragGPT, an innovative and comprehensive fragment-based drug design large language model. By employing FU-SMILES, FragGPT proficiently handles fragment generation tasks, efficiently mitigating th error accumulation issues associated with atom-by-atom generation, thereby enhancing the efficiency of molecule construction.
Following a methodology akin to general language models, FragGPT undergoes initial pretraining on an extensive molecular dataset to enhance its generalization capabilities, followed by fine-tuning tailored for specific downstream tasks. To fulfill the requirement for drug molecules that need to meet multiple biophysical properties, the proximal policy optimization (PPO) algorithm37 is utilized to steer the fine-tuning process of our model across specific case studies. We propose a comprehensive evaluation reward model for the generated molecules, encompassing several key metrics such as docking score, synthetic accessibility (SA) score,38 penalized![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) logP (plogP) score, and quantitative estimation of drug-likeness (QED) score. To evaluate the efficacy of FragGPT, we conducted assessments across a wide range of drug design scenarios, achieving performance comparable to SOTA methods across all tasks. Additionally, to examine the performance of FragGPT in real-world drug design settings, we executed case studies covering diverse aspects such as de novo design, fragment linker design, R-group exploration, PROTAC design, side chain optimization, and scaffold hopping. Our experimental findings highlight that FragGPT not only accelerates drug design in various scenarios but also demonstrates significant effectiveness in handling multi-constraint generation tasks.
logP (plogP) score, and quantitative estimation of drug-likeness (QED) score. To evaluate the efficacy of FragGPT, we conducted assessments across a wide range of drug design scenarios, achieving performance comparable to SOTA methods across all tasks. Additionally, to examine the performance of FragGPT in real-world drug design settings, we executed case studies covering diverse aspects such as de novo design, fragment linker design, R-group exploration, PROTAC design, side chain optimization, and scaffold hopping. Our experimental findings highlight that FragGPT not only accelerates drug design in various scenarios but also demonstrates significant effectiveness in handling multi-constraint generation tasks.
Results
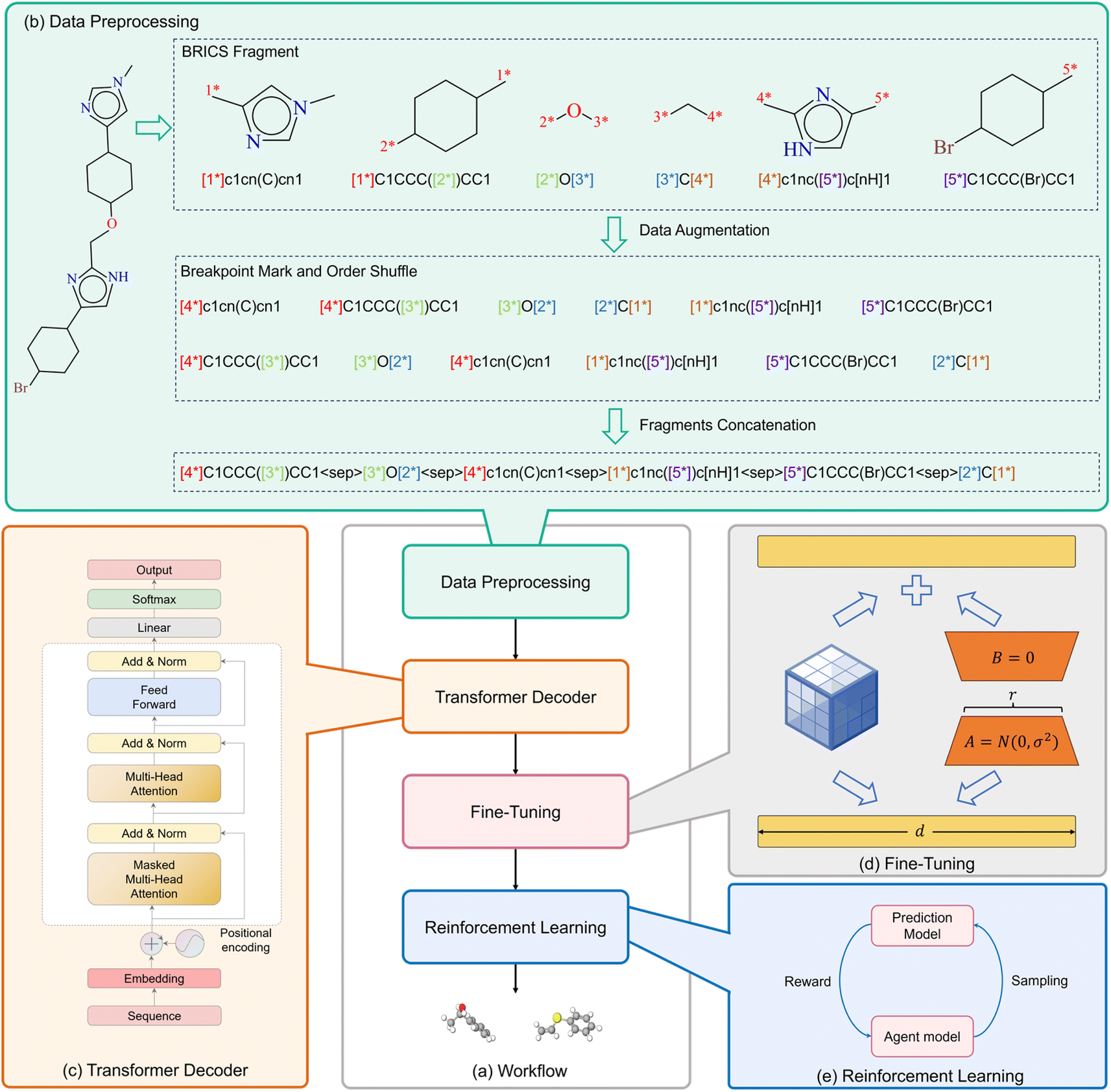
The comprehensive workflow of FragGPT is outlined in Fig. 1. Initially, molecules are converted into the FU-SMILES format. Subsequently, the BRICS algorithm is used to segment molecular representations in SMILES format, accompanied by the inclusion of markers indicating connection points. Following this segmentation, data augmentation techniques are implemented, ultimately leading to the concatenation process that generates the FU-SMILES representation of the molecule. The obtained FU-SMILES are then fed into our backbone model for pretraining. To economize computational resources in downstream applications, we implement low-rank adaptation (LoRA)39 for fine-tuning. Finally, within the contexts of specific drug design scenarios, the PPO algorithm is deployed to strategically optimize the generated molecules across a diverse set of properties.
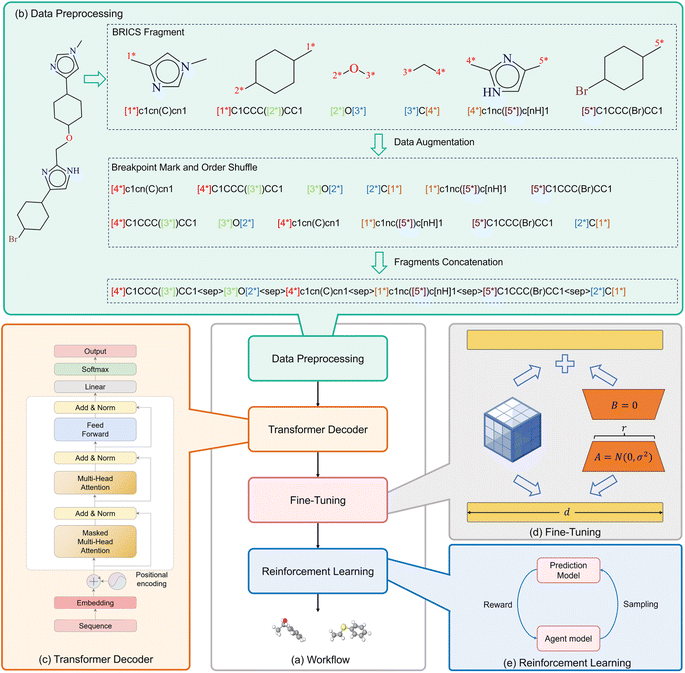 |
| | Fig. 1 The overview of FragGPT. (a) The workflow of FragGPT. (b) Data processing of FU-SMILES. (c) The backbone of FragGPT. The SMILES sequence is derived through the augmentation of molecular fragment data processing. Following this, position encoding and word embedding are applied to the SMILES sequence to obtain token embedding, which is subsequently fed into GPT2 for pre-training, resulting in the acquisition of the pre-trained model FragGPT. (d) Fine-tuning architecture. (e) RL for the optimization of multiple properties. | |
To enforce constraints on the generation of molecular pharmacological properties, we augment the pre-training process by incorporating information about the absorption, distribution, metabolism, excretion, toxicity (ADMET) properties of molecules, leading to the development of FragGPT-ADMET. Unlike FragGPT, FragGPT-ADMET requires specifying a set of ideal ADMET values during the generation process. Typically, we provides the ADMET properties of a reference molecule, enabling the model to generate molecules with similar properties, thus adhering to he specified constraints. For a thorough understanding of the methodology, please refer to the Methods section.
FragGPT underwent a rigorous evaluation involving various metrics. Initially, benchmark assessments are conducted across multiple test datasets, covering five distinct tasks: de novo design, linker design, R-group exploration, side chain optimization, and scaffold hopping. Subsequently, we employed reinforcement learning (RL) based on FragGPT to delve deeper into specific case studies.
De novo design
Molecular de novo generation stands as one of the fundamental tasks in molecular design. To assess its performance on this task, we sampled 30000 molecules and evaluated them on the MOSES40 benchmark, utilizing multiple metrics, including validity, uniqueness, novelty, SNN, Frag, and IntDiv. Our model was benchmarked against several baseline models including cMOlGPT,41 MOlGPT,2 develop,42 VAE,43 AAE,44 JTN-VAE,45 and LatentGAN.46 The evaluation results are shown in the Table 1. Since the original literature for cMOlGPT did not report the results for IntDiv and novelty scores, these two metrics o were excluded from the comparison.
Table 1 The evaluation results for the de novo design task
| Model |
FragGPT |
CharRNN |
VAE |
AAE |
LatentGAN |
JT-VAE |
MolGPT |
cMolGPT |
| Validity↑ |
0.983 |
0.975 |
0.977 |
0.937 |
0.897 |
1.000
|
0.994 |
0.988 |
| Unique@1K↑ |
1.000 |
1.000 |
1.000 |
1.000 |
1.000 |
1.000 |
1.000 |
1.000 |
| Unique@10K↑ |
0.999 |
0.999 |
0.998 |
0.997 |
0.997 |
0.999 |
1.000
|
0.999 |
| Novelty↑ |
0.994
|
0.842 |
0.695 |
0.793 |
0.949 |
0.914 |
0.797 |
— |
| IntDiv1↑ |
0.862
|
0.856 |
0.856 |
0.856 |
0.857 |
0.855 |
0.857 |
— |
| IntDiv2↑ |
0.861
|
0.851 |
0.852 |
0.852 |
0.851 |
0.849 |
0.851 |
— |
| Frag/Test↑ |
0.994 |
1.000
|
0.999 |
0.991 |
0.999 |
0.997 |
— |
1.000
|
| SNN/Test↓ |
0.547 |
0.601 |
0.626 |
0.608 |
0.538
|
0.548 |
— |
0.619 |
As shown in Table 1, FragGPT notably excelled in all other models in terms of uniqueness, novelty and diversity. This exceptional performance may be attributed to its extensive data-driven training and the sufficient utilization of the comprehensive and diverse molecular representations and properties. Most models, particularly MOlGPT, cMOlGPT and FragGPT, demonstrated high validity, surpassing 98%. However, LatentGAN lagged behind in validity due to its reliance on molecular latent vector for training, which posed challenges in reconstructing molecules from the latent space. Besides, all models generated molecules with nearly 100% uniqueness and frag scores.
Beyond traditional novelty metrics, we utilized multiple additional metrics including IntDiv and SNN to highlight the models' capability to generate molecules with diverse and distinct structures. Notably, FragGPT outperformed the other models in novelty, IntDiv and SNN, further demonstrating its efficacy in generating novel molecules. In comparison, VAE and AAE showed lower novelty among the models, likely due to their design strategy of reducing the latent space dimensions, leading to higher similarity to the training dataset and lower novelty.47
Linker design
As for linker design, we systematically evaluated FragGPT across three benchmark datasets: ZINC, CASF, and PDBbind, utilizing various evaluation metrics including validity, uniqueness, novelty, SA, plogP, QED, and recovery. The detailed results are shown in Table 2.
Table 2 The evaluation results for the linker design task
| Metric |
FragGPT |
FragGPT-LoRA |
DeLinker |
DiffLinker |
3DLinker |
DEVELOP |
|
ZINC
|
| Validity↑ |
90.75% |
97.34% |
98.40% |
94.80% |
98.67%
|
— |
| Uniqueness↑ |
65.47%
|
37.93% |
44.20% |
50.90% |
29.42% |
— |
| Novelty↑ |
98.61%
|
98.16% |
39.50% |
47.70% |
32.48% |
— |
| Recovery↑ |
21.25% |
24.75% |
79.00% |
77.50% |
93.58%
|
— |
| SA↓ |
3.14 |
2.93
|
3.10 |
3.24 |
— |
— |
| plogP↑ |
0.74 |
0.75
|
0.32 |
−0.24 |
— |
— |
| QED↑ |
0.56 |
0.66
|
0.64 |
0.65 |
— |
— |
|
|
CASF
|
| Validity↑ |
90.00% |
91.36% |
95.50%
|
68.40% |
— |
— |
| Uniqueness↑ |
24.00% |
23.00% |
51.90% |
57.10%
|
— |
— |
| Novelty↑ |
99.21%
|
99% |
51.00% |
56.90% |
— |
— |
| Recovery↑ |
25.42% |
26.00% |
53.70%
|
48.80% |
— |
— |
| SA↓ |
3.91 |
3.84
|
4.05 |
4.12 |
— |
— |
| plogP↑ |
−0.36
|
−0.36
|
−0.91 |
−0.41 |
— |
— |
| QED↑ |
0.43
|
0.42 |
0.36 |
0.40 |
— |
— |
|
|
PDBbind
|
| Validity↑ |
93.20% |
94.56% |
96.90%
|
— |
— |
93.10% |
| Uniqueness↑ |
39.60% |
35.30% |
86.10% |
— |
— |
77.30% |
| Novelty↑ |
98.33%
|
99.00% |
84.00% |
— |
— |
88.70% |
| Recovery↑ |
19.80% |
14.10% |
1.90% |
— |
— |
22.40% |
| SA↓ |
3.73 |
3.60
|
4.05 |
— |
— |
4.05 |
| plogP↑ |
−0.89 |
−0.83
|
−2.00 |
— |
— |
−1.93 |
| QED↑ |
0.41 |
0.45
|
0.37 |
— |
— |
0.37 |
FragGPT exhibited an overall satisfactory performance in terms of validity, achieving a validity rate above 90% through autoregressive fragment generation without conducting valence checks during the pre-training and fine-tuning phases. Fine-tuning FragGPT with LoRA further enhanced its validity, comparable to those of DeLinker and 3DLinker. DeLinker, employing a masking mechanism to enforce simple valence rules, achieved top results on the CASF and PDBbind datasets, closely trailing 3Dlinker on ZINC. The validity of the molecules generated by DiffLinker exhibited significant variation across the ZINC and CASF datasets, possibly owing to its implicit organization of atom coordinates.
Remarkably, FragGPT surpassed all models in generating over 98% novel molecules across datasets, exceeding the second-best model by a substantial margin of approximately 50% on ZINC, 42% on CASF, and 9% on PDBbind, showcasing its exceptional capability to generate novel molecules and explore a wider chemical space.
However, FragGPT displayed a relatively weaker performance in recovery metrics compared to other models, partly attributed to mismatches between fragment tokens and the specific connections of test molecules. Nevertheless, FragGPT still achieved notable recovery rates even without fine-tuning, and its recovery performance was only slightly inferior to that of DEVELOP on PDBbind, reflecting a balance between recovery and a reasonable training data segmentation strategy.
The performance of FragGPT on three drug-likeness metrics corroborated our initial hypothesis. FragGPT, especially FragGPT-LoRA, excelled in drug-like properties across all datasets, achieving superior SA, QED, and plogP score performances. This success can be attributed to our autoregressive fragment-by-fragment molecule generation strategy, which effectively circumvented the generation of chemically infeasible structures, especially in complex ring systems. FragGPT's outstanding performance in SA, QED, and plogP scores surpassed all other atom-by-atom generation models across all test sets, highlighting its efficacy in generating molecules with desired properties.
R-group exploration
We conducted an evaluation of FragGPT using the CASF and PDBbind benchmarks, and compared its performance against Delinker and DEVELOP, as summarized in Table 3. Similar to the linker design task, the absence of atomic valence checks slightly reduced the validity of the molecules generated by FragGPT compared to DeLinker and DEVELOP. DeLinker exhibited outstanding performance on CASF and PDBbind, achieving 100% validity, while FragGPT's pre-training model achieved over 90% validity on these two test sets, indicating its commendable performance.
Table 3 The evaluation results for the R-group design task
| Metric |
FragGPT |
FragGPT-LoRA |
DeLinker |
DEVELOP |
|
CASF
|
| Validity↑ |
91.12% |
91.09% |
100.00%
|
99.80% |
| Uniqueness↑ |
35.66% |
40.80% |
74.20%
|
39.70% |
| Novelty↑ |
98.18% |
98.56%
|
55.10% |
43.40% |
| Recovery↑ |
39.40% |
40.00% |
33.60% |
58.70%
|
| SA↓ |
3.26 |
3.21
|
— |
3.39 |
| plogP↑ |
−0.26 |
−0.24
|
— |
−0.54 |
| QED↑ |
0.39 |
0.54
|
— |
0.52 |
|
|
PDBbind
|
| Validity↑ |
95.86% |
95.52% |
100.00%
|
99.50% |
| Uniqueness↑ |
40.00% |
43.00% |
87.80%
|
76.20% |
| Novelty↑ |
99.19% |
99.61%
|
71.10% |
78.20% |
| Recovery↑ |
16.78%
|
13.00% |
1.00% |
15.30% |
| SA↓ |
3.48 |
3.41
|
— |
3.87 |
| plogP↑ |
−0.81 |
−0.78
|
— |
−1.57 |
| QED↑ |
0.45 |
0.52
|
— |
0.42 |
Due to the constrained modification space and comparatively larger building blocks in the R-group exploration task, our uniqueness performance on CASF was intermediate between DeLinker and DEVELOP, which slightly trailing on PDBbind. However, in terms of novelty, FragGPT exhibited robust capabilities, generating over 95% novel molecules and surpassing other models on both test datasets. Particularly note-worthy, FragGPT's pre-trained model achieved a remarkable 98.18% novelty on the CASF test dataset, surpassing the second-highest model, DeLinker, by 43.08%.
In terms of recovery, FragGPT exhibited comparable performance to other models. On the CASF test dataset, FragGPT demonstrated superior performance in the R-group exploration task, achieving a 25.42% higher recovery rate compared to the linker design task. This discrepancy may stem from the distinct linking methodologies employed in these two tasks. Linker design involves the intricate connection between two breakpoints, followed by the integration of the generated linker fragments, while R-group exploration simplifies this process by only considering the connection of a single breakpoint. The variability in molecules generated based on different linking points, even with identical linker fragments, may explain the lower recovery observed in the linker design task.
Regarding drug likeness properties, FragGPT excelled in SA, QED, and plogP on both test datasets, surpassing the other two models. Its consistently outstanding performance across both linker design and R-group exploration tasks underscores FragGPT's capacity in generating chemically reasonable molecules with substantial potential for lead design.
Scaffold hopping
Given the scarcity of scaffold hopping benchmark datasets, we evaluated the pre-trained FragGPT models on the PDBbind test dataset and compared them with DiffHopp. The results are summarized in Table 4. DiffHopp achieved the highest molecular validity performance of 91.4%, while FragGPT displayed a slightly lower validity of 85.30%, still outperforming DiffHopp-EGNN, GVP-inpainting, and EGNN-inpainting. Additionally, FragGPT exhibited significantly higher levels of Uniqueness and novelty compared to all other models. Conversely, DiffHopp-EGNN only generated 0.64% unique molecules, indicating potential mode collapse. The observed decline in Uniqueness for DiffHopp and DiffHopp-EGNN may be attributed to their more complex molecular representations. DiffHopp employed graph information to model protein pockets, atom features, and ligand coordinates, aiming to capture a broader range of spatial structural features. Generating molecules based on atom types and coordinate information posed greater challenges compared to utilizing only 2D molecule graph information. This spatial characteristic also impacted the performance of QED, where FragGPT exhibited weaker performance than DiffHopp but was comparable to other models.
Table 4 The evaluation results for the scaffold hopping task on the PDBbind dataset
| Metric |
FragGPT |
DiffHopp |
DiffHopp-EGNN |
GVP-inpainting |
EGNN-inpainting |
| Validity↑ |
85.30% |
91.40%
|
75.70% |
65.20% |
79.30% |
| Uniqueness↑ |
82.80%
|
59.20% |
0.64% |
66.80% |
66.70% |
| Novelty↑ |
99.80%
|
99.80%
|
100.00% |
99.70% |
99.90% |
| QED↑ |
0.48 |
0.61
|
0.51 |
0.55 |
0.47 |
Side chain optimization
Finally, we assessed the side chain growth capabilities on the PDBbind test dataset, with results summarized in Table 5. Given the scarcity of related studies or benchmark datasets for this task, we exclusively compared the generated molecules with the test dataset. We analyzed the validity, uniqueness, novelty, recovery, SA, plogP, and QED metrics of the molecules generated by FragGPT, FragGPT-LoRA (fine-tuned on the PDBbind training set), and FragGPT-ADMET. The pre-trained FragGPT model, without the constraints imposed by the PDBbind training set, achieved the highest scores in validity (92.81%), uniqueness (72.73%), and novelty (99.99%) among the three models. However, its recovery rate was comparatively lower, standing at 3.9%. Upon fine-tuning with the PDBbind training set, FragGPT-LoRA demonstrated a slight decrease in validity and uniqueness but an increase in recovery. This observation could be attributed to the limited size of the PDBbind training dataset, comprising less than 20000 entries, which may reduce the model's search space.
Table 5 The evaluation results for the side chain optimization task
| Model |
FragGPT |
FragGPT-LoRA |
Test/PDBbind |
| Validity↑ |
92.81%
|
77.63% |
— |
| Uniqueness↑ |
72.73%
|
43.97% |
— |
| Novelty↑ |
99.99%
|
99.98% |
— |
| Recovery↑ |
3.90% |
11.80%
|
— |
| SA↓ |
3.04 |
3.00
|
3.30 |
| plogP↑ |
−0.20
|
−0.89
|
−2.23 |
| QED↑ |
0.54 |
0.62
|
0.56 |
FragGPT demonstrated superior performance in the SA, QED, and plogP evaluations in comparison to the molecules from the PDBbind dataset. FragGPT achieved a higher SA score than the test set by a margin of approximately 0.26 and significantly outperformed it in plogP, with a difference of 2.03 higher. However, regarding the QED score, FragGPT's performance was comparable to that of the test set. After fine-tuning with the training set, FragGPT-LoRA exhibited substantial enhancement in both SA and QED scores, significantly surpassing the scores of the molecules in the PDBbind test set. In summary, FragGPT and FragGPT-LoRA demonstrated notable improvements in molecular quality compared to the molecules generated by the PDBbind test set.
Conditioned generation using FragGPT-ADMET
We introduce FragGPT-ADMET, an extension of FragGPT, that integrates 56 ADMET properties as conditional regulators for molecular generation, aiming to enrich molecular attributes. To assess its effectiveness, we evaluated FragGPT-ADMET's performance in optimizing both single and multiple ADMET properties across two distinct tasks: de novo design and linker design. Specifically, our experiments focused on analyzing the impact of three key ADMET properties: logP, stress response-antioxidant response element (SR-ARE) and inhibitor of cytochrome P450 2C9 protein (CYP2C9), under single- and multi-constraint conditions.
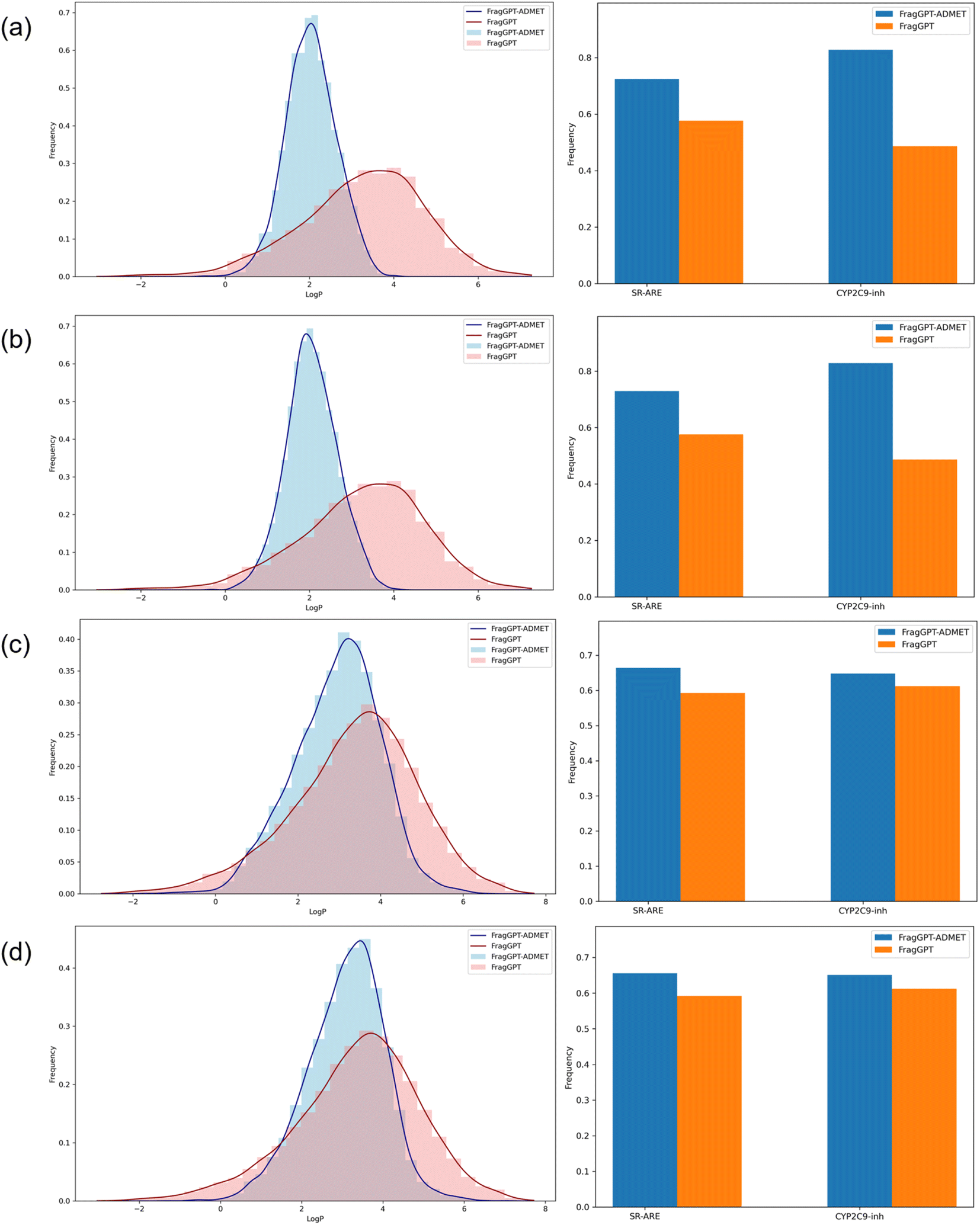
As depicted in Fig. 2, our findings revealed that FragGPT-ADMET outperformed FragGPT in generating molecules with improved properties across all three dimensions, both in linker design and de novo design tasks. Subsequently, we tested the proportion of molecules that met the specified criteria under multiple constraints. These multiple constraints include a logP value between 1 and 3, as well as meeting the criteria for SR-ARE and CYP2C9 inhibition. As summarized in Table 6, FragGPT-ADMET achieved success rates 2.6 times higher in linker design and 1.2 times higher in de novo design compared to FragGPT. Integrating these results with Fig. 2, we observed a more pronounced enhancement of FragGPT-ADMET in the de novo design task, likely due to the limited chemical space in linker design, which restricts molecule generation to linker structures defined by terminal groups. In essence, FragGPT-ADMET exhibited superior conditional generation capabilities.
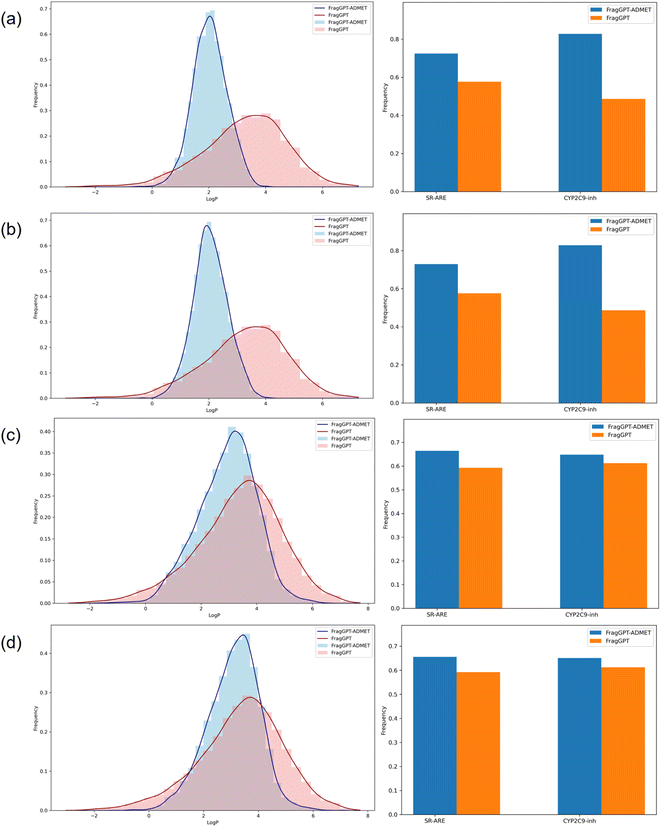 |
| | Fig. 2 The performance of the conditioned generation task. (a) De novo design with single constraint, (b) de novo design with multi-constraints, (c) linker design with single constraint, and (d) linker design with multi-constraints. FragGPT-ADMET (blue) and FragGPT-ADMET (red). | |
Table 6 | The success rate for the de novo and linker design tasks with multi-constraints
| Task |
De novo design |
Linker design |
| FragGPT |
24.55% |
39.89% |
| FragGPT-ADMET |
63.09% |
46.27% |
Drug design in real-world scenarios
In this section, we applied FragGPT in conjunction with RL to address four specific molecular design challenges in real-world scenarios, including linker design, R-group exploration, scaffold hopping, and PROTAC design. Our primary objective was to generate molecules with optimized docking scores, QED, and SA, thereby demonstrating the exceptional generalization capacity of FragGPT. For each design task, we set specific targets and utilized four authentic reference molecules for comparative analysis, with detailed information provided in the Methods section.
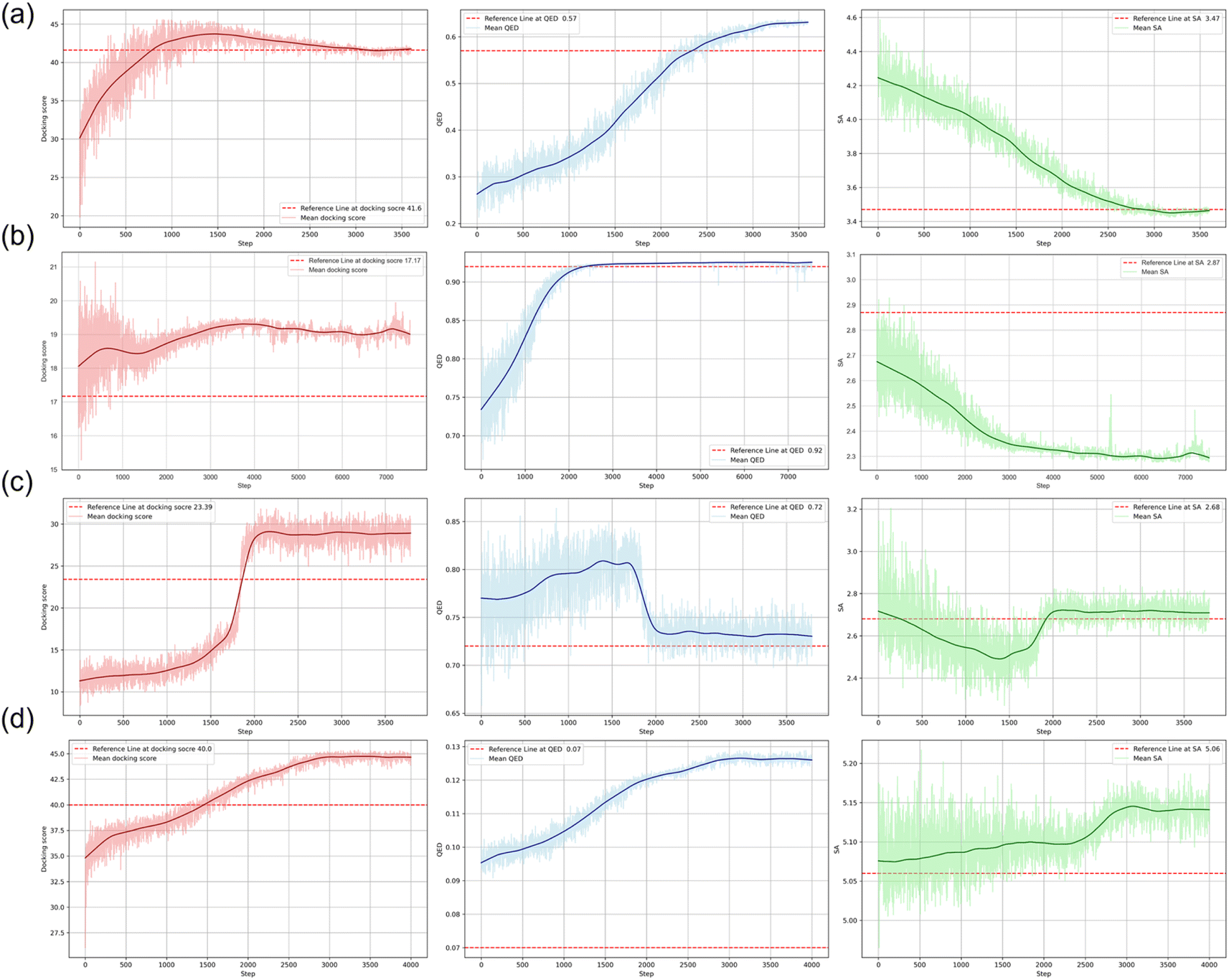
Molecular design tasks were conducted by utilizing RL tailored to the specific objectives across diverse scenarios. Fig. 3 depicts the progression of the RL optimization steps and the mean docking scores, QED, and SA values for the molecules generated at each step. Throughout all tasks, a consistent improvement in these three molecular properties was observed as the RL iterations progressed under the guidance of FragGPT. In each subplot, the red dashed line represents the values of the reference molecules.
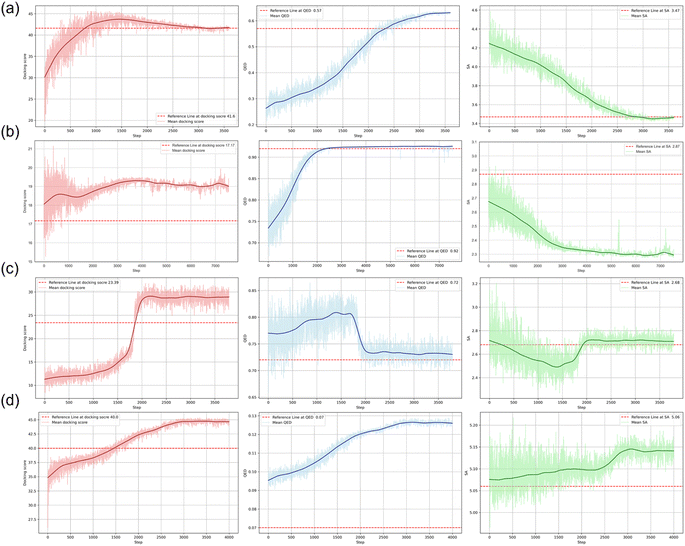 |
| | Fig. 3 The correlation between the average molecular properties and the progression of optimization steps during RL. (a) Linker design, (b) R-group exploration, (c) scaffold hopping, and (d) PROTAC design. The curves denote the mean values of the docking scores (red), QED (blue), and SA (green). The red dashed lines denote the values for the reference molecules. | |
In the linker design task, RL was employed to refine the linker structure of the reference molecule, aiming to optimize their three key properties. As depicted in Fig. 3(a), a continual improvement in the average molecular properties was observed through RL, ultimately reaching a level comparable to the reference molecule. Initially, the docking score increased, followed by a slight decrease, converging closely to the docking score of the reference molecule. Concurrently, QED demonstrated steady enhancement, while SA gradually declined, eventually surpassing the reference value. The initial rise and subsequent minor decline in docking score were attributed to model adjustments for balancing the three properties. The R-group exploration task involved RL optimization of the R-group structure of the reference molecule to enhance their three properties. As illustrated in Fig. 2(b), continuous enhancement was observed across all three molecular properties via RL, ultimately exceeding the reference molecule values. In the scaffold hopping and PROTAC design tasks, FragGPT demonstrated exceptional performance. As depicted in Fig. 3(c and d), the model-generated molecules exhibited a more significant enhancement in docking and QED scores compared to the reference molecules. Given the significantly longer length of PROTAC, the average SA scores for both generated and reference PROTACs were higher than the molecules in the other three cases. Consequently, the shift range of SA score was limited and occupied a relatively small part of the optimization process.
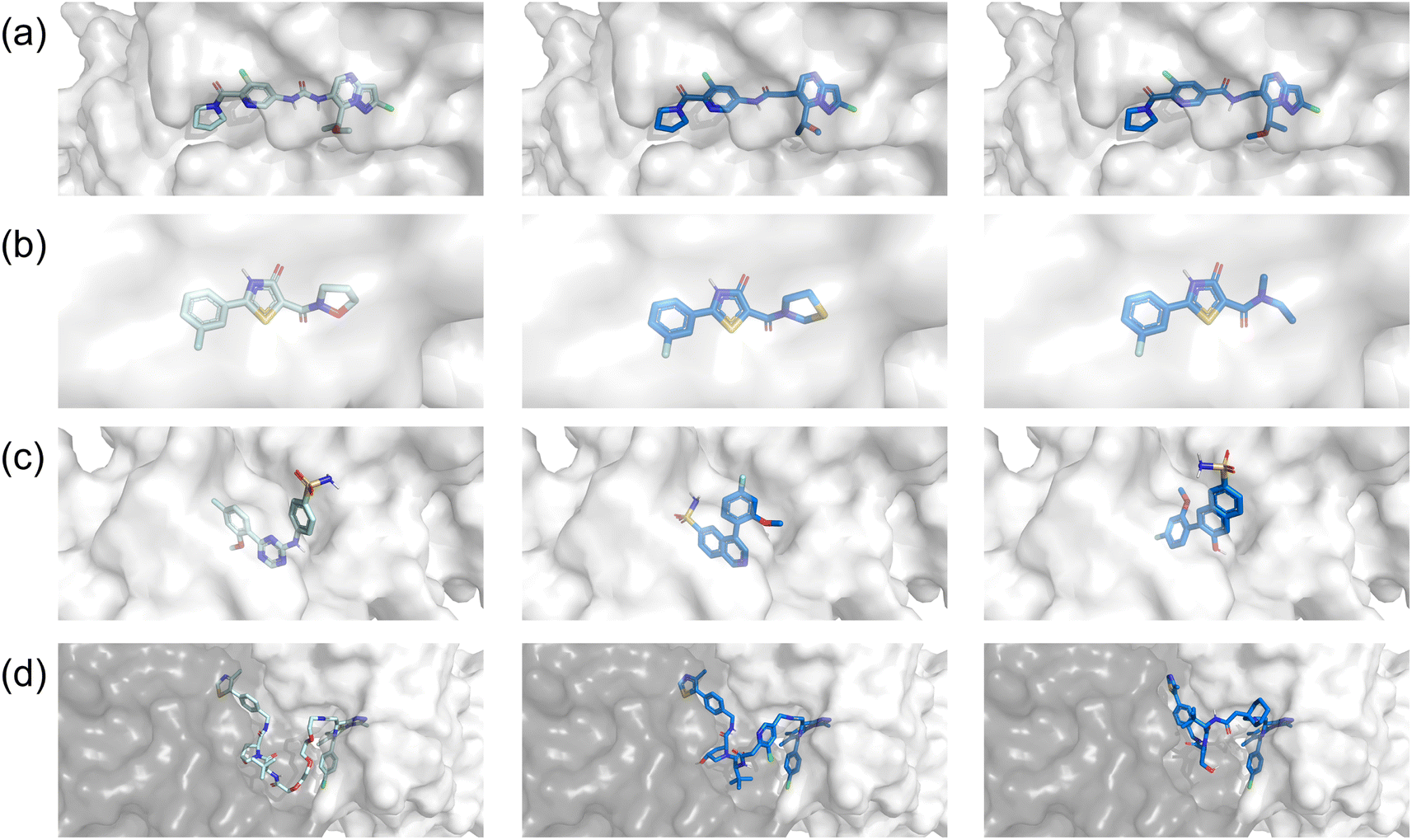
According to the foregoing results, we hypothesize that the variance in performance may stem from the relevance of each task to the corresponding objectives. As shown in Fig. 4, the fragments to be modified in linker design and R-group exploration were quite small, and thus the molecules generated by FragGPT remained highly similar docking conformations to the reference molecule, with minimal changes to the unaltered fragments. This feature not only stayed consistent with our initial design but also reduced the complexity of optimizing molecule properties, which was confirmed by the RL performance of FragGPT in these two cases, as seen in Fig. 3(a and b). In contrast, for scaffold hopping, while the generated molecules occupy the same protein pocket as the reference, the non-fixed orientations of the excised fragments lead to a more significant variation in the generated molecule structures compared to the other three cases. This may be the reason why the molecular properties in this case were much more difficult to be predicted or optimized.
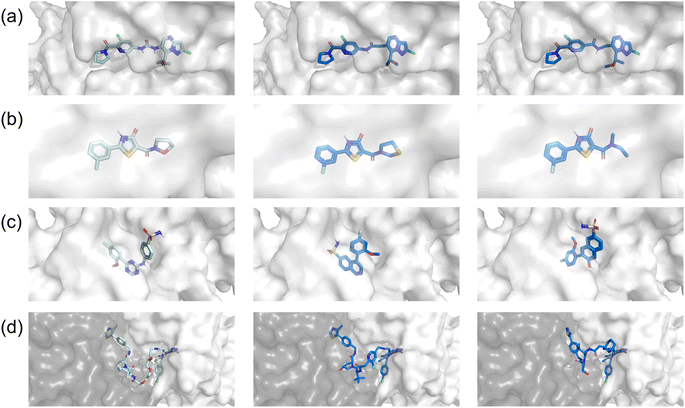 |
| | Fig. 4 The docking conformations of the reference molecules and the corresponding chosen molecules generated by FragGPT with improved docking score, SA and QED scores in four real-world scenarios: (a) linker design, (b) R-group exploration, (c) scaffold hopping, and (d) PROTAC design. The pale blue conformation shown in the first row refers to the reference molecule in each case while the other two dark blue conformations refer to the generated molecules. | |
Besides, FragGPT exhibited a remarkable capability in producing fragments that were highly analogous yet superior to the reference fragment. For example, the linker in the second row of Fig. 4(a) differed from the reference linker with only one atom, while the R-group in the second row of Fig. 4(b) precisely resembled the reference, comprising a five-atom heterocyclic ring. Remarkably, even in the PROTAC design case, FragGPT generated PROTACs with conformations quite similar to the reference PROTAC, leveraging the flexibility of its linkers to outperform the latter in terms of docking and QED scores. The visualization of these docking conformations, together with the above RL optimization steps, demonstrated the ability of FragGPT to generate molecules with comprehensive chemical properties and controllable conformational transformation.
Discussion
In this study, we present a novel molecular representation, FU-SMILES, alongside FragGPT, a pioneering comprehensive large language model for drug design that relies on this representation. Through rigorous benchmarking experiments across 5 generation scenarios, FragGPT exhibited remarkable performance, distinctiveness, and innovation. Notably, FragGPT-generated molecules surpassed all SOTA models in terms of SA and QED metrics, highlighting its ability to capture critical drug-like properties. Moreover, we simulated real-world drug design scenarios and employed RL optimization to design molecules with superior properties compared to the reference molecules. These results demonstrate FragGPT's proficiency in generating molecules with chemically plausible structures and desired properties, aligned with predefined optimization objectives. As a result, FragGPT emerges as a unified and robust molecular generation framework with significant potential across diverse applications.
Methods
Datasets
Pre-training.
The training data set comprised 78 million molecules, originating from PubChem35 and ChEMBL.48 We make 56 ADMET predictions for these molecules, thereby enriching the pre-training with pharmacochemical property information.
De novo design.
We used the Moses40 data set for fine-tuning, which contains a total of 1743265 training molecules and 19367 validation molecules.
Linker and R-group.
We used the ZINC-250K11 data set that contains 156922 training molecules and 400 validation molecules.
Scaffold hopping and side chain optimization.
We used the PdbBind49 data set for fine-tuning. The training set includes 17393 small molecules and the validation set included 1933 molecules.
Linker design.
We tested on ZINC-250K,11 casf-2016,12 and PdbBind.49 The processing of fragment–linker pairs in the test set followed the approach reported by Imrie et al.8 The ZINC-250K data set contains 400 linker test pairs, casf-2016 contains 309 linker pairs, and PdbBind contains 321 linker tests.
R-group exploration.
We tested on the casf-2016 and PdbBind datasets. Like the linker, the fragment processing followed the method of Imrie et al.8 casf-2016 contains 237 R-group test pairs, and PdbBind contains 295 R-group test pairs. To process the test sets, we adopt the Diffhopp method,50 which divides each molecule into two parts: the R group (>1) and the Murcko scaffold.51 The growth based on the scaffold serves as the side chain modification task, and the generation based on the side chain is used as the scaffold hopping task. The scaffold hopping task includes 82 pairs of test data, and the side chain task includes a similar number of 82 pairs. Among the 82 pairs used for scaffold hopping, 66 pairs of data have an R number less than or equal to 2, while 16 pairs exhibit an R number greater than 2.
FU-SMILES
FU-SMILES cleverly characterizes molecular fragments in a very simple way, enabling the model to seamlessly adapt to diverse generation scenarios. Fig. 1(b) shows the brief flow of data processing of FU-SMILES. Molecular generative models handle generated data very differently from language models. Language models assemble words corresponding to tokens into complete sentences based on the token generation order, whereas molecular generation necessitates consideration of the crucial connectivity issue, not merely the token order. How to effectively assemble the fragments generated by the model into a valid molecule is the key to ensure the model's proper functionality. We found that a molecule can be divided into several fragments. To reconstruct a molecule from its fragments, one approach is to record the disconnection site information. Based on this, we first use BRISC to fragment the small molecule X and obtain molecular fragments. We then identify the two endpoints of the disconnection point using [i*], resulting in the fragment Xfrag with connection information. Notably, these identifiers must appear in pairs, where the serial number i <= n (n is the number of molecular fragments). Finally, we connect the molecular fragments using the special mark delimiter 〈sep〉> to obtain Xseq.| | 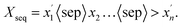 | (2) |
At the same time, we found that the two ends of the disconnection sites in assembled molecular fragments and bonds is independent of the value of i or the order of the fragments within a molecular fragment sequence. The value of i can be considered as an unordered categorical information used to distinguish different pairs of break points. Based on this, we performed the first data augmentation method: randomly transforming the i value within (1 ∼ n), so that the break point identification of each fragment has a probability of any value in (1 ∼ n) (ensuring that the serial number i <= n appears only once). Through this transformation, for the molecular fragment group obtained through n break keys, there are a total of n factorial combinations of possibilities. By adjusting the order of break point numbering, the model can more effectively learn the relationship between the data. Furthermore, the assembly of fragments into valid molecules relies solely on their identification information. Unlike language models that operate with sequential sequences where varying orders can yield different meanings, different fragment sequences may represent the same molecule. Therefore, we conducted a second data augmentation method: by randomly shuffling molecular fragments, allowing the fragments to be in any position in the sequence, achieving the disordering of fragments in the sequence. Assuming the molecule consists of three fragments, namely fragment A, fragment B, and fragment C. After data augmentation, there are six possible combinations, namely {ABC, ACB, BCA, BAC, CBA, CAB}, and during the training phase, one of these possibilities is randomly selected as input. Both data augmentations are carried out simultaneously to further enrich the diversity of data and enhance the robustness of the model.
Backbone
FragGPT.
We follow the GPT2 architecture, which is a general generative language model. Its core is the attention mechanism that can comprehensively consider the relationships among each token in the sequence data and update its representation based on the degree of relevance. This mechanism can effectively capture the contextual information of the text and guide the model generation.| | 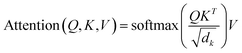 | (3) |
For a SMILES data set, the language modeling task is used as the training target.23 The standard goal of language modeling is to maximize the likelihood:
| | | F(D) = ∑logP(di|di−k…,d1;θ). | (4) |
GPT2 builds a neural network θ through the transformer decoder to model the conditional probability P, where k is the prefix or contextual information generated by the molecule. Here Xseq is the input of the model, which is initially encoded through TokenEncode. Among them, TokenEncode utilizes a tokenizer to obtain the token encoding of Xseq, followed by position encoding and word embedding on the token to derive H0. Subsequently, H0 is fed into the GPTBlock block of l layer, and the predicted molecular fragment sequence is decoded using TokenDecoder. Among them, TokenDecoder is Hl that is generated through the MLP layer and is the score of the model for each possible fragment in the vocabulary at each output position. The final fragment sequence is determined through softmax activation, and these fragments are then assembled to form the final molecule generated by the model.
| | | H0 = TokenEncode(Xseq), | (5) |
| | | P(Xi) = TokenDecode(Hl). | (7) |
FragGPT-ADMET.
First, we predicted the properties of the SMILES molecule data and obtained 56 ADMET properties. These ADMET properties were then encoded using BertModel to get S0.| | | Sadmet = {s1,s2,…,sk}, | (8) |
| | | S0 = Admet Encoder(Sadmet). | (9) |
Then we concatenate the ADMET feature S0 and the molecular fragment sequence feature H0, yielding Hs as the input of GPTBlock, which is expressed as:
| | | Hs = concatenation(S0,H0). | (10) |
Hs is propagated through the
l layer GPTBlock block, resulting in an output
Hsl that incorporates both the ADMET properties and the characteristics of small molecules. For decoding purposes, we exclusively extract the small molecule features
Hl from
Hsl to obtain the final molecule. Assuming that the feature dimension of
L1 is the sum of
S0 and
H0, with
S0 spliced before
H0 as a prefix, we extract the features
Hl that have the same dimensions as
H0 at the end, and use them as the small molecule features.
| | | Hsl = GPTBlock(Hsl−1), | (11) |
| | | P(Xi) = TokenDecode(Hl). | (13) |
Fine-tuning
We use LoRA39 to fine-tune the pre-trained model on a target dataset. The core strategy of LoRA is to inject trainable low-rank decomposition matrices into specific layers of the GPT architecture, thereby reducing the number of trainable parameters for downstream tasks. For each layer, a linear layer is introduced to compress the features from dimension d to r, and then expand thm back from dimension r to d (adding the feature dimension of the next layer of dimension d). Finally, the original features of the model are then combined with the LoRA-derived features to produce the final output. Here, r is the rank in LoRA, where r ≪ d. Assuming that the pre-training model weight parameter matrix is W0 ∈ Rd*k, and Wl is the LoRA weight parameter matrix, the parameter update of LoRA can be expressed through eqn (15):| | | W0 + Wl = W0 + L1L2,L1 ∈ Rd*rL2 ∈ Rr*d | (14) |
During the training process, the parameters W0 are frozen, and only the parameters in L1 and L2 are updated. To maintain the original output of the network at the start of training while ensuring better convergence during learning, we follow the LoRA parameter initialization method. The parameters in L1 are initialized with a Gaussian distribution, and those in L2 are initialized to 0. If both matrices are initialized to 0 at the same time, all neurons will be initially equivalent and may easily cause the gradient to disappear. If all initialization is Gaussian, an excessively large offset will be obtained in the initial stage of model training, and too much noise will be introduced, which could hinder model convergence. By using these methods, we have fine-tuned our model on datasets such as MOSES and ZINC, enhancing its adaptability to specific data.
Reinforcement learning
The concise process of RL is illustrated in Fig. 1(e), where the model is continually refined through feedback from the scorer. However, fully updating model parameters in RL can easily lead to model collapse. To address this, we use the LoRA method for fine-tuning. We freeze the pre-trained model during parameters update, focusing solely on updating the LoRA parameters. Then we designed a reward function (reward model) based on proximal policy (PPO) with multiple objectives. We have set up a public reward function, a docking reward function, and a medicinal property reward function. The public reward function scores whether a legal molecule is generated and provides a score S(comment). The docking reward function uses karmadock to dock the generated molecule, and its docking score is compared with that of the reference molecule to obtain S(docking). For drug properties, we calculated the QED and SA of the generated molecule and compared them with those of the reference molecule to obtain S(drug). The final score S(m) is the sum of these three reward function scores:| | | S(m) = S(comment) + S(docking) + S(drug). | (15) |
The process of establishing the reward function involves inputting a specific task and calculating a penalty term for the difference between generated and reference molecules. The penalty term is sued to punish or reward RL for any deviation from the defined optimization goals within each training batch. The aim is to ensure that the model generates molecules that align with the set optimization goals. Finally, the model is optimized based on the reward index of the current batch of data to guide the training of LoRA accordingly.
Experimental setup
For the pre-training model, a 12-layer GPT2 model with a hidden dimension of 768 and an attention head of 8 is used as the backbone architecture. In order to make the model convergence more robust, we use the cosine annealing algorithm52 to dynamically adjust the optimization learning rate, with an initial learning rate of 0 and a final learning rate of 1 × 10−4, and use AdamW for parameter optimization. During the training phase, the hyperparameter batch size was set to 32 × 8, and 3 epochs were trained on 8 A100 GPUs. In the LoRA fine-tuning phase, we set the dimension r of the update mean matrix to 16, the scaling factor to 32, and trained for 10 epochs. The scaling factor is to adjust the amplitude of the update matrix, with a higher factor exerting a greater influence on model parameters. RL uses the same LoRA settings as the fine-tuning stage. Benchmarking involved fine-tuning models: de novo design on Moses, side chain optimizaiton and scaffold hopping on PdbBind, and linker design and R-group exploration on ZINC. Except for the de novo design task, which generates 30000 molecules for testing on Moses, the other tasks generate 250 molecules for each test pair.
Baselines
In our study, we evaluated a diverse array of models for different molecular design tasks. For de novo design, we examined CharRNN,42 VAE,43 AAE,44 LatentGAN,46 JT-VAE,45 MolGPT,2 and cMolGPT.41 For linker design, our comparisons included DiffLinker,10 DeLinker,8 3DLinker,53 and DEVELOP.14 For R-group design, we assessed DeLinker and DEVELOP. Since scaffold hopping design lacks established benchmarks, we solely compared with DiffHopp.50 To assess the effectiveness of our models in the de novo design task, we generated 30000 molecules for evaluation. For the other tasks, we sampled 250 molecules per test pair. The evaluation metrics, including validity, uniqueness and novelty were aligned with the findings reported in the original papers.2,41 Additionally, for traceability, SA, plogP and QED, we relied on the results reported in the FFLOM paper.21
Metrics
Evaluation of medicinal properties.
SA score. This parameter is used to evaluate the complexity of synthesized compounds,54 where lower the SA scores, the easier the molecules are to be synthesized. QED score. It is a probability value that reflects the likelihood of a compound being a potential drug candidate, with a score closer to 1 indicating a higher potential. plogP score. This score, penalized by ring size and synthetic reachability, is predicted using the model reported by You et al.55 A higher plogP score indicates better overall properties. For the de novo task, we used the MOSES evaluation metric.
Validity.
RDKit's molecular structure parser is usually used to determine the validity of a molecule. Validity refers to the valid molecule Gv in the generated set Gm.| | 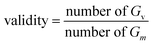 | (16) |
Uniqueness@k.
In the generated set Gm, we calculated the proportion of Gu after removing duplicated molecules for the first K = 1000 and K = 10000.| | 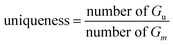 | (17) |
Novelty.
As for the proportion of molecules Gn in the generated set Gm that are not present in the training set, a lower novelty score may indicate overfitting of the model.| | 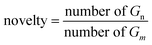 | (18) |
Internal diversity (IntDiv).
By calculating the power (r) mean of the Tanimoto similarity (TM) across all SMILES molecules in the generated set Gm, the internal diversity of the generated molecules is evaluated. We evaluated the internal diversity when r is 1 and 2 simultaneously.| | 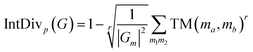 | (19) |
Fragment similarity (Frag).
This parameter uses cosine similarity to measure the BRICS fragment pairs of molecules within the generation set G and the training set T.| | | Frag(Gm,Tm) = 1 − cos(F(Gm),F(Gt)) | (20) |
Nearest neighbor similarity (SNN).
By calculating the average Tanimoto similarity TM(ma,mb) between a molecule ma in the generated set Gm and the nearest neighbor molecule mb in the training set Tm. If the generated molecule is far away from the training set, the similarity to its nearest neighbor will be lower.| | 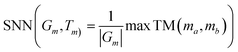 | (21) |
For the linker, R-group, and sidechain tasks, in addition to the validity, uniqueness, novelty, and drug feasibility evaluations, traceability evaluations were also performed. Recovery refers to the proportion of test data in the test set(test) that can regenerate the same test case set(G) as the ground truth.
| | 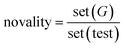 | (22) |
In the case study, we also calculated docking scores and applied the structural filtering strategy proposed by Imrie
et al.8 to the generated molecules.
Docking score.
We use karmadock56 to calculate the docking scores for generated molecules, serving as a component in developing a comprehensive molecule evaluation reward model within the RL framework. Karmadock, proposed by Zhang et al.,56 can quickly and accurately predict protein-ligand binding conformation and affinities. Given the need for rapid yet accurate evaluation models in RL, Karmadock stands out as a fast alternative while maintaining comparable accuracy to traditional docking methods.
Reference molecules in real-world drug design
Linker design.
MALT1, a crucial regulator of the immune system, plays a pivotal role in lymphocyte antigen-dependent responses and orchestrates the NF-κB signaling pathway. Inspired by the discovery of the nanomolar selective allosteric inhibitors MLT-748 and MLT-747 of MALT1 reported by Quancard et al.,57 we took MLT-747 as a reference molecule for the linker design task. The properties of MLT-747 are as follows: a docking score of 41.64, along with the QED and SA values of 0.57 and 3.47, respectively.
R-group exploration.
The association of the TRPM8 channel with cold pain induced by oxaliplatin and nerve damage was elucidated by Zhao et al., revealing its therapeutic potential for mitigating migraine and inflammation-induced cold hyperalgesia.58 Additionally, Bianchini et al. developed potent antagonists targeting TRPM8 for ocular pain relief, with compound 51 (N-alkoxyamide derivative) demonstrating notable pharmacological efficacy.59 Building upon this groundwork, our investigation focused on the R-group substitution based on the scaffold of compound 51. Compound 51 served as the reference with a docking score of 17.17, QED of 0.92, and SA of 2.87.
Scaffold hopping.
Hu et al. identified 11 scaffold hopping fragment pairs from the structures of known CDK9 inhibitors, excluding those designated as CDK9 hinge region.60 By augmenting the number of terminal fragments, they generated thousands of novel molecules containing the input fragments. Notably, one of these compounds, compound 4, displayed similar hydrogen bonds and end fragments to those of a known inhibitor BAY-114357. BAY-114357 served as the reference, with a docking score of 23.39, QED of 0.72, and SA of 2.68.
PROTAC design.
BRD4, a crucial factor in transcriptional and epigenetic regulation, harbors bromodomains essential for embryogenesis and cancer progression. Gadd et al. introduced MZ1, a PROTAC degrader targeting BRD4, which effectively induced protein degradation, impeded BRD4's binding to the acetylated markers and disrupted gene transcriptional regulation.61 In this study, we took MZ1 as a reference PROTAC and modified its linker using FragGPT. The properties of MZ1 are as follows: a docking score of 40.00, QED of 0.07, and SA of 5.06.
Data availability
The code and data used in the study are publicly available from the GitHub repository: https://github.com/pengbingxin/FragGPT-Interface.
Author contributions
J. Y., T. J. H, J. K. W., J. Y. J., and Y. C. designed the research study. Y. C. and B. X. P. developed the method and wrote the code. B. X. P., J. K. W. and J. Y. J. performed the analysis. B. X. P., J. K. W., J. Y. J., J. Y, and T. J. H, wrote the paper. All authors read and approved the manuscript.
Conflicts of interest
The authors declare that they have no competing interests.
Acknowledgements
This work was financially supported by National Key Research and Development Program of China (2022YFF1203003), National Natural Science Foundation of China (22220102001, 22303083), China Postdoctoral Science Foundation (2023M733128, 2023TQ0285), and Postdoctoral Fellowship Program of CPSF (GZB20230657).
References
- D. Weininger, J. Chem. Inf. Comput. Sci., 1988, 28, 31–36 CrossRef CAS.
- V. Bagal, R. Aggarwal, P. Vinod and U. D. Priyakumar, J. Chem. Inf. Model., 2021, 62, 2064–2076 CrossRef PubMed.
-
J.-N. Wu, T. Wang, Y. Chen, L.-J. Tang, H.-L. Wu and R.-Q. Yu, arXiv, 2023, preprint, arXiv:2301.01829, DOI:10.48550/arXiv.2301.01829.
- J. Wang, C.-Y. Hsieh, M. Wang, X. Wang, Z. Wu, D. Jiang, B. Liao, X. Zhang, B. Yang and Q. He, Nat. Mach. Intell., 2021, 3, 914–922 CrossRef.
- T. Blaschke, J. Arús-Pous, H. Chen, C. Margreitter, C. Tyrchan, O. Engkvist, K. Papadopoulos and A. Patronov, J. Chem. Inf. Model., 2020, 60, 5918–5922 CrossRef CAS PubMed.
-
T. Fu, C. Xiao, X. Li, L. M. Glass and J. Sun, arXiv, 2021, preprint, arXiv:2010.02318, DOI:10.48550/arXiv.2010.02318.
- Ł. Maziarka, A. Pocha, J. Kaczmarczyk, K. Rataj, T. Danel and M. Warchoł, J. Cheminf., 2020, 12, 1–18 Search PubMed.
- F. Imrie, A. R. Bradley, M. van der Schaar and C. M. Deane, J. Chem. Inf. Model., 2020, 60, 1983–1995 CrossRef CAS PubMed.
-
D. P. Kingma and M. Welling, arXiv, 2013, preprint, arXiv:1312.6114, DOI:10.48550/arXiv.1312.6114.
-
I. Igashov, H. Stärk, C. Vignac, V. G. Satorras, P. Frossard, M. Welling, M. Bronstein and B. Correia, arXiv, 2022, preprint, arXiv:2210.05274, DOI:10.48550/arXiv.2210.05274.
- T. Sterling and J. J. Irwin, J. Chem. Inf. Model., 2015, 55, 2324–2337 CrossRef CAS PubMed.
- M. Su, Q. Yang, Y. Du, G. Feng, Z. Liu, Y. Li and R. Wang, J. Chem. Inf. Model., 2018, 59, 895–913 CrossRef PubMed.
- S. Axelrod and R. Gomez-Bombarelli, Sci. Data, 2022, 9, 185 CrossRef CAS PubMed.
- F. Imrie, T. E. Hadfield, A. R. Bradley and C. M. Deane, Chem. Sci., 2021, 12, 14577–14589 RSC.
-
A. E. Klon, Fragment-Based Methods in Drug Discovery, Springer, 2015 Search PubMed.
- O. Ichihara, J. Barker, R. J. Law and M. Whittaker, Mol. Inf., 2011, 30, 298–306 CrossRef CAS PubMed.
- S. R. Langdon, P. Ertl and N. Brown, Mol. Inf., 2010, 29, 366–385 CrossRef CAS PubMed.
- H.-J. Böhm, A. Flohr and M. Stahl, Drug Discovery Today: Technol., 2004, 1, 217–224 CrossRef PubMed.
- R. I. Troup, C. Fallan and M. G. Baud, Explor Target Antitumor Ther., 2020, 1, 273 Search PubMed.
- J. Li and J. Liu, ChemistrySelect, 2020, 5, 13232–13247 CrossRef CAS.
- J. Jin, D. Wang, G. Shi, J. Bao, J. Wang, H. Zhang, P. Pan, D. Li, X. Yao and H. Liu, J. Med. Chem., 2023, 66, 10808–10823 CrossRef CAS PubMed.
- A. Radford, J. Wu, R. Child, D. Luan, D. Amodei and I. Sutskever, OpenAI blog, 2019, 1, 9 Search PubMed.
-
A. Radford, K. Narasimhan, T. Salimans and I. Sutskever, Improving language understanding by generative pre-training, 2018, preprint at https://s3-us-west-2.amazonaws.com/openai-assets/research-covers/language-unsupervised/language_understanding_paper.pdf.
- T. Brown, B. Mann, N. Ryder, M. Subbiah, J. D. Kaplan, P. Dhariwal, A. Neelakantan, P. Shyam, G. Sastry and A. Askell, Adv. Neural Inf. Process Syst., 2020, 33, 1877–1901 Search PubMed.
- L. Ouyang, J. Wu, X. Jiang, D. Almeida, C. Wainwright, P. Mishkin, C. Zhang, S. Agarwal, K. Slama and A. Ray, Adv. Neural Inf. Process Syst., 2022, 35, 27730–27744 Search PubMed.
-
H. Touvron, L. Martin, K. Stone, P. Albert, A. Almahairi, Y. Babaei, N. Bashlykov, S. Batra, P. Bhargava and S. Bhosale, arXiv, 2023, preprint, arXiv:2307.09288, DOI:10.48550/arXiv.2307.09288.
-
Z. Liu, H. Hu, Y. Lin, Z. Yao, Z. Xie, Y. Wei, J. Ning, Y. Cao, Z. Zhang, L. Dong, F. Wei and B. Guo, Swin Transformer V2: Scaling Up Capacity and Resolution, 2022 IEEE/CVF Conference on Computer Vision and Pattern Recognition (CVPR), 2022, pp. 11999–12009 Search PubMed.
-
L. Dong, S. Xu and B. Xu, Speech-Transformer: A No-Recurrence Sequence-to-Sequence Model for Speech Recognition, 2018 IEEE International Conference on Acoustics, Speech and Signal Processing (ICASSP), IEEE Press, Calgary, AB, Canada, 2018, pp. 5884–5888 Search PubMed.
-
F. Sun, J. Liu, J. Wu, C. Pei, X. Lin, W. Ou and P. Jiang, arXiv, 2019, preprint, arXiv:1904.06690, DOI:10.48550/arXiv.1904.06690.
- P. Schwaller, T. Laino, T. Gaudin, P. Bolgar, C. A. Hunter, C. Bekas and A. A. Lee, ACS Cent. Sci., 2019, 5, 1572–1583 CrossRef CAS PubMed.
-
D. Xue, H. Zhang, D. Xiao, Y. Gong, G. Chuai, Y. Sun, H. Tian, H. Wu, Y. Li and Q. Liu, bioRxiv, 2020, preprint, DOI:10.1101/2020.12.23.424259.
-
F. Wu, D. Radev and S. Z. Li, Molformer: motif-based transformer on 3D heterogeneous molecular graphs, Proceedings of the Thirty-Seventh AAAI Conference on Artificial Intelligence and Thirty-Fifth Conference on Innovative Applications of Artificial Intelligence and Thirteenth Symposium on Educational Advances in Artificial Intelligence, AAAI Press, 2023, p. 593 Search PubMed.
-
S. Chithrananda, G. Grand and B. Ramsundar, arXiv, 2020, preprint, arXiv:2010.09885, DOI:10.48550/arXiv.2010.09885.
-
S. Wang, Y. Guo, Y. Wang, H. Sun and J. Huang, SMILES-BERT: Large Scale Unsupervised Pre-Training for Molecular Property Prediction, Proceedings of the 10th ACM International Conference on Bioinformatics, Computational Biology and Health Informatics, Association for Computing Machinery, New York, NY, USA, 2019, pp. 429–436 Search PubMed.
-
W. Ahmad, E. Simon, S. Chithrananda, G. Grand and B. Ramsundar, arXiv, 2022, preprint, arXiv:2209.01712, DOI:10.48550/arXiv.2209.01712.
- D. Flam-Shepherd, K. Zhu and A. Aspuru-Guzik, Nat. Commun., 2022, 13, 3293 CrossRef CAS PubMed.
-
J. Schulman, F. Wolski, P. Dhariwal, A. Radford and O. Klimov, arXiv, 2017, preprint, arXiv:1707.06347, DOI:10.48550/arXiv.1707.06347.
- P. Ertl and A. Schuffenhauer, J. Cheminf., 2009, 1, 8 Search PubMed.
-
E. J. Hu, Y. Shen, P. Wallis, Z. Allen-Zhu, Y. Li, S. Wang, L. Wang and W. Chen, arXiv, 2021, preprint, arXiv:2106.09685, DOI:10.48550/arXiv.2106.09685.
- D. Polykovskiy, A. Zhebrak, B. Sanchez-Lengeling, S. Golovanov, O. Tatanov, S. Belyaev, R. Kurbanov, A. Artamonov, V. Aladinskiy and M. Veselov, Front. Pharmacol, 2020, 11, 565644 CrossRef CAS PubMed.
- Y. Wang, H. Zhao, S. Sciabola and W. Wang, Molecules, 2023, 28(11), 4430 CrossRef CAS PubMed.
- K. Preuer, P. Renz, T. Unterthiner, S. Hochreiter and G. Klambauer, J. Chem. Inf. Model., 2018, 58, 1736–1741 CrossRef CAS PubMed.
- R. Gómez-Bombarelli, J. N. Wei, D. Duvenaud, J. M. Hernández-Lobato, B. Sánchez-Lengeling, D. Sheberla, J. Aguilera-Iparraguirre, T. D. Hirzel, R. P. Adams and A. Aspuru-Guzik, ACS Cent. Sci., 2018, 4, 268–276 CrossRef PubMed.
- D. Polykovskiy, A. Zhebrak, D. Vetrov, Y. Ivanenkov, V. Aladinskiy, P. Mamoshina, M. Bozdaganyan, A. Aliper, A. Zhavoronkov and A. Kadurin, Mol. Pharm., 2018, 15, 4398–4405 CrossRef CAS PubMed.
-
W. Jin, R. Barzilay and T. Jaakkola, arXiv, 2018, preprint, arXiv:1802.04364, DOI:10.48550/arXiv.1802.04364.
- O. Prykhodko, S. V. Johansson, P.-C. Kotsias, J. Arús-Pous, E. J. Bjerrum, O. Engkvist and H. Chen, J. Cheminf., 2019, 11, 1–13 Search PubMed.
-
K. Madhawa, K. Ishiguro, K. Nakago and M. Abe, arXiv, 2019, preprint, arXiv:1905.11600, DOI:10.48550/arXiv.1905.11600.
- B. Zdrazil, E. Felix, F. Hunter, E. J. Manners, J. Blackshaw, S. Corbett, M. de Veij, H. Ioannidis, D. M. Lopez and J. F. Mosquera, Nucleic Acids Res., 2024, 52, D1180–D1192 CrossRef PubMed.
- Z. Liu, M. Su, L. Han, J. Liu, Q. Yang, Y. Li and R. Wang, Acc. Chem. Res., 2017, 50, 302–309 CrossRef CAS PubMed.
-
J. Torge, C. Harris, S. V. Mathis and P. Lio, arXiv, 2023, preprint, arXiv:2308.07416, DOI:10.48550/arXiv.2308.07416.
- G. W. Bemis and M. A. Murcko, J. Med. Chem., 1996, 39, 2887–2893 CrossRef CAS PubMed.
-
I. Loshchilov and F. Hutter, arXiv, 2018, preprint, arXiv:1711.05101, DOI:10.48550/arXiv.1711.05101.
-
Y. Huang, X. Peng, J. Ma and M. Zhang, arXiv, 2022, preprint, arXiv:2205.07309, DOI:10.48550/arXiv.2205.07309.
- P. Ertl and A. Schuffenhauer, J. Cheminf., 2009, 1, 1–11 Search PubMed.
- J. You, B. Liu, Z. Ying, V. Pande and J. Leskovec, Adv. Neural Inf. Process Syst., 2018, 31, 6412–6422 Search PubMed.
- X. Zhang, O. Zhang, C. Shen, W. Qu, S. Chen, H. Cao, Y. Kang, Z. Wang, E. Wang and J. Zhang, Nat. Comput. Sci., 2023, 3, 789–804 CrossRef CAS PubMed.
- J. Quancard, T. Klein, S.-Y. Fung, M. Renatus, N. Hughes, L. Israël, J. J. Priatel, S. Kang, M. A. Blank and R. I. Viner, Nat. Chem. Biol., 2019, 15, 304–313 CrossRef CAS PubMed.
- C. Zhao, Y. Xie, L. Xu, F. Ye, X. Xu, W. Yang, F. Yang and J. Guo, Nat. Commun., 2022, 13, 3113 CrossRef CAS PubMed.
- G. Bianchini, M. Tomassetti, S. Lillini, A. Sirico, S. Bovolenta, L. Za, C. Liberati, R. Novelli and A. Aramini, J. Med. Chem., 2021, 64, 16820–16837 CrossRef CAS PubMed.
- L. Hu, Y. Yang, S. Zheng, J. Xu, T. Ran and H. Chen, J. Chem. Inf. Model., 2021, 61, 4900–4912 CrossRef CAS PubMed.
- M. S. Gadd, A. Testa, X. Lucas, K.-H. Chan, W. Chen, D. J. Lamont, M. Zengerle and A. Ciulli, Nat. Chem. Biol., 2017, 13, 514–521 CrossRef CAS PubMed.
|
| This journal is © The Royal Society of Chemistry 2024 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence bc,
Xiangyang
Ji
d,
Chang-Yu
Hsieh
bc,
Xiangyang
Ji
d,
Chang-Yu
Hsieh