
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicencePhoto-controlled order-to-order host–guest self-assembly transfer for an afterglow effect with water resistance†
Mouwei
Liu
a,
Bin
Wu
a,
Glib V.
Baryshnikov
 b,
Shen
Shen
a,
Hao
Sun
a,
Xinyan
Gu
a,
Hans
Ågren
c,
Yifei
Xu
a,
Qi
Zou
*d,
Da-Hui
Qu
d and
Liangliang
Zhu
*a
b,
Shen
Shen
a,
Hao
Sun
a,
Xinyan
Gu
a,
Hans
Ågren
c,
Yifei
Xu
a,
Qi
Zou
*d,
Da-Hui
Qu
d and
Liangliang
Zhu
*a
aDepartment of Macromolecular Science, State Key Laboratory of Molecular Engineering of Polymers, Fudan University, Shanghai 200438, China. E-mail: zhuliangliang@fudan.edu.cn
bDepartment of Science and Technology, Laboratory of Organic Electronics, Linköping University, Norrköping, 60174 Sweden
cDepartment of Physics and Astronomy, Uppsala University, Box 516, Uppsala, SE-751 20, Sweden
dKey Laboratory for Advanced Materials and Feringa Nobel Prize Scientist Joint Research Center, Frontiers Science Center for Materiobiology and Dynamic Chemistry, School of Chemistry and Molecular Engineering, East China University of Science and Technology, Shanghai 200237, China. E-mail: zouqi@ecust.edu.cn
First published on 2nd July 2024
Abstract
Due to the general incompleteness of photochemical reactions, the photostationary structure in traditional photo-controlled host–guest self-assembly transfer is usually disordered or irregular. This fact readily affects the photoregulation or improvement of related material properties. Herein, a photoexcitation-induced aggregation molecule, hydroxyl hexa(thioaryl)benzene (HB), was grafted into β-cyclodextrin to form a host–guest system. Upon irradiation, the excited state conformational change of HB can drive an order-to-order phase transition of the system, enabling the transfer of the initial linear nanostructure to a photostationary worm-like nanostructure with orderliness and crystallinity capability. Along with the photoexcitation-controlled phase transition, an afterglow effect was obtained from the films prepared by doping the host–guest system into poly(vinyl alcohol). The afterglow effect had a superior water resistance, which successfully overcame the general sensitivity of doped materials with the afterglow effect to water vapor. These results are expected to provide new insights for pushing forward chemical self-assembly from the light perspective, towards materials with superior and stable properties under light treatment.
Introduction
Using host–guest recognition for constructing supramolecular systems with different topologies is a typically effective way to construct functional nanomaterials.1–7 Modulation of host–guest assemblies using various factors, such as host–guest species and ratios, solvent composition, temperature, and so forth, has been well explored in related structure and functional tuning.8,9 The use of light to change supramolecular host–guest assemblies provides excellent flexibility, as it can be immediately applied and transported to a precise location in the form of different wavelengths, thus exhibiting good spatial precision and on-demand tunability.10–12Conventional photoresponsive host–guest systems are constructed as follows: guest molecules are first encapsulated into host molecules, and then the guest molecules undergo photochemical isomerization under irradiation to drive the change of the whole assembly (e.g., a typical example is the cis–trans isomerization of azobenzene). However, conventional light-controlled host–guest systems focus on switching between assembly and disassembly, rather than order-to-order phase transitions.13–17 As ordered structures often exhibit unique functionalities,18–22 in this regard, it has remained an intriguing challenge for implementing photo-controlled host–guest systems with remarkable ordered nanostructures.
As a result, we explored the causes of the above problems in detail. Firstly, traditional photoresponsive host–guest systems were based on ground-state self-assembly with photochemical principles. The efficiency of chemical reactions such as photoisomerization cannot reach 100% and side reactions are likely to occur,23 so the transformation of the assemblies upon light exposure is usually incomplete and defective. Conversely, the photoresponse of hexa(thioaryl)benzene, employed herein, is a photophysical process, relying on the change in molecular conformation to drive the change of the assembled system. The long lifetime of the excited state of hexa(thioaryl)benzene ensures that the molecules have sufficient time to collide and interact before relaxing, and thus the self-assembly occurs, namely, the excited-state self-assembly.24 Secondly, the difference in molecular size change can have a significant impact on the control of self-assembly and phase transition. The huge geometrical variations in the conventional photoresponsive moieties lead to very large differences in the corresponding assemblies, which makes the assembly transformation easily uncontrollable. For example, the large geometrical change of azobenzene accompanying photoisomerization is revealed by the substantial variation in the distance between the carbon atoms in the para positions of the phenyl rings that decreases from 9.9 to 5.5 Å.25 In contrast, hexa(thioaryl)benzenes are typically characterized by significant differences in ground-state and excited-state conformation. Upon irradiation, their multiple C–S–C bonds rotate synchronously. The dihedral angle between the side-chain benzene ring and the central benzene ring changes from 120° to 90°, and the side-chain spacing is simply reduced from 5.0 to 4.1 Å.26 Thirdly, the photochemical cases are essentially bistable processs regulated by the two ratios of the isomers,27–29 and the activation energy of isomer transformation is often large.30 Consequently, the switching of the mixing ratio of the two isomers corresponds to the assemblies, and the entire process is discontinuous. Therefore, the phase change upon irradiation is difficult to predict and design.31–36 Instead, for our model, due to only involving conformational changes, the energy barrier required for light control is relatively low, so the entire process is continuous and more controllable over irradiation time.37–41
Considering the significantly different properties of hexa(thioaryl)benzene from conventional photoresponsive moieties, we expect that such a driving force can fully utilize photons and play a powerful role in maintaining the orderliness of host–guest self-assembly transfer. Cyclodextrins (CyDs) have excellent molecular binding ability to various substrates in aqueous solution, and many molecular-scale nanostructures with unique topological features can be derived from them.42,43 We here covalently linked hydroxyl hexa(thioaryl)benzene (HB) and β-CyD to construct a host–guest system HB-CyD with a photoexcitation induced assembly (PEIA) effect (see chemical structures in Fig. 1a). The ordered phase transition from a one-dimensional linear nanostructure to a worm-like nanostructure could be achieved by unique photoirradiation without other stimuli and components. The fine structure and kinetic processes of this host–guest self-assembly transfer as UV irradiation were comprehensively investigated using circular dichroism (CD), a cryo-Transmission Electron Microscope (cryo-TEM), and grazing-incidence small-angle X-ray scattering (GISAXS). In terms of excellent phosphorescence properties of the HB luminophore, poly(vinyl alcohol) (PVA) thin films doped with the worm-like nanostructure of HB-CyD after irradiation have a largely prolonged photoluminescence lifetime to show an afterglow effect characteristic, which also exerts excellent resistance to water vapor owing to the protective effect brought by the specific host–guest self-assembly fashion.
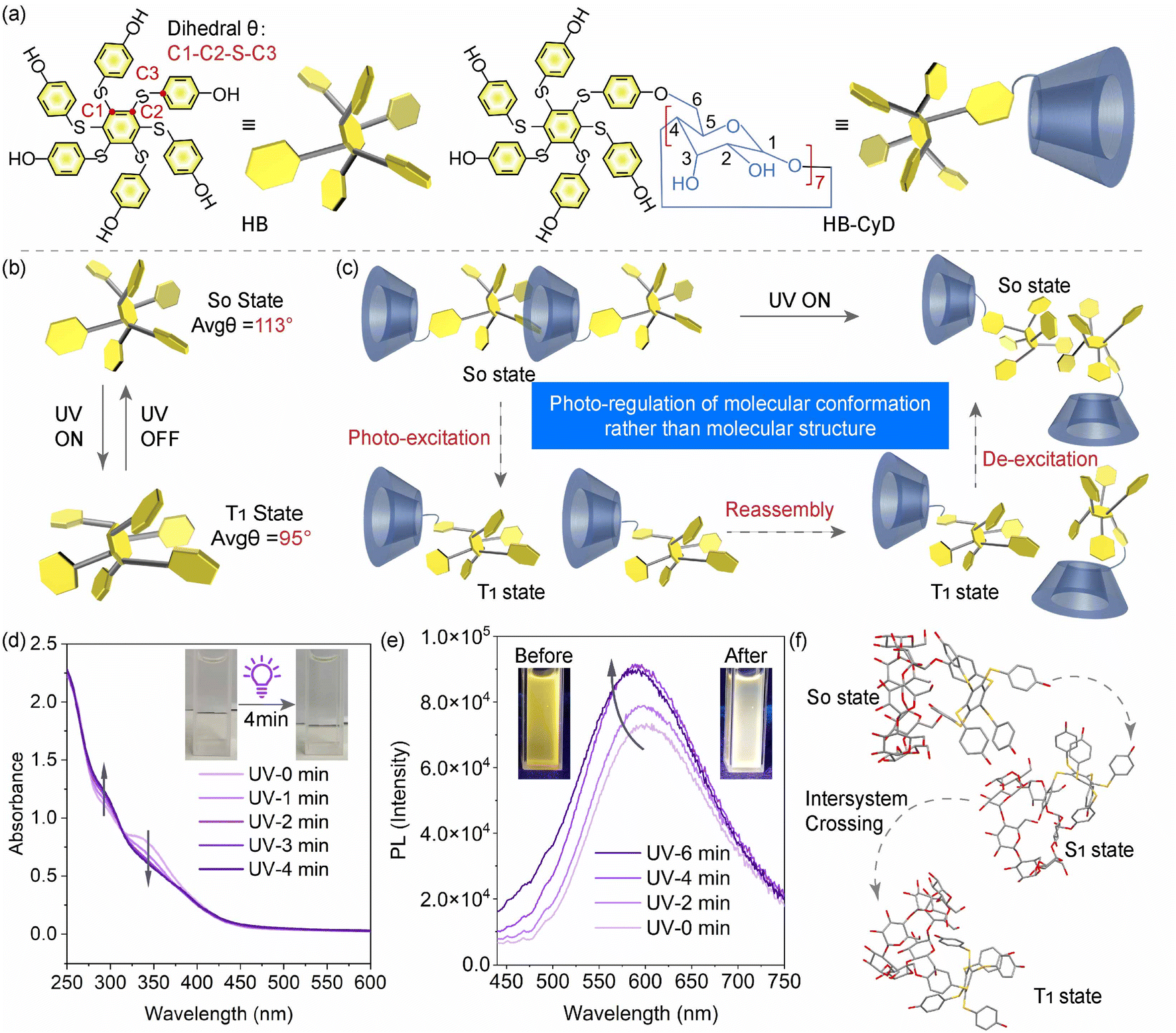 | ||
| Fig. 1 Molecular-level study. (a) Chemical structures of HB and HB-CyD. Illustration of the conformational change upon photoexcitation of (b) HB and (c) HB-CyD. (d) UV-vis absorbance spectral changes of aqueous HB-CyD solution upon UV irradiation for 0–4 minutes at 298 K (concentration: 0.05 mg mL−1). Inset: the corresponding photographs under ambient light before and after UV irradiation. (e) Emission spectra of HB-CyD (concentration: 1 mg mL−1, λex = 365 nm, 298 K) upon irradiation with UV light. Inset: the corresponding photographs under UV light before and after UV irradiation. (f) Calculated geometries of HB-CyD at the ground (S0 state) and the excited states (S1 and T1 states), respectively. | ||
Results
Molecular-level study
First, we investigated the photophysical properties of HB before and after grafting it to β-CyD. Since β-CyD has no absorption in the UV-visible region, the UV-vis absorption wavelengths of HB and HB-CyD in solution are essentially the same (Fig. S1†). HB-CyD powder shows stronger absorption in the 500–800 nm spectral region than HB powder (Fig. S1†). The emission (λmax = 603 nm) of HB-CyD powder also shows an apparent red shift compared with the maximum emission of HB (λmax = 519 nm) and a slight increase in PL lifetime (Fig. S2†). This is due to the fact that after attaching to β-CyD, the HB skeleton is squeezed by the hydrogen bonding environment of β-CyD with a tighter stacking, which restricts the molecular vibration and inhibits its non-radiative transition to a certain extent. These factors can lead to spectral redshift and increased luminescence lifetime. At the same time, the PL quantum yield decreased from 5.82% to 0.86% with the introduction of cyclodextrin (Fig. S3 and S4†).Theoretical calculations show that there is a significant difference between the ground state and excited state conformations of HB, i.e., the average dihedral torsion angle θ in the ground state conformation is much larger than that in the excited state conformation (113° for the S0 state and 95° for the T1 state, Fig. 1b), laying the basis for the possible PEIA for the phase transition of the host–guest self-assembly of HB-CyD (Fig. 1c and f). It was found that the UV absorptions at 290 nm and 345 nm of aqueous HB-CyD solution showed an increasing and decreasing trend, respectively (Fig. 1d), and the absorption did not change further once a photostationary state was reached within ∼4 minutes (Fig. S5†). The emission of aqueous HB-CyD solution is also gradually enhanced upon irradiation along with a microsecond-level PL lifetime (Fig. 1e and S6†), indicating an RTP characteristic. The emission of the HB-CyD solution can return to the initial state with simultaneous ultrasonic heating (Fig. S7†) and becomes weaker with longer UV irradiation because prolonged irradiation causes degradation of hexa(thioaryl)benzene (Fig. S8†). However, the emission is unchanged when UV light is applied to the solution at 77 K (Fig. S9†). This is because the molecular conformation is locked at low temperatures, making it difficult to deform even upon UV irradiation. These behaviors are in agreement with PEIA cases reported previously.26,37,38,40,41 The formation of singlet oxygen, indicated by the increase in the emission of SOSG (Singlet Oxygen Sensor Green) at about 533 nm wavelength, reflects the triplet-state behavior of HB (Fig. S10†).44–46 The NMR spectra of HB were unchanged during UV irradiation (Fig. S11†), signifying that the skeleton cannot undergo any molecular structural variation. The FTIR (Fourier transform infrared spectrometer) and Raman spectra of HB-CyD before and after irradiation were also consistent during irradiation (Fig. S12†). These results suggested that the photoexcitation-based control of HB-CyD is a typical physical process rather than a photo-chemical reaction. In contrast, no PEIA phenomenon can be observed in HB, which can be proved by undetected DLS (dynamic light scattering) and neglectable optical signal changes upon photoirradiation (Fig. S13†), simply because the solubility difference between the ground and excited states of HB is negligible.
Host–guest self-assembly
The chemical shift between 6 and 7 ppm is assigned to the hydrogen signal on the benzene ring on the HB skeleton in 1H NMR spectra (Fig. 2a). The NMR peaks gradually broaden with increasing irradiation time, indicating the aggregation of the HB unit in HB-CyD (Fig. 2a). In addition, the absence of other new stray peaks also suggests that no photochemical reaction occurs during the irradiation of the aqueous HB-CyD solution but is a purely physical process in which the assemblies are changed. To investigate the host–guest interaction between β-CyD and the HB skeleton, 2D NMR NOESY experiments of HB-CyD were carried out. The resonance in the HB part showed a cross peak that of the internal proton H3 of the β-CyD cavity (Fig. 2b), indicating that the HB skeleton enters the β-CyD cavity from the edge of the larger side of the cyclodextrin.47 If only HB and β-CyD are individually mixed in water, the two compounds essentially fail to interact with each other because of an extremely low binding constant (Fig. S14†). Due to the insolubility of HB in water, HB was dissolved in an aqueous sodium hydroxide solution at pH = 10 to form sodium phenol (abbreviated as HB-ONa). Even so, HB-ONa exhibited no interaction with cyclodextrin (Fig. S15†). The fact features the subtle design of HB covalently grafting into β-CyD. | ||
| Fig. 2 Host–guest self-assembly. (a) 1H NMR spectra of HB-CyD in the solution of 10% deuterated DMSO and 90% deuterated H2O upon UV irradiation (concentration: 4 mg mL−1). (b) 2D NOESY spectra of HB-CyD in the solution of 10% deuterated DMSO and 90% deuterated H2O before irradiation. (c) The circular dichroism spectra of aqueous HB-CyD solution upon UV irradiation for 3 min and with the addition of 1-adamantanol after UV irradiation. The inset: circular dichroism spectra of aqueous HB-CyD solution (concentration: 1 mg mL−1) upon UV irradiation for 3 min, 4 min, and 5 min, respectively. (d) Illustration of the assembled conformation of HB-CyD before and after UV irradiation. (e) Dynamic light scattering spectra of HB-CyD in the solution of 10% DMSO and 90% H2O as irradiation time increases (concentration: 0.1 mg mL−1). (f) Schematic diagram of the assembly structural changes before and after irradiation. | ||
To monitor the fine-structural change of HB-CyD upon irradiation, detailed characterization is performed by measuring CD spectra. Since the cyclodextrin cavities are chiral, a guest with UV-visible absorbance can produce an induced circular dichroism (ICD) signal via chirality transfer when it is passed into a cyclodextrin cavity. According to the general Harata–Kodaka's rule, a negative Cotton effect occurs (Fig. 2c) when the guest transition moment is located out-side but near the chiral body and parallel to the central axis. In contrast, a positive Cotton effect occurs when it is perpendicular. The ICD signal converges to zero when the angle between the guest transition moment and the body axis is 54.7°.48–52 Therefore, the change in the Cotton effect directly reflects the angle and positional relationship between the guest molecules and cyclodextrin.53–57 Before irradiation, the aqueous solution of HB-CyD produces a broad negative Cotton effect in the 320–450 nm region. Considering the relative size of the HB skeleton and β-CyD, the transition dipole moment of the HB molecule should be located close to the outer side of the narrow edge of the β-CyD cavity. The angle between the direction of the transition dipole moment and the central axis of the β-CyD should be less than 54.7°. Since the direction of the HB's transition dipole moment is calculated to be parallel to the plane of the central benzene ring (Fig. S16†), we deduced the assembly structure before irradiation as shown in Fig. 2d.
Upon irradiation, the broad negative CD signal peak gradually split into a pair with a positive peak at 400 nm and a negative one at 380 nm (Fig. 2c). Such a pair with a crossover point suggests that an overall positive Cotton effect appears, behaving as a positive ICD followed by a negative ICD on going from longer to shorter wavelengths.58 The split pair is an exciton-coupled ICD signal, which originates from the strong coupling of the electric transition dipole moments of the chromophores close to each other.54,59,60 Thus, the photostationary Cotton effect indicates that the HB skeletons are close to each other on the edges of β-CyD after irradiation. The positive signals indicate that HB's transition moment is at an angle of slightly more than 54.7° with the central axis of β-CyD. This is in accordance with the fact that after irradiation, the excited-state conformation of the HB skeleton shrinks relative to the ground-state conformation, thus resulting in the disassembly of HB from the β-CyD cavity. Subsequently, HB returns to the ground-state conformation with stacking with each other upon de-excitation, owing to the hydrophobicity of the skeleton and π–π interactions. The disappearance of the cross peaks after irradiation confirms the above inference (Fig. S17†). Then, the hydrophilic cyclodextrins wrap around the periphery of the stacked HB molecules (as illustrated in Fig. 2d).
However, if only HB and β-CyD are individually co-mixed, the ICD signals are nearly zero (Fig. S18†), indicating that there is basically no host–guest interaction in between, which further demonstrates the necessity of the covalent linkage of HB and β-CyD. Based on the above we deduced that the thermodynamic energy change of the process is shown in Fig. S19.† Moreover, we also investigated the ICD signal changes of aqueous HB-CyD solutions with different concentrations (Fig. S20 and S21†) and found that the signal intensity was linear with the concentration within a wide concentration range, suggesting a simple and flexible manipulability for preparing the host–guest system. This is also supported by the fact that the ICD signal change during irradiation at high concentrations is essentially similar to that at low concentrations (Fig. S22†).
While considering a larger-scale exploration, a dynamic light scattering (DLS) experiment was performed to show the kinetic radius of HB-CyD aggregates. It is found that the aggregates increased slightly during UV irradiation at low concentrations (Fig. 2e), whereas they were essentially unchanged at high concentrations (Fig. S23†). Together with the NMR study mentioned above and the CD study, we deduced that the self-assembly structure may have transformed, as shown in Fig. 2f. At low concentrations, the structural variability in between dominates to exhibit a difference in the radius of light scattering. However, at high concentrations, the aggregation size becomes so large that the resolution of the light-scattering radii is weakened.
Nanoscale phase transition study
To study the photo-controlled phase transition of HB-CyD at the nanoscale, cryo-TEM is used to observe the nanomorphology of HB-CyD before and after irradiation in aqueous solution in situ. Before irradiation, the nanomorphology of HB-CyD exhibits a one-dimensional linear structure with a length of about a few hundred nanometers (Fig. 3a). After irradiation, the nanoassembly becomes much shorter and curved to yield a worm-like structure, with a distance between the head and tail of about 16 nm (Fig. 3c). We also captured an intermediate state in which the two structures coexist (Fig. 3b), demonstrating that the transition from a one-dimensional linear structure to a worm-like structure upon irradiation is progressive. To account for the overall orderliness and homogeneity of the assembled structure before and after irradiation, grazing-incidence small-angle X-ray scattering (GISAXS) experiments were carried out on the dried film prepared from aqueous HB-CyD solution with different irradiation periods (Fig. 3d). In the absence of light irradiation, the scattering peak appeared at a peak position of 0.403 nm−1, which proved that the micro-zone size and spacing of the HB-CyD assembly had good regularity and homogeneity, and its long-period structure was calculated to be 15.6 nm according to Bragg's formula (Fig. 3e). After 2 minutes of irradiation, the peak position of the small-angle scattering shifted to 0.340 nm−1, and the long-period structure was enlarged to 18.5 nm (Fig. 3e) due to the simultaneous presence of worm-like and one-dimensional linear structures. The presence of both structures leads to a looser stacking than when only one-dimensional linear structures are present. Continuing the irradiation for 4 and 6 minutes, the scattering peak positions at 0.465 nm−1 and 0.470 nm−1, respectively, corresponding to the long-periodic structures with lessened dimensions (d = 13.5 nm and d = 13.4 nm, respectively) in turn (Fig. 3e). This is because both of them change to shorter worm-like structures, and the stacking becomes tight again.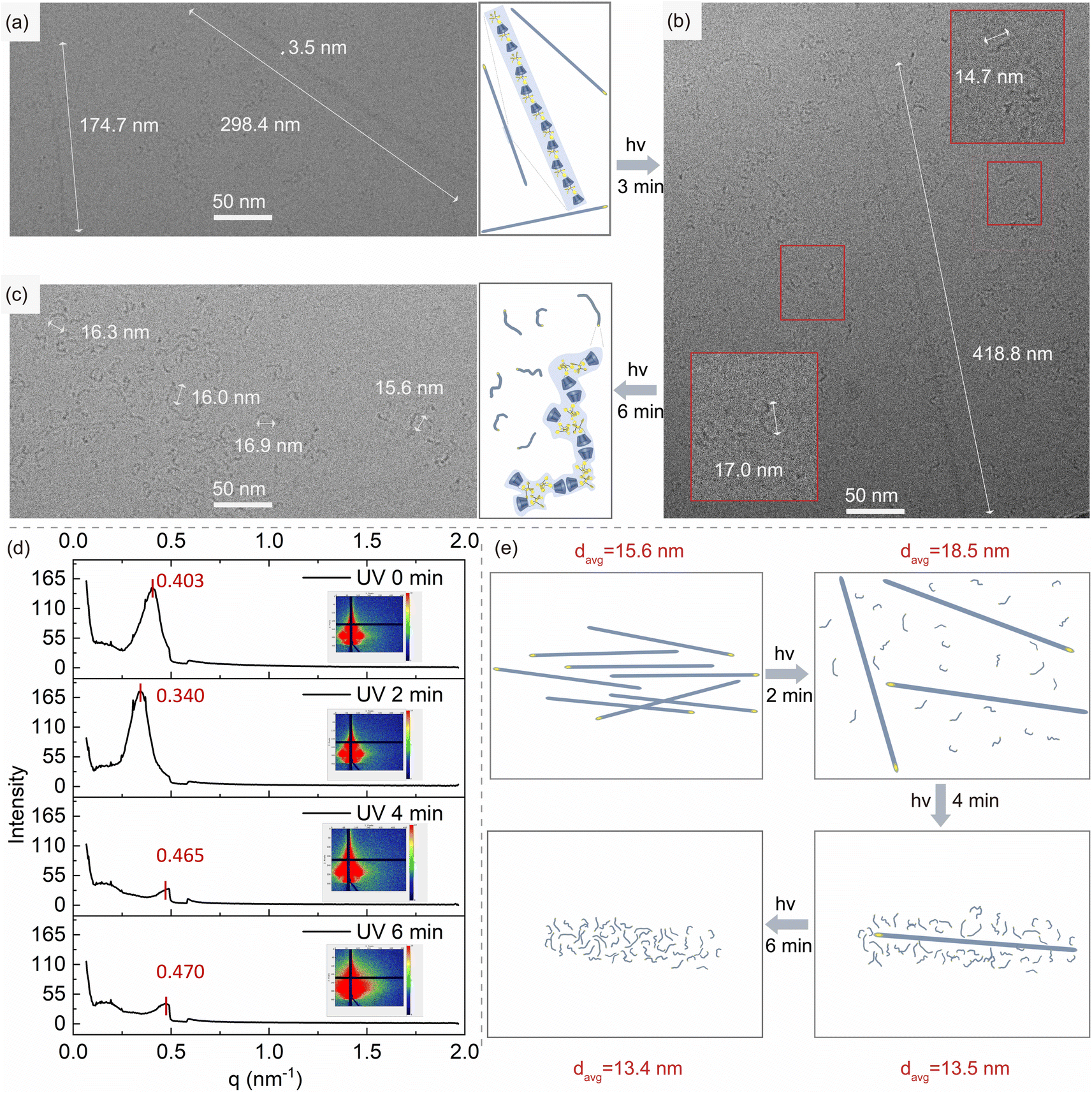 | ||
| Fig. 3 Nanoscale phase transition study. The cryo-TEM image of HB-CyD prepared from aqueous solution (a) before and after UV irradiation for (b) 3 min and (c) 6 min, respectively, as well as the corresponding structural diagram of HB-CyD assembly. (d) GISAXS integral curve and corresponding two-dimensional spectra (inset) of HB-CyD thin films upon irradiation with different periods. (e) Schematic diagram of the stacking of HB-CyD assemblies before and after irradiation at different periods. | ||
Crystalline structure via high-level self-assembly
Through an optical microscope, we can see that there are many large crystals before irradiation (Fig. 4a), which is caused by further assembly from the one-dimensional linear structure prepared from a highly concentrated sample. However, after irradiation for 2 minutes (Fig. 4b) and 4 minutes (Fig. 4c), the formed crystals are small and fine, simply due to the coexistence of two kinds of host–guest structures, enabling the high-level structure to become loose. After irradiation for 6 minutes (Fig. 4d), the system is filled with worm-like structures, and it can undergo further self-assembly into regular and large crystal particles. The photorefractive phenomenon can be observed from both the initial and the photostationary state because of their order crystalline characteristic. The same trend was also observed in the sample prepared from a lower concentration (Fig. S24†). In addition, through Atomic Force Microscopy (AFM) with a smaller scale (Fig. 4e and f and S25†), it is found that compared with the sample without UV irradiation, there are many regular small crystal particles with about hundreds of nanometers in size in the sample after irradiation, indicating that the worm-like structure with more than ten nanometers in size after irradiation is easier to stack to form small crystal particles. All these results suggest a nanoscale order-to-order host–guest structural photocontrol process, relying on the unique PEIA effect of HB and the host–guest design. | ||
| Fig. 4 Crystalline structure via high-level self-assembly. Morphology for aqueous HB-CyD solution with a higher concentration (1 mg mL−1) under optical micro-scope observation (a) before and after irradiation for (b) 2 min, (c) 4 min, and (d) 6 min, respectively. The photorefractive phenomenon can be seen from both the initial and the photostationary states. AFM images (height mode) of HB-CyD prepared from the aqueous solution with a lower concentration (0.1 mg mL−1) (e) before and (f) after UV irradiation. | ||
Functional film study
PVA is often used for the preparation of afterglow effect (a long-lasting emission effect that can still be observed with the naked eye after removing the excitation light source) photoluminescent films due to its confinement ability to luminophores by suppressing their non-radiative electronic pathways.61–63 However, previous studies show that planar luminophores doped in PVA can have good afterglow effect properties. In contrast, it is challenging for luminophores with twisted structures to obtain the afterglow effect due to molecules' relatively free vibration and rotation.64 Besides, due to the presence of a large number of hydrophilic hydroxyl groups in PVA, water vapor shows a quenching effect on the afterglow effect of films.65 Considering the order-to-order phase transition of HB-CyD before and after irradiation, we doped HB-CyD before and after irradiation with different periods with PVA and dried it from an aqueous solution to obtain films (Fig. 5a). The emission of these films remained at 560 nm, yet the PL lifetime increased from 2.61 ms to 53.80 ms upon irradiation at different periods (Fig. S26†), which can finally be witnessed by the naked eye after ceasing the excitation light source. We named the PVA film doped with HB-CyD without irradiation HB-CyD@PVA and the PVA film doped with HB-CyD after irradiation for 6 minutes HB-CyD-UV@PVA (as illustrated in Fig. 5a). HB-CyD@PVA films show little or no change in emission or lifetime after in situ irradiation (Fig. S27†). This is because the conformation of HB-CyD is already locked in the film, and it is tough to change the conformation by applying UV irradiation. In contrast, for an aqueous solution of only HB, a co-mixed solution of HB and β-CyD and a co-mixed solution of HB-ONa and β-CyD were employed for PVA doping, and the emissions of these films were almost the same. However, the PL lifetimes of these films were merely several milliseconds, no matter whether irradiated or not (Fig. S28–S30†), which cannot be observed by the naked eye after ceasing the excitation light source. These results well demonstrated the trade-off process to access the afterglow effect characteristic through delicate host–guest molecular design. | ||
| Fig. 5 Functional film study. (a) Schematic diagram of the fabrication of a functional PVA film by doping aqueous HB-CyD solution before and after irradiation. (b) The pattern of dragonfly and flower made of HB-CyD@PVA and HB-CyD-UV@PVA, respectively and the corresponding afterglow photos. (c) Emission spectra and (d) corresponding phosphorescence lifetime decay at 550 nm of the thin film of HB-CyD-UV@PVA treated under different humidity conditions for 2 days. (e) Schematic illustrating the application process of the water resistance of the HB-CyD-UV@PVA film with the excitation of a 365 nm lamp on and off. | ||
To further demonstrate the advantages of such a photocontrol strategy, we fabricated a pattern consisting of two components, in which the “flower bush” is made of the HB-CyD-UV@PVA film, while the “dragonfly” is made of the HB-CyD@PVA film. The whole pattern emits yellow phosphorescence under an excitation light source of 365 nm, and only the “flower bush” part can still be seen after the light source is withdrawn (Fig. 5b). Then, we placed the PVA film doped with HB-CyD after irradiation under different humidity conditions for 2 days. The emission lifetime was 51.01 ms, 44.79 ms, and 44.73 ms at 20%, 35%, and 50% humidity, respectively, indicating that the afterglow effect was still maintained. Meanwhile, the emission intensity of the film basically did not change (Fig. 5c and d). Only when the humidity reached 70%, the emission intensity reduced to a certain degree, and the PL lifetime declined to 28.72 ms, but it can still be witnessed after ceasing the excitation. These results illustrate the superior water resistance of the film, owing to the unique photo-regulated host–guest self-assembled structure. In another showcase of water resistance, we made a leaf pattern with the PVA film doped with HB-CyD after irradiation, and the afterglow effect was not burned out after spraying a large amount of water (Fig. 5e), indicating the stability of the photo-regulated host–guest self-assembled structure as well as the afterglow effect properties. Compared with the host–guest structure of HB-CyD without irradiation, the HB skeleton in the structure after irradiation is more tightly stacked with each other, and the β-CyD cavity can be better wrapped in the outer layer of the stacked HB molecules (as illustrated in Fig. 5a). In addition, the presence of a large number of hydroxyl groups on β-CyD enables it to interact with PVA via multiple H-bonding. These facts play significant roles in immobilizing the film system to behave with the stable afterglow effect characteristic with water resistance.
Conclusions
By covalently linking photoexcitation-induced aggregation active HB molecules with β-cyclodextrin, a host–guest system, HB-CyD, was constructed. Upon irradiation, the HB skeleton's excited-state conformation shrinks compared to its ground state, resulting in the HB skeleton detaching from the β-cyclodextrin cavity. Upon de-excitation, HB returns to ground-state conformation and stacks with each other due to its hydrophobicity and π–π interactions. Therefore, the assembly structure changes dramatically, driving the host–guest system's nanoscale order-to-order phase transition from a one-dimensional linear structure to a worm-like structure. On this basis, a PVA film doped with HB-CyD after irradiation exhibited stable afterglow effect characteristics with remarkable water resistance, successfully solving the general problem that doped materials with an afterglow effect are sensitive to water vapor. This work realizes the photo-controlled order-to-order phase transition in a simple system, which may be helpful for the construction of novel materials with predictable structures and properties under phototreatment.Data availability
Data are available on reasonable request from the authors.Author contributions
L. Z. and M. L. conceived the idea and designed the study. M. L. synthesized, purified and crystallized all compounds and performed general photophysical measurements with the assistance of Q. X. and B. W. G. B. and H. Å. provided theoretical calculations and analysed the mechanism. The manuscript was written jointly by L. Z. and M. L. and revised with help from Q. Z. All authors discussed the results and commented on the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the National Natural Science Foundation of China (NSFC, Grant No. 22275038, 22205038 and 22175064).Notes and references
- X. Yan, H. Peng, Y. Xiang, J. Wang, L. Yu, Y. Tao, H. Li, W. Huang and R. Chen, Recent Advances on Host–Guest Material Systems toward Organic Room Temperature Phosphorescence, Small, 2022, 18, 2104073 CrossRef CAS PubMed.
- H. Yang, B. Yuan, X. Zhang and O. A. Scherman, Supramolecular Chemistry at Interfaces: Host–Guest Interactions for Fabricating Multifunctional Biointerfaces, Acc. Chem. Res., 2014, 47, 2106–2115 CrossRef CAS PubMed.
- L.-J. Chen and H.-B. Yang, Construction of Stimuli-Responsive Functional Materials via Hierarchical Self-Assembly Involving Coordination Interactions, Acc. Chem. Res., 2018, 51, 2699–2710 CrossRef CAS PubMed.
- T. Aida, E. W. Meijer and S. I. Stupp, Functional Supramolecular Polymers, Science, 2012, 335, 813–817 CrossRef CAS PubMed.
- D. J. van Dijken, J. M. Beierle, M. C. A. Stuart, W. Szymański, W. R. Browne and B. L. Feringa, Autoamplification of Molecular Chirality through the Induction of Supramolecular Chirality, Angew. Chem., Int. Ed., 2014, 53, 5073–5077 CrossRef CAS PubMed.
- I. Aprahamian, The Future of Molecular Machines, ACS Cent. Sci., 2020, 6, 347–358 CrossRef CAS.
- C. Cheng, P. R. McGonigal, S. T. Schneebeli, H. Li, N. A. Vermeulen, C. Ke and J. F. Stoddart, An artificial molecular pump, Nat. Nanotechnol., 2015, 10, 547–553 CrossRef CAS PubMed.
- S. Fu, X. Su, M. Li, S. Song, L. Wang, D. Wang and B. Z. Tang, Controllable and Diversiform Topological Morphologies of Self-Assembling Supra-Amphiphiles with Aggregation-Induced Emission Characteristics for Mimicking Light-Harvesting Antenna, Adv. Sci., 2020, 7, 2001909 CrossRef CAS PubMed.
- W. Zhang, Y.-M. Zhang, S.-H. Li, Y.-L. Cui, J. Yu and Y. Liu, Tunable Nanosupramolecular Aggregates Mediated by Host–Guest Complexation, Angew. Chem., Int. Ed., 2016, 55, 11452–11456 CrossRef CAS PubMed.
- J. Liu, S. Wang, T. Huang, P. Manchanda, E. Abou-Hamad and S. P. Nunes, Smart covalent organic networks (CONs) with “on-off-on” light-switchable pores for molecular separation, Sci. Adv., 2020, 6, eabb3188 CrossRef CAS PubMed.
- F. Nan, X. Li, S. Zhang, J. Ng and Z. Yan, Creating stable trapping force and switchable optical torque with tunable phase of light, Sci. Adv., 2022, 8, eadd6664 CrossRef CAS PubMed.
- Z. Liang, H. Joh, B. Lian and D. E. Fan, Light-stimulated micromotor swarms in an electric field with accurate spatial, temporal, and mode control, Sci. Adv., 2023, 9, eadi9932 CrossRef CAS PubMed.
- Y. Wang, N. Ma, Z. Wang and X. Zhang, Photocontrolled Reversible Supramolecular Assemblies of an Azobenzene-Containing Surfactant with α-Cyclodextrin, Angew. Chem., Int. Ed., 2007, 46, 2823–2826 CrossRef CAS PubMed.
- G. Yu, C. Han, Z. Zhang, J. Chen, X. Yan, B. Zheng, S. Liu and F. Huang, Pillar[6]arene-Based Photoresponsive Host–Guest Complexation, J. Am. Chem. Soc., 2012, 134, 8711–8717 CrossRef CAS PubMed.
- J. Yang, G. Yu, D. Xia and F. Huang, A pillar[6]arene-based UV-responsive supra-amphiphile: synthesis, self-assembly, and application in dispersion of multiwalled carbon nanotubes in water, Chem. Commun., 2014, 50, 3993–3995 RSC.
- H.-L. Sun, Y. Chen, J. Zhao and Y. Liu, Photocontrolled Reversible Conversion of Nanotube and Nanoparticle Mediated by β-Cyclodextrin Dimers, Angew. Chem., Int. Ed., 2015, 54, 9376–9380 CrossRef CAS.
- H.-L. Sun, Y. Chen, X. Han and Y. Liu, Tunable Supramolecular Assembly and Photoswitchable Conversion of Cyclodextrin/Diphenylalanine-Based 1D and 2D Nanostructures, Angew. Chem., Int. Ed., 2017, 56, 7062–7065 CrossRef CAS.
- H. Yan, S. H. Park, G. Finkelstein, J. H. Reif and T. H. LaBean, DNA-Templated Self-Assembly of Protein Arrays and Highly Conductive Nanowires, Science, 2003, 301, 1882–1884 CrossRef CAS.
- D. Han, S. Pal, J. Nangreave, Z. Deng, Y. Liu and H. Yan, DNA Origami with Complex Curvatures in Three-Dimensional Space, Science, 2011, 332, 342–346 CrossRef CAS PubMed.
- D. Han, S. Pal, Y. Yang, S. Jiang, J. Nangreave, Y. Liu and H. Yan, DNA Gridiron Nanostructures Based on Four-Arm Junctions, Science, 2013, 339, 1412–1415 CrossRef CAS PubMed.
- F. Hong, F. Zhang, Y. Liu and H. Yan, DNA Origami: Scaffolds for Creating Higher Order Structures, Chem. Rev., 2017, 117, 12584–12640 CrossRef CAS PubMed.
- P. Zhan, M. J. Urban, S. Both, X. Duan, A. Kuzyk, T. Weiss and N. Liu, DNA-assembled nanoarchitectures with multiple components in regulated and coordinated motion, Sci. Adv., 2019, 5, eaax6023 CrossRef CAS PubMed.
- J. K. Yu, C. Bannwarth, R. Liang, E. G. Hohenstein and T. J. Martínez, Nonadiabatic Dynamics Simulation of the Wavelength-Dependent Photochemistry of Azobenzene Excited to the nπ* and ππ* Excited States, J. Am. Chem. Soc., 2020, 142, 20680–20690 CrossRef CAS PubMed.
- X. Jia and L. Zhu, Photoexcitation-Induced Assembly: A Bottom-Up Physical Strategy for Driving Molecular Motion and Phase Evolution, Acc. Chem. Res., 2023, 56, 655–666 CrossRef CAS PubMed.
- F. A. Jerca, V. V. Jerca and R. Hoogenboom, Advances and opportunities in the exciting world of azobenzenes, Nat. Rev. Chem, 2022, 6, 51–69 CrossRef PubMed.
- X. Jia, C. Shao, X. Bai, Q. Zhou, B. Wu, L. Wang, B. Yue, H. Zhu and L. Zhu, Photoexcitation-controlled self-recoverable molecular aggregation for flicker phosphorescence, Proc. Natl. Acad. Sci. U. S. A., 2019, 116, 4816–4821 CrossRef CAS PubMed.
- M. Baroncini, J. Groppi, S. Corra, S. Silvi and A. Credi, Light-Responsive (Supra)Molecular Architectures: Recent Advances, Adv. Opt. Mater., 2019, 7, 1900392 CrossRef.
- J. Griffiths, II. Photochemistry of azobenzene and its derivatives, Chem. Soc. Rev., 1972, 1, 481–493 RSC.
- S. K. M. Nalluri, J. Voskuhl, J. B. Bultema, E. J. Boekema and B. J. Ravoo, Light-Responsive Capture and Release of DNA in a Ternary Supramolecular Complex, Angew. Chem., Int. Ed., 2011, 50, 9747–9751 CrossRef CAS.
- A. Cembran, F. Bernardi, M. Garavelli, L. Gagliardi and G. Orlandi, On the Mechanism of the cis−trans Isomerization in the Lowest Electronic States of Azobenzene: S0, S1, and T1, J. Am. Chem. Soc., 2004, 126, 3234–3243 CrossRef CAS.
- M. Li and W.-H. Zhu, Sterically Hindered Diarylethenes with a Benzobis(thiadiazole) Bridge: Enantiospecific Transformation and Reversible Photosuperstructures, Acc. Chem. Res., 2022, 55, 3136–3149 CrossRef CAS PubMed.
- H.-B. Cheng, S. Zhang, J. Qi, X.-J. Liang and J. Yoon, Advances in Application of Azobenzene as a Trigger in Biomedicine: Molecular Design and Spontaneous Assembly, Adv. Mater., 2021, 33, 2007290 CrossRef CAS.
- J. Zhang, Q. Zou and H. Tian, Photochromic Materials: More Than Meets The Eye, Adv. Mater., 2013, 25, 378–399 CrossRef CAS PubMed.
- L. Zhu, H. Tran, F. L. Beyer, S. D. Walck, X. Li, H. Ågren, K. L. Killops and L. M. Campos, Engineering Topochemical Polymerizations Using Block Copolymer Templates, J. Am. Chem. Soc., 2014, 136, 13381–13387 CrossRef CAS PubMed.
- N. M.-W. Wu, M. Ng, W. H. Lam, H.-L. Wong and V. W.-W. Yam, Photochromic Heterocycle-Fused Thieno[3,2-b]phosphole Oxides as Visible Light Switches without Sacrificing Photoswitching Efficiency, J. Am. Chem. Soc., 2017, 139, 15142–15150 CrossRef CAS PubMed.
- X.-M. Chen, X.-F. Hou, H. K. Bisoyi, W.-J. Feng, Q. Cao, S. Huang, H. Yang, D. Chen and Q. Li, Light-fueled transient supramolecular assemblies in water as fluorescence modulators, Nat. Commun., 2021, 12, 4993 CrossRef CAS.
- B. Yue, X. Feng, C. Wang, M. Zhang, H. Lin, X. Jia and L. Zhu, In Situ Regulation of Microphase Separation-Recognized Circularly Polarized Luminescence via Photoexcitation-Induced Molecular Aggregation, ACS Nano, 2022, 16, 16201–16210 CrossRef CAS PubMed.
- B. Yue, X. Jia, G. V. Baryshnikov, X. Jin, X. Feng, Y. Lu, M. Luo, M. Zhang, S. Shen, H. Ågren and L. Zhu, Photoexcitation-Based Supramolecular Access to Full-Scale Phase-Diagram Structures through in situ Phase-Volume Ratio Phototuning, Angew. Chem., Int. Ed., 2022, 61, e202209777 CrossRef CAS PubMed.
- J. Gu, B. Yue, G. V. Baryshnikov, Z. Li, M. Zhang, S. Shen, H. Ågren and L. Zhu, Visualizing Material Processing via Photoexcitation-Controlled Organic-Phase Aggregation-Induced Emission, Research, 2021, 2021, 9862093 CAS.
- T. Weng, Q. Zou, M. Zhang, B. Wu, G. V. Baryshnikov, S. Shen, X. Chen, H. Ågren, X. Jia and L. Zhu, Enhancing the Operability of Photoexcitation-Controlled Aggregation-Induced Emissive Molecules in the Organic Phase, J. Phys. Chem. Lett., 2021, 12, 6182–6189 CrossRef CAS PubMed.
- S. Shen, G. Baryshnikov, B. Yue, B. Wu, X. Li, M. Zhang, H. Ågren and L. Zhu, Manipulating crystals through photoexcitation-induced molecular realignment, J. Mater. Chem. C, 2021, 9, 11707–11714 RSC.
- Y.-M. Zhang, Y.-H. Liu and Y. Liu, Cyclodextrin-Based Multistimuli-Responsive Supramolecular Assemblies and Their Biological Functions, Adv. Mater., 2020, 32, 1806158 CrossRef CAS.
- G. Wenz, B.-H. Han and A. Müller, Cyclodextrin Rotaxanes and Polyrotaxanes, Chem. Rev., 2006, 106, 782–817 CrossRef CAS PubMed.
- P. R. Ogilby, Singlet oxygen: there is indeed something new under the sun, Chem. Soc. Rev., 2010, 39, 3181–3209 RSC.
- K. Apel and H. Hirt, REACTIVE OXYGEN SPECIES: Metabolism, Oxidative Stress, and Signal Transduction, Annu. Rev. Plant Biol., 2004, 55, 373–399 CrossRef CAS PubMed.
- A. Gollmer, J. Arnbjerg, F. H. Blaikie, B. W. Pedersen, T. Breitenbach, K. Daasbjerg, M. Glasius and P. R. Ogilby, Singlet Oxygen Sensor Green®: Photochemical Behavior in Solution and in a Mammalian Cell, Photochem. Photobiol., 2011, 87, 671–679 CrossRef CAS PubMed.
- H.-J. Schneider, F. Hacket, V. Rüdiger and H. Ikeda, NMR Studies of Cyclodextrins and Cyclodextrin Complexes, Chem. Rev., 1998, 98, 1755–1786 CrossRef CAS PubMed.
- K. Harata, Induced circular dichroism of cycloamylose complexes with meta- and para-disubstituted benzenes, Bioorg. Chem., 1981, 10, 255–265 CrossRef CAS.
- H. Kazuaki and U. Hisashi, The Circular Dichroism Spectra of the β-Cyclodextrin Complex with Naphthalene Derivatives, Bull. Chem. Soc. Jpn., 1975, 48, 375–378 CrossRef.
- M. Kodaka and T. Fukaya, Induced Circular Dichroism Spectrum of α-Cyclodextrin Complex with Heptylviologen, Bull. Chem. Soc. Jpn., 1989, 62, 1154–1157 CrossRef CAS.
- M. Kodaka, A general rule for circular dichroism induced by a chiral macrocycle, J. Am. Chem. Soc., 1993, 115, 3702–3705 CrossRef CAS.
- M. Kodaka, Sign of circular dichroism induced by .beta.-cyclodextrin, J. Phys. Chem., 1991, 95, 2110–2112 CrossRef CAS.
- A. Z. Yu, E. A. Yurii, E. V. Kompantseva and E. N. Vergeichik, Induced optical activity in cyclodextrin complexes, Russ. Chem. Rev., 1992, 61, 563 CrossRef.
- S. Allenmark, Induced circular dichroism by chiral molecular interaction, Chirality, 2003, 15, 409–422 CrossRef CAS PubMed.
- R. Krishnan, A. M. Rakhi and K. R. Gopidas, Study of β-Cyclodextrin–Pyromellitic Diimide Complexation. Conformational Analysis of Binary and Ternary Complex Structures by Induced Circular Dichroism and 2D NMR Spectroscopies, J. Phys. Chem. C, 2012, 116, 25004–25014 CrossRef CAS.
- W. Zhi, Z. Pu, C. Ma, K. Liu, X. Wang, J. Huang, Y. Xiao and Y. Yan, α-Cyclodextrin-Catalyzed Symmetry Breaking and Precise Regulation of Supramolecular Self-Assembly Handedness with Harata–Kodaka’s Rule, ACS Nano, 2021, 15, 19621–19628 CrossRef CAS PubMed.
- X. Wang, W. Zhi, C. Ma, Z. Zhu, W. Qi, J. Huang and Y. Yan, Not by Serendipity: Rationally Designed Reversible Temperature-Responsive Circularly Polarized Luminescence Inversion by Coupling Two Scenarios of Harata–Kodaka’s Rule, JACS Au, 2021, 1, 156–163 CrossRef CAS PubMed.
- E. Krieg, M. M. C. Bastings, P. Besenius and B. Rybtchinski, Supramolecular Polymers in Aqueous Media, Chem. Rev., 2016, 116, 2414–2477 CrossRef CAS PubMed.
- X. Cheng, Q. Wang, C. Lu and Q. Meng, Watching the Conformational Changes of Maleonitriledithiolate Chromophores Inside the Inclusion Complexes with Cyclodextrins: Probed by ICD Spectra and DFT Calculations, J. Phys. Chem. A, 2010, 114, 7230–7240 CrossRef CAS PubMed.
- G. Pescitelli, ECD exciton chirality method today: a modern tool for determining absolute configurations, Chirality, 2022, 34, 333–363 CrossRef CAS PubMed.
- J. He, Y. He, Y. Chen, X. Zhang, C. Hu, J. Zhuang, B. Lei and Y. Liu, Construction and multifunctional applications of carbon dots/PVA nanofibers with phosphorescence and thermally activated delayed fluorescence, Chem. Eng. J., 2018, 347, 505–513 CrossRef CAS.
- Q. Cao, K.-K. Liu, Y.-C. Liang, S.-Y. Song, Y. Deng, X. Mao, Y. Wang, W.-B. Zhao, Q. Lou and C.-X. Shan, Brighten Triplet Excitons of Carbon Nanodots for Multicolor Phosphorescence Films, Nano Lett., 2022, 22, 4097–4105 CrossRef CAS PubMed.
- Z. Tian, D. Li, E. V. Ushakova, V. G. Maslov, D. Zhou, P. Jing, D. Shen, S. Qu and A. L. Rogach, Multilevel Data Encryption Using Thermal-Treatment Controlled Room Temperature Phosphorescence of Carbon Dot/Polyvinylalcohol Composites, Adv. Sci., 2018, 5, 1800795 CrossRef PubMed.
- H. Wu, W. Chi, Z. Chen, G. Liu, L. Gu, A. K. Bindra, G. Yang, X. Liu and Y. Zhao, Achieving Amorphous Ultralong Room Temperature Phosphorescence by Coassembling Planar Small Organic Molecules with Polyvinyl Alcohol, Adv. Funct. Mater., 2019, 29, 1807243 CrossRef.
- H. Wu, L. Gu, G. V. Baryshnikov, H. Wang, B. F. Minaev, H. Ågren and Y. Zhao, Molecular Phosphorescence in Polymer Matrix with Reversible Sensitivity, ACS Appl. Mater. Interfaces, 2020, 12, 20765–20774 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4sc03451a |
| This journal is © The Royal Society of Chemistry 2024 |