
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceIn situ self-assembled near-infrared phototherapeutic agent: unleashing hydrogen free radicals and coupling with NADPH oxidation†
Yingcui
Bu
ab,
Zhi-Peng
Yu
a,
Zhou
Lu
 d,
Haoran
Wang
e,
Yu
Deng
a,
Xiaojiao
Zhu
*a and
Hongping
Zhou
*ac
d,
Haoran
Wang
e,
Yu
Deng
a,
Xiaojiao
Zhu
*a and
Hongping
Zhou
*ac
aSchool of Chemistry and Chemical Engineering, Institute of Physical Science and Information Technology, Anhui University, Key Laboratory of Functional Inorganic Materials Chemistry of Anhui Province, Key Laboratory of Chemistry for Inorganic/Organic Hybrid Functionalized Materials of Anhui Province, Key Laboratory of Structure and Functional Regulation of Hybrid Materials (Anhui University) Ministry of Education, Hefei, 230601, P.R. China. E-mail: zhpzhp@263.ne; xiaojzhu@ahu.edu.cn
bSchool of Materials and Chemistry, Anhui Agricultural University, P.R. China
cSchool of Chemical and Environmental Engineering, Anhui Polytechnic University, P.R. China
dAnhui Key Laboratory for Control and Applications of Optoelectronic Information Materials, School of Physics and Electronic Information, Anhui Normal University, P.R. China
eSchool of Chemistry and Chemical Engineering, Nanjing University of Science and Technology, P.R. China
First published on 8th July 2024
Abstract
Investigation of electron transfer (ET) between photosensitizers (PSs) and adjacent substrates in hypoxic tumors is integral to highly efficient tumor therapy. Herein, the oxygen-independent ET pathway to generate hydrogen free radicals (H˙) was established by the in situ self-assembled phototherapeutic agent d-ST under near-infrared (NIR)-light irradiation, coupled with the oxidation of reduced coenzyme NADPH, which induced ferroptosis and effectively elevated the therapeutic performance in hypoxic tumors. The higher surface energy and longer exciton lifetimes of the fine crystalline d-ST nanofibers were conducive to improving ET efficiency. In hypoxic conditions, the excited d-ST can effectively transfer electrons to water to yield H˙, during which the overexpressed NADPH with rich electrons can power the electron flow to facilitate the generation of H˙, accompanied by NADP+ formation, disrupting cellular homeostasis and triggering ferroptosis. Tumor-bearing mouse models further showed that d-ST accomplished excellent phototherapy efficacy. This work sheds light onto the versatile electron pathways between PSs and biological substrates.
Introduction
Current research indicates that the non-oxygen-dependent photodynamic therapy (PDT) system, which involves electron transfer (ET) between photosensitizers (PSs) and substrates (water and biomolecules) has witnessed great advances in addressing the Achilles heel of hypoxic tumor therapy.1–5 For instance, Feng demonstrated ET between water (the most abundant component in living organisms) and photocatalytically active organic molecules under visible light to yield hydrogen radicals (H˙), which can trigger severe cancer cell apoptosis in hypoxic conditions, indicating great potential for translation into clinical tumor therapy.6 Nevertheless, there is little related research into the photo-generation of H˙, let alone the rational molecular design strategy of PSs to yield H˙ activated by near-infrared (NIR) light illumination.7 Hence, the development of a highly-efficient H˙-generator for PDT remains challenging but very noteworthy.Inspired by the photocatalytic behaviors of organic molecules in an aqueous system, it is proposed that the highly-efficient NIR photo-generation of H˙ essentially demands careful molecular design, mainly including the following four aspects. (i) Considering that NIR light delivers minor photo-damage and high tissue penetration, the PSs should be activated in the NIR bio-window.8–11 (ii) From a mechanistic perspective, the long enough exciton lifetime and high charge separation (CS) efficiency of the PSs guarantee high carrier transport efficacy on the water/PSs interface.12–14 (iii) To facilitate ET from the PSs to water, close contact with water molecules and the lower redox potential of PSs with regard to E (H2O/H˙) should be fulfilled.15–17 (iv) For the sake of a high yield of H˙, the construction of a redox system would orchestrate the ET process between PSs and substrates.18,19 To date, despite various efforts having been made, PSs that meet these requirements for generating H˙ have scarcely been reported.
The molecular self-assembly strategy, which would shape the spatial arrangement into a more ordered structure, thus favoring the formation of a CS state and prolonging the lifetime of the CS state, has achieved great renown for regulating the ET process in PDT.20–22 For example, Peng and Fan designed an OH˙ photogenerator based on the ET process between self-assembled PSs and water in cancer cells.23 Of note, various intracellular ET processes involving redox biomolecule pairs (such as the NAD(P)+/NAD(P)H pair), would allow the platform to correlate with the ET process between PSs and water. Additionally, the depletion of the redox biomolecule pairs can in turn result in disruption of intracellular redox homeostasis and exacerbate cancer cell death.24,25 In this regard, building self-assembled molecular structures coupled with endogenous redox biomolecule pairs would provide an innovative avenue to accomplish a highly efficient H˙-generator for PDT.
Along these lines, we present the in situ assembled phototherapeutic agent d-ST, in which the ET pathway to generate H˙ under NIR light was established, coupled with the oxidation of reduced coenzyme NADPH. A lower dimer binding energy (ΔG) and larger molecular dipole enable the moderately hydrophilic d-ST to form nanofibers in aqueous solution. The finely crystalline ordered assembly of d-ST possessing a higher surface energy and longer exciton lifetime was conducive to improving ET efficiency. Detailed mechanistic studies revealed that, under oxygen-free conditions, the excited d-ST can transfer the electrons to water to generate H˙, during which the overexpressed NADPH with rich electrons can power the electron flow to facilitate the generation of H˙, accompanied by NADP+ formation, disrupting cellular homeostasis and triggering ferroptosis. In vivo experiments demonstrated that d-ST with superior biocompatibility accomplished excellent phototherapy efficacy, exhibiting great potential in clinical applications.
Results and discussion
Molecular assembly regulation
Considering that a bow-shaped molecular backbone can result in a larger dipole moment along with a tendency toward orderly aggregation,26–29d-ST, as depicted in Scheme 1, was carefully constructed to enable ordered self-assembly in an aqueous environment. The bilateral fused onium salt (FOA) containing N,N-dimethyl (an electron-donating group) and cationic salts (electron acceptor groups), which was equipped for NIR-region absorption and mitochondrial targeting properties, was centered in a thiophene bridge which possesses a lone pair of electrons and thus would be prone to interacting with polar water molecules.30 In addition, to demonstrate the effect of the shape of the molecule on the assembly behavior, d-CT with a phenol-bridge bearing a Z-shaped molecular backbone was prepared (Scheme S1†). The synthetic methods were meticulously devised to ensure high yields and the purity of the final products, which were systematically characterized by 1H NMR, 13C NMR, and high-resolution mass spectrometry (HRMS) (Fig. S1–S6†). | ||
| Scheme 1 A novel photochemical therapeutic platform toward hypoxic tumors. (a) In situ self-assembly behavior using single organic molecule d-ST in aqueous solution. (b) ET mechanism between PSs and substrates, and high-efficiency anti-hypoxic tumor therapy upon NIR-light excitation. | ||
The self-assembly behaviors of d-ST in aqueous solution were explored. The transmission electron microscopy (TEM) in Fig. S7a† and 1a revealed that the morphology of d-ST (10 μM) underwent a significant transition from uniformly dispersed nanodots (∼6 nm) to nanofibers (∼190 nm) after 20 min of self-assembly in water. Additionally, almost all the dispersed nanodots vanished and nanofibers occupied the whole view after extending for another 10 min, indicating the feasibility of the self-assembled behavior of d-ST in aqueous solution. Gratifyingly, these nanofibers can be stabilized in aqueous solution for 6 h or even longer, suggesting their potential for practical biological applications. As exhibited in the high resolution TEM in Fig. 1b, a certain crystallinity of d-ST with a clear d-spacing of 0.31 nm was observed, which could be due to the strong intermolecular π–π stacking.31–35 By contrast, d-CT (10 μM) presented distinct assembly behaviors, as shown in Fig. S7b† and 1c. Afterwards, to investigate the distinguishable self-assembled behaviors between d-ST and d-CT, the relaxed potential surface energy scans of rotatable single C–C bonds between B and the FOA-unit (denoted as the S-FOA rotamer for d-ST and the C-FOA rotamer for d-CT) were calculated using density functional theory (DFT). According to the energy–torsion angle curves (E–θ curves) in Fig. 1d, the energy difference between the conformation with the lowest E (0°) and second lowest E (190°) was 4.23 kJ mol−1 for d-ST, much higher than the 0.88 kJ mol−1 energy difference between the conformation with the lowest E (150°) and second lowest E (330°) for d-CT. Accordingly, the calculated energy barrier of the S-FOA rotamer in d-ST is 28.13 kJ mol−1, higher than the 16.31 kJ mol−1 energy barrier of the C-FOA rotamer in d-CT. Moreover, as shown in Fig. 1e, the dimer Gibbs free energy (ΔG) of the corresponding optimal confirmation of d-ST and d-CT was calculated to be −2241 kJ mol−1 and −964 kJ mol−1, respectively. Fig. S8† demonstrates that in monomers the substituent of thiophene groups can significantly expand the molecular dipole from 1.2379 debye (D) to 1.8291 D, while in dimers it expands from 3.9537 to 12.9237 D. Collectively, d-ST with better planarity, lower ΔG and a larger molecular dipole, did not tend to undergo conformational conversion, which provided further powerful evidence for its ordered aggregation in solution, which would be beneficial for a subsequent photo-induced ET process.36,37
 | ||
| Fig. 1 (a and c) TEM images of the self-assembled structures of d-ST and d-CT in H2O (10 μM) after 30 min. (b) The streak orientation and lattice distance of d-ST (10 μM) by high-resolution TEM. (d) Molecular energy and corresponding geometries in different conformational states of d-ST and d-CT. (e) Proposed packing models of d-ST (10 μM) and d-CT (10 μM) with the corresponding binding energy (ΔG). (f) Absorption spectra of d-ST and d-CT in DMSO and water. (g) Absorption spectra of d-ST with different concentrations (10–30 μM). (h) Molar absorption coefficient (ε) of d-ST. | ||
Notably, as displayed in Fig. 1f, d-ST in an aqueous environment showed quite different photo-physical properties from those in DMSO, in which the absorption spectra of d-ST in aqueous solution were mainly distributed around the NIR region, together with a significantly broadened peak and a red-shift of about 100 nm. The concentration-dependent absorption spectra were implemented to calculate d-ST with larger molar extinction coefficients (ε × 104) (Fig. 1g and h). All the above highlighted the potential of subsequent biotherapy applications for ordered-assembly d-ST.
Photo-generation of H˙ and oxidation of NADPH
To study the ET process between self-assembled d-ST and water, the NIR-light-activated radical species were confirmed by the electron paramagnetic resonance (EPR) trapping technique with 5,5-dimethyl-1-pyrroline N-oxide (DMPO) as the spin-trapping agent. As shown in Fig. 2a, DMPO-H˙ adducts with the characteristic nine-line signal (hyperfine splitting constants: aN = 16.03 G, aβH = 21 G, g = 2.00)38 were observed in the d-ST group after 1 min of NIR-light irradiation, while no signal was detected in the dark or in the d-CT group (Fig. S9†). The additional six-line signal in the ESR spectrum was assigned to DMPO-R˙ adducts (R: carbon-centered radical). For an in-depth study of the interfacial ET, cyclic voltammetry was conducted to determine the redox potential. As illustrated in Fig. S10,† the reduction potential of d-ST (Ered = −0.324 V) was lower than that of d-CT (Ered = −0.004 V), confirming the dynamic feasibility of ET from excited d-ST to substrate water. Furthermore, HRMS analysis of the TEMPO-H˙ adduct (m/z 158.1545) (Fig. 2b), as well as the hydrogenation of 4-vinylbenzoic acid (PVBA) from m/z 147.0435 to 149.0489 (Fig. S11†), provided strong evidence for the ET process of water to form H˙.7 To further facilitate the generation of H˙, the redox pair NADPH/NADP+, which is overexpressed in cancer cells (5–10 times higher than that in normal cells),39,40 was elaborately selected to couple with d-ST. As exhibited in Fig. 2c and S12,† under light exposure of 90 s, in the NADPH (50 μM) + system, a pronounced decrease in the absorption spectrum of NADPH was observed, while it remained unchanged in the control (dark and NADPH alone) and d-CT groups. Then, Cy3 as a commercial H˙ indicator with red emission was utilized to detect H˙ when coupled with NADPH. Fig. 2d and e indicate that the fluorescence signal of Cy3 treated with d-ST was notably decreased by ∼25% upon light irradiation, and even decreased by ∼75% in the d-ST and NADPH system upon light irradiation. It could be concluded that NADPH/NADP+ as a vital electron pool can fuel the ET between PSs and water to yield more H˙, further supported by the quenching experiment results of Cy3 in dark conditions (Fig. S13†). In addition, based on theoretical calculation (Fig. S14†), NADPH can be oxidized by the excited triplet state (T1) (1.11 eV) of d-ST to form NADP+, which accelerated the cellular redox homeostasis imbalance and laid the foundation for an effective hypoxic treatment. | ||
| Fig. 2 (a) Electron spin resonance spectra of DMPO/d-ST under NIR-light illumination for 1 min. (b) HRMS analysis of d-ST containing TEMPO under laser irradiation for 1 min. (c) Photo-oxidation of NADPH (50 μM) without and with d-ST (10 μM) recorded by absorption spectra. (d) Detection of H˙ production in different treatment groups (Cy3, Cy3 + d-ST, Cy3 + d-ST + NADPH (50 μM)) upon irradiation for 2 min in vitro by virtue of commercial fluorescent dye Cy3. (Inset: detection of H˙ production in Cy3 + d-ST + NADPH upon different irradiation times (0–2 min)). (e) Schematic diagram of the photo-induced ET process. All light illumination used in the experiments was from a 660 nm laser at a power of 200 mW. | ||
Surface potential and photophysical behavior of the assembled structure
The in-depth mechanism of H˙ generation in the self-assembled d-ST was further comprehensively investigated. As exhibited in Fig. 3a and b, atomic force microscopy (AFM) revealed that d-ST with a helically-assembled nanofiber structure had a height profile of 9 nm, while d-CT emerged as aggregated nanodots of 43 nm in height, consistent with the TEM results. Then, Kelvin probe force microscopy (KPFM) was employed to analyze their surface potential (SP), in which the assembled d-ST possessed a much larger SP of 15.32 mV/9 nm than that of d-CT (4.76 mV/43 nm) (Fig. 3c and d), benefiting exciton dissociation and thus being conducive to the photo-induced CS process.14,41–43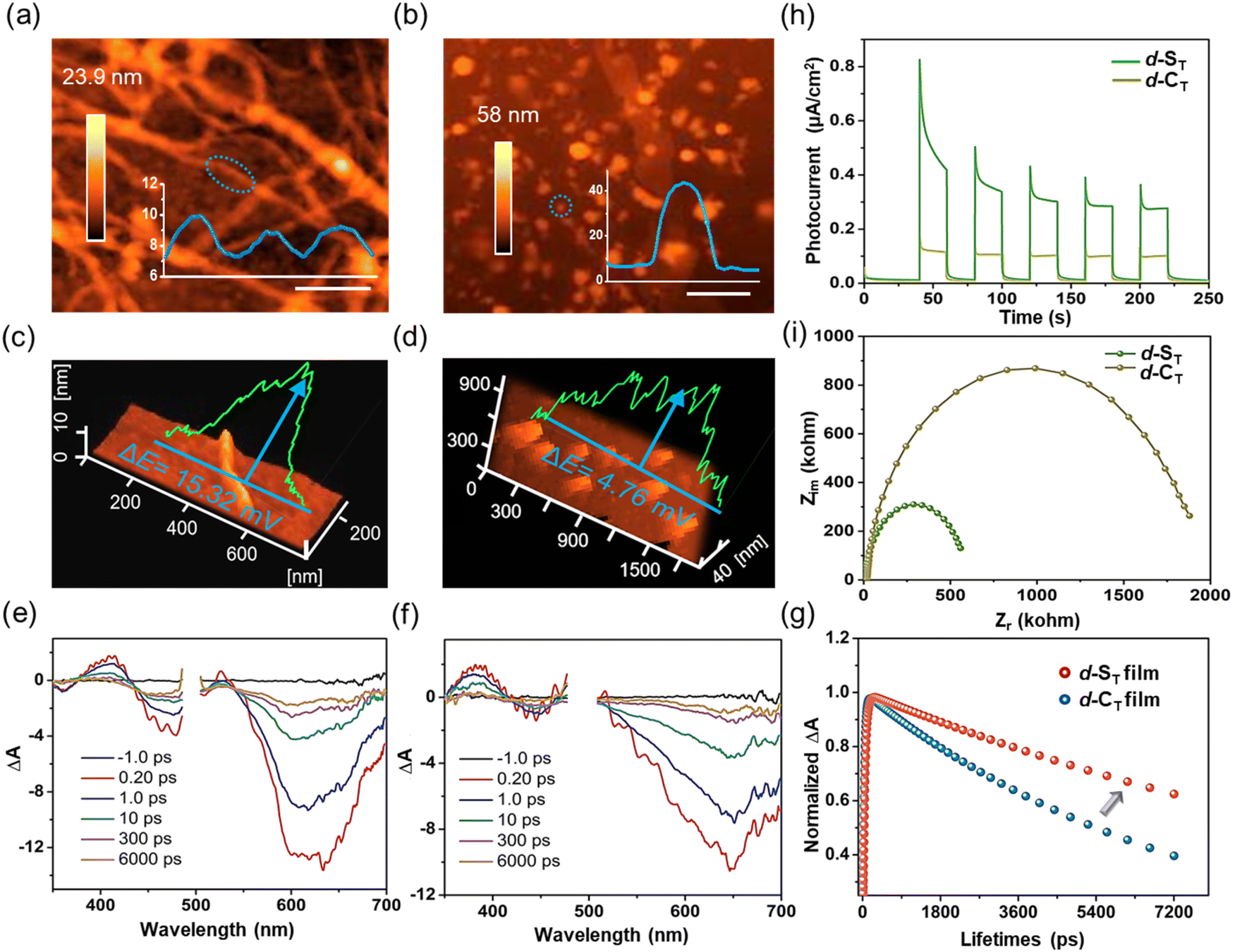 | ||
| Fig. 3 (a and b) AFM images with sectional analysis of assembled d-ST (10 μM) and d-CT (10 μM). (c and d) The surface potential of assembled d-ST (10 μM) and d-CT (10 μM) was detected with KPFM. (e and f) The transient absorption spectra of d-ST film and d-CT film (A: absorption). (g) The dynamics measured by the femtosecond transient absorption technique for excited-state absorption of d-ST film and d-CT film. (h) Photocurrent and (i) electrochemical impedance spectra of assembled d-ST and d-CT. | ||
Subsequently, the stability of the excited excitons was investigated by femtosecond transient absorption (fs-TA) spectroscopy. Further, displayed in Fig. 3e and f are the spectra for the samples in thin film form, where ground-state bleach (GSB) peaks at 610 nm for d-ST and 650 nm for d-CT could clearly be observed upon pulse excitation. By global fitting, an increase to longer lifespans of over 200 times was captured in d-ST and in d-CT, and the lifetime (15.21 ns) of d-ST was twice that of d-CT (7.74 ns). Even after 1000 ns, the long-lived species signal of d-ST had not completely disappeared, which was conducive to improving the ET efficiency of the excited state (Fig. 3g). By contrast, the TA of d-ST and d-CT in an isolated molecular state presented similar lifetime values with a picosecond timescale (23.86 ps for non-assembled d-ST and 30.05 ps for non-assembled d-CT) (Fig. S15a and S15b†). Furthermore, photocurrent and impedance tests were accomplished to investigate the surface potential and exciton lifetime. As shown in Fig. 3h and i, the transient photocurrent responses of assembled d-ST in five on–off photo-irradiation cycles exhibited much higher photo-induced electron transfer than d-CT. According to the fitted electrochemical impedance spectra, the electron transfer resistance of assembled d-ST was lower than that of d-CT, indicating the superior ET kinetics of assembled d-ST.44–46
NIR-triggered antihypoxia performance in cell level
Subsequently, the internalization capability of d-ST/d-CT into HepG2 cells was evaluated by confocal laser scanning microscopy (CLSM) based on their in vitro fluorescence emission behaviors (Fig. S16†). As displayed in Fig. S17,† HepG2 cells pretreated with d-ST/d-CT for only 10 min emitted bright green fluorescence, suggesting that they can be taken in by HepG2 cells in a short time. Co-localization experiments demonstrated that d-ST and d-CT can target mitochondria (Fig. S18†), providing a strong guarantee for the precise killing of cancer cells. Then, as shown in Fig. 4a and S19† (red arrow in the enlarged area), the green fluorescence of d-ST with a self-assembled nanofiber morphology was spotted in HepG2 cells after 30 min of co-incubation, which was further confirmed with the commercial dye Mito-Tracker (mitochondria-targeting dye) and DAPI (nuclear dye) in Fig. S20† (red arrow in the enlarged area). As depicted in Fig. 4b for the TEM images of d-ST in HepG2 cells, the highly-ordered nanofibers in the cell medium also confirmed the in situ self-assembly behavior of d-ST, which ensured the subsequent NIR-triggered phototherapy. | ||
| Fig. 4 (a) CLSM images of HepG2 cells following 30 min of incubation with d-ST (10 μM). (b) TME images of HepG2 cells treated with PBS (control) and d-ST under laser irradiation. The red arrows in (a) and (b) represent the d-ST nanofibers. (c) Intracellular hypoxia imaging by virtue of ROS-ID as an anaerobic indicator. (d) Intracellular H˙ generation with Cy3 as a fluorescence probe by CLSM images. (e) Cell viabilities of HepG2 cells treated with d-ST (10 μM) and d-CT (10 μM) within a 2% O2 environment under laser irradiation. (f) CLSM imaging of live/dead cell staining pretreated with d-ST and d-CT in hypoxic conditions for different irradiation times (0–6 min). (g) Bio-imaging of live/dead cell staining treated with d-ST in cancer cells (A549 and HeLa cells) in hypoxic conditions. Scale bar: 20 μm. All light illumination used in the experiments was from a 660 nm laser at a power of 200 mW. Data represent the mean ± SD of biological replicates (n = 3); significances are determined by a two-tailed, unpaired Student's t-test. | ||
To demonstrate the great advantage of in situ self-assembled NIR phototherapeutic agents with H˙ generation in hypoxic environments, the killing effects of d-ST and d-CT in normoxic (21% O2) and hypoxic (2% O2) conditions were tested. First, the intuitive anticancer potency in the cell level was assessed by methylthiazolyltetrazolium (MTT) assays. As displayed in Fig. S21,† negligible dark toxicity was recorded among various cancer cell lines, especially for HepG2 cells even at a concentration up to 25 μM (Fig. S22†), implying their good biosafety. Next, an intracellular hypoxic model was carefully built, as evidenced by the 8.8-fold increase in the fluorescence of the anaerobic fluorescent indicator ROS-ID (Fig. 4c), the nitro group of which was reduced to an amino group under hypoxic conditions with the occurrence of bright red fluorescence. Then, in hypoxic conditions, the generation of H˙ in HepG2 cells was detected using Cy3 as the fluorescence probe. As depicted in Fig. 4d, bright red fluorescence could be observed in the groups (Cy3, Cy3 + light and Cy3 + d-ST group), whereas the fluorescence signal of Cy3 was notably attenuated by a factor of 82% in the Cy3 + d-ST + light group, confirming the yield of H˙ even in a hypoxic environment. Fig. S23 and S24† show that reactive oxygen species O2˙− and 1O2 were negligible when HepG2 cells were incubated with d-ST under irradiation in hypoxic conditions, as detected by the O2˙−-specific fluorescent probe DHE and 1O2-specific fluorescent probe SOSG. Moreover, live/dead cell staining experiments dealing with d-ST in cancer cells lines (HepG2, HeLa, and A549 cells) using an AM/PI kit provided more intuitive results for the antihypoxia methodology of H˙ (Fig. 4f and g). The half maximal inhibitory concentration (IC50) value of d-ST subjected to NIR-light irradiation (IC50 irrad) in hypoxic conditions was calculated to be 2.4 μM according to the MTT assay, while the IC50 dark (without light) under the same conditions was 36 μM. Such a higher IC50 value in dark conditions suggested its superior biosafety. Then, in hypoxic conditions, the photo-cytotoxicity index (PI) (the ratio of IC50 dark/IC50 irrad) of d-ST-treated HepG2 cells was calculated to be 15.00. As a comparison, the PI value of d-CT was as low as 2.04, which indicated poor efficiency for killing cancer cells in hypoxic conditions. Furthermore, similar results were observed in A549 and HeLa cell lines (Fig. 4e and Table 1), suggesting the superior potential of the in situ self-assembled d-ST with H˙ generation in the ablation of hypoxic tumors.
| Compounds | Cell lines | Darka (μM) | Irradiationa (μM) | PIa |
|---|---|---|---|---|
| a Under hypoxia (O2: 2%); 2 min of NIR-light irradiation with a laser power of 200 mW (660 nm); photocytotoxicity index (PI), the ratio of IC50 dark/IC50 irrad. | ||||
| d-CT | HepG2 | 37.5 | 18.4 | 2.04 |
| A549 | 36.3 | 19.0 | 1.91 | |
| HeLa | 38.2 | 18.8 | 2.03 | |
| d-ST | HepG2 | 36.0 | 2.4 | 15.00 |
| A549 | 38.1 | 2.2 | 17.32 | |
| HeLa | 35.1 | 2.6 | 13.50 | |
H˙-induced ferroptosis under hypoxia
For a deeper insight into the cell death mechanism mediated by H˙, high-resolution microscopy imaging of the fine structure of the mitochondria was performed in HepG2 cells treated in different groups, (i): control (Mito-Tracker Green FM); (ii): with ferroptosis-inducer erastin; (iii): d-ST in the dark; (iv): d-CT in the dark; (v): d-ST + light; (vi): d-CT + light; (vii): d-ST + Vc + light; (viii): d-CT + Vc + light; cells with added Vc and Fer-1 under irradiation; (ix): d-ST + Vc + Fer-1 + light; (x): d-CT + Vc + Fer-1 + light. Fig. 5a indicates that most mitochondria in groups (i), (iii) and (iv) were in a tubular shape: from the magnified areas, we can clearly observe the related morphological characteristics, such as mitochondrial cristae (indicated by yellow arrows). After irradiation for 1 min, many bubble structures in the cytoplasm can be found in cells treated with d-ST (red arrows in group (v)), which fitted well with the previous reports about reactive oxygen species inducing cell apoptosis.47 Conversely, photo-irradiation of d-CT led to only minor morphological changes in group (vi). In particular, distinct mitochondria shrinkage was easily captured (white arrows in group (v)), which was similar to the mitochondria shrinkage and fragmentation in cells undergoing ferroptosis induced by erastin in group (ii). Furthermore, specific inhibitors of the cell death pathway were pre-incubated to decipher their respective contributions. As manifested in Fig. 5a (group (vii)), while dealing with a ROS-induced apoptosis/necrosis inhibitor (vitamin C; Vc), the major morphological analysis of cells co-cultured with d-ST based on the mitochondrion length-to-width ratio (L/W) disclosed that this shrinkage led to more round (1.0, L/W < 1.5) and intermediate (1.5, L/W < 2.0) mitochondria (ferroptosis characteristics, indicated by yellow arrows). Interestingly, when treated with Vc and ferroptosis inhibitor (ferrostatin-1; Fer-1),48 the mitochondrial cristae were still clearly visible in group (ix). Moreover, d-CT-cultured HepG2 cells displayed almost no effect on mitochondrial viability whether with Vc or Fer-1 in groups (viii) and (x) due to their lower ability to damage cells. On the basis of the above studies, we propose that the main mechanism of killing cancer cells induced by the generated H˙ upon hypoxia was via the ferroptosis process (Fig. 5d).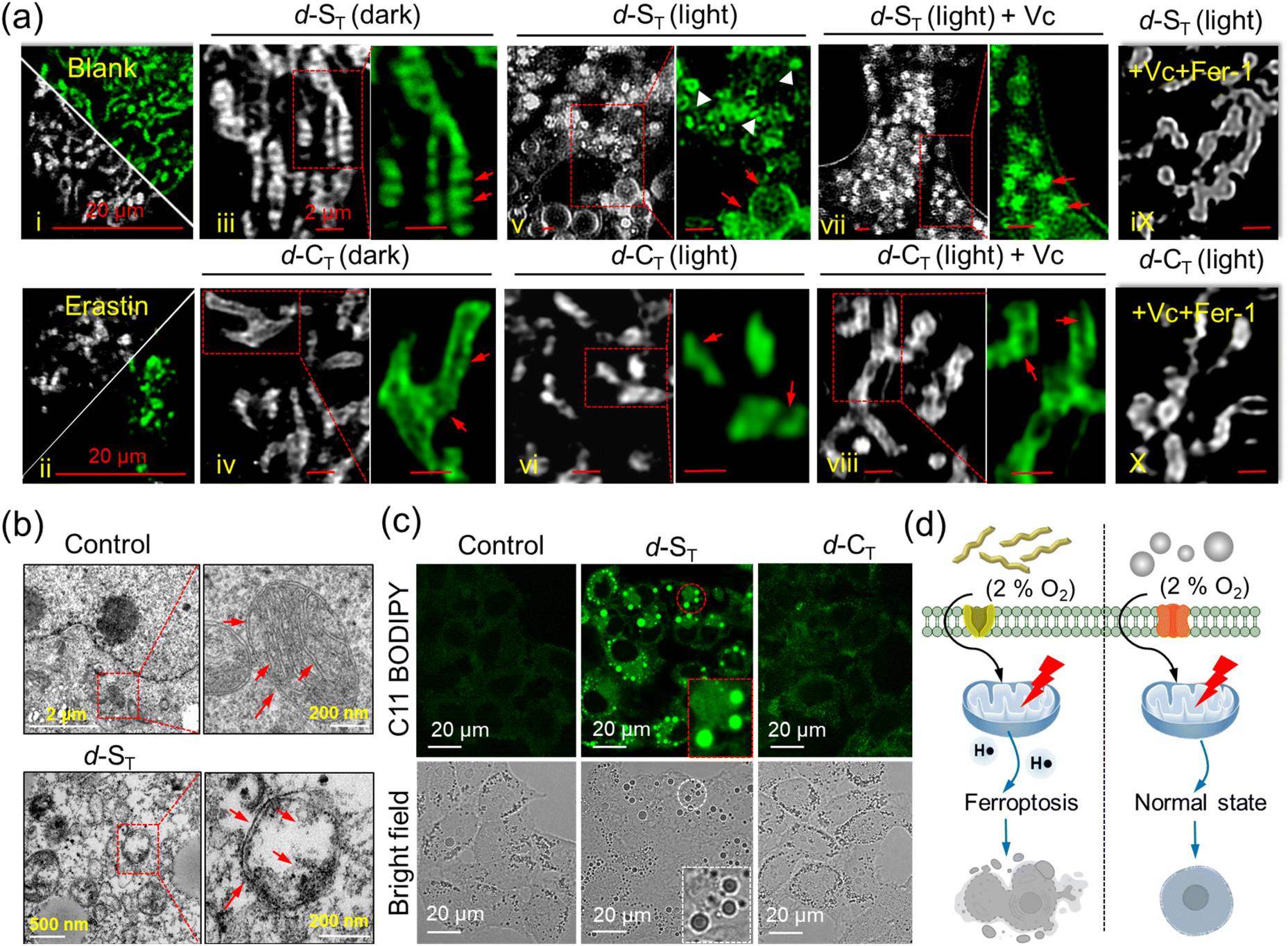 | ||
| Fig. 5 (a) Fine structures of mitochondria in HepG2 cells dealt with in different groups, (i): control (Mito-Tracker Green FM); (ii): with ferroptosis-inducer erastin (10 μM, 24 h); (iii): d-ST in the dark; (iv): d-CT in the dark; (v): d-ST + light; (vi): d-CT + light; (vii): d-ST + Vc + light; (viii): d-CT + Vc + light; cells with added Vc and Fer-1 under irradiation (ix): d-ST + Vc + Fer-1 + light; (x): d-CT + Vc + Fer-1 + light. Scale bar: 2 μm. (b) Ferroptosis revealed by TME images of HepG2 cells treated with d-ST and d-CT. Red arrowheads show the normal mitochondria in the control and shrunken mitochondria in d-ST, respectively. (c) Confocal imaging of HepG2 cells incubated with d-ST and d-CT and further stained with C11-BODIPY under light and hypoxia. (d) Comparison of light-induced cell viability treated with d-ST and d-CT in hypoxic conditions. Scale bar: 20 μm. All light illumination used in the experiments was with a 660 nm laser at a power of 200 mW. | ||
The morphological changes in the mitochondria were also monitored with TME images (Fig. 5b). Compared to the normal microscopic structure in the untreated or d-CT group, a large number of light-induced shrunken mitochondria with increased membrane density were captured in the d-ST group, which further manifested the occurrence of ferroptosis.
Moreover, the mitochondrial dysfunction indicator JC-1 which evaluates the changes in mitochondria membrane potential (MMP) was applied. Fig. S25† shows that bright red fluorescence gradually evolved to green fluorescence, which indicated that the mitochondrial membrane was seriously damaged upon irradiation.49 Lipid peroxide accumulation, a typical characteristic of ferroptosis, was detected under hypoxia using a commercial lipid peroxide probe C11-BODIPY. As displayed in Fig. 5c and S26,† bio-imaging disclosed obvious green fluorescence in cells incubated with d-ST alone, indicating the accumulation of lipid peroxide.50,51 Moreover, when HepG2 cells were treated with d-ST, the intracellular adenosine triphosphate (ATP) level decreased dramatically in a concentration-dependent manner under NIR-light irradiation for 1 min (Fig. S27†). Therefore, one could clearly see that the photo-reaction pathway based on the ET process of in situ self-assembled d-ST witnesses a great advance for photodynamic therapy in hypoxic tumors.
High-efficiency therapeutic performance against hypoxic tumors
The in vivo anti-tumor effect of d-ST (10 μM, 200 μL) was explored on tumor-bearing female ICR mouse models. Seven days after injection of H22 cells into the mice, the volume of the original tumor went up to ∼100 mm3 (Fig. 6a). To more intuitively differentiate the anticancer effect for deep-seated hypoxic tumors, the mice were randomly divided into four groups (n = 3) and received different treatments ((i): control; (ii): laser; (iii): d-ST; (iv): d-ST + laser) by intratumoral (i.t.) administration. As demonstrated in Fig. S28,† after 18 days of treatment, compared to groups (i) (injected with PBS alone), (ii) (NIR-light irradiation alone) and (iii) (d-ST without irradiation), the tumor presented nearly 100% inhibition in d-ST + laser group (iv) with almost complete ablation, which agreed well with the representative mouse tumor images (Fig. S29†) and relative tumor volumes (Fig. 6b). Relative body weight (Fig. 6c) over the whole therapeutic course did not show noticeable changes, indicating the superior biocompatibility of d-ST. Moreover, H&E (hematoxylin-eosin) staining showed that the d-ST + laser group caused significant destruction of H22 tumor tissues (Fig. 6d), whereas there was no obvious inflammation or histological changes in the main organs (heart, liver, spleen, lung, kidneys) (Fig. 6e). All the above offered strong evidence for the potency of d-ST in photodynamic therapy for hypoxic tumors and superior bio-safety in biomedical applications. | ||
| Fig. 6 (a) The scheme of NIR-light activated photodynamic therapy on an in vivo tumor-bearing mouse model. (b) Relative tumor volume (c) and relative body weight of different groups. (d) H&E staining of tumor tissues in different groups. (e) H&E staining for pathological analysis (heart, liver, spleen, lung, kidneys) in different groups. Scale bar: 200 μm. | ||
Conclusions
In summary, we carefully regulated the assembly behaviors of a single molecule and obtained d-ST nanofibers with certain crystallinity. Compared with disordered d-CT, the ordered d-ST nanofibers had a higher SP and a more favorable exciton lifetime, which could effectively promote the ET process and generate H˙ upon NIR photo-excitation. Encouragingly, d-ST could precisely target mitochondria and intercept the ET process in the mitochondrial respiratory chain through coupling with the oxidation of NADPH and subsequently induce ferroptosis under hypoxic conditions. This work achieved NIR-irradiation triggering the yield of H˙ based on in situ assembled phototherapeutic nanoagents, which would open up new possibilities for phototherapy and beyond.Data availability
All experimentally relevant data can be obtained from the manuscript and ESI.†Author contributions
Yingcui Bu: conceptual design, investigation, synthesis of compounds, processing of test data, writing-original draft, and funding acquisition. Zhou Lu and Zhipeng Yu: processing of data on femtosecond (fs) transient absorption (TA) spectroscopy, funding acquisition, and validation. Haoran Wang: DFT calculation. Yu Deng: processing of test data. Xiaojiao Zhu and Hongping Zhou: method inquiry, project administration, funding acquisition, writing-review, and editing. All authors approved the final version of the manuscript.Conflicts of interest
The authors declare no conflict of interest.Acknowledgements
We thank the financial support from the National Natural Science Foundation of China (51972001, 52372073, 22375003, 22073001, and 22005002), Changjiang Scholars and Innovative Research Team in University, and Anhui Agricultural University Scientific Research Start-up Fund (rc382306). The University Annual Scientific Research Plan of Anhui Province (2022AH010013 and 2023AH050064).Notes and references
- Y.-Y. Zhao, L. Zhang, Z. Chen, B.-Y. Zheng, M. Ke, X. Li and J.-D. Huang, J. Am. Chem. Soc., 2021, 143, 13980–13989 CrossRef CAS PubMed.
- K. Wen, H. Tan, Q. Peng, H. Chen, H. Ma, L. Wang, A. Peng, Q. Shi, X. Cai and H. Huang, Adv. Mater., 2022, 34, 2108146 CrossRef CAS.
- J. An, S. Tang, G. Hong, W. Chen, W. M. Chen, J. Song, Z. Li, X. Peng, F. Song and W.-H. Zheng, Nat. Commun., 2022, 13, 2225 CrossRef CAS.
- K.-X. Teng, L.-Y. Niu and Q.-Z. Yang, Chem. Sci., 2022, 13, 5951–5956 RSC.
- M. Li, T. Xiong, J. Du, R. Tian, M. Xiao, L. Guo, S. Long, J. Fan, W. Sun, K. Shao, X. Song, J. W. Foley and X. Peng, J. Am. Chem. Soc., 2019, 141, 2695–2702 CrossRef CAS PubMed.
- J. Peng, K. Du, J. Sun, X. Yang, X. Wang, X. Zhang, G. Song and F. Feng, Angew. Chem., Int. Ed., 2023, 62, e202214991 CrossRef CAS PubMed.
- J. Sun, X. Cai, C. Wang, K. Du, W. Chen, F. Feng and S. Wang, J. Am. Chem. Soc., 2021, 143, 868–878 CrossRef CAS PubMed.
- J. Zhuang, B. Wang, H. Chen, K. Zhang, N. Li, N. Zhao and B. Z. Tang, ACS Nano, 2023, 17, 9110–9125 CrossRef CAS PubMed.
- E. D. Cosco, et al. , Nat. Chem., 2020, 12, 1123–1130 CrossRef CAS PubMed.
- Y. Bu, T. Xu, X. Zhu, J. Zhang, L. Wang, Z. Yu, J. Yu, A. Wang, Y. Tian, H. Zhou and Y. Xie, Chem. Sci., 2020, 11, 10279–10286 RSC.
- X. Ma, Y. Huang, W. Chen, J. Liu, S. H. Liu, J. Yin and G.-F. Yang, Angew. Chem., Int. Ed., 2023, 62, e202216109 CrossRef CAS PubMed.
- D. Liu, A. M. El-Zohry, M. Taddei, C. Matt, L. Bussotti, Z. Wang, J. Zhao, O. F. Mohammed, M. Di Donato and S. Weber, Angew. Chem., Int. Ed., 2020, 59, 11591–11599 CrossRef CAS PubMed.
- S. Kazemi, Y. Jang, A. Liyanage, P. A. Karr and F. D'Souza, Angew. Chem., Int. Ed., 2022, 61, e202212474 CrossRef CAS PubMed.
- A. Pannwitz and O. S. Wenger, J. Am. Chem. Soc., 2017, 139, 13308–13311 CrossRef CAS PubMed.
- K.-X. Teng, L.-Y. Niu and Q.-Z. Yang, J. Am. Chem. Soc., 2023, 145, 4081–4087 CrossRef CAS.
- X. Yu, S. Wang, X. Zhang, A. Qi, X. Qiao, Z. Liu, M. Wu, L. Li and Z. L. Wang, Nano Energy, 2018, 46, 29–38 CrossRef CAS.
- S. Wang, M. Rong, H. Li, T. Xu, Y. Bu, L. Chen, X. Chen, Z.-P. Yu, X. Zhu, Z. Lu and H. Zhou, Small, 2022, 18, 2104857 CrossRef CAS.
- W. Chen, Z. Wang, M. Tian, G. Hong, Y. Wu, M. Sui, M. Chen, J. An, F. Song and X. Peng, J. Am. Chem. Soc., 2023, 145, 8130–8140 CrossRef CAS PubMed.
- K.-X. Teng, L.-Y. Niu, N. Xie and Q.-Z. Yang, Nat. Commun., 2022, 13, 6179 CrossRef CAS PubMed.
- M. R. Wasielewski, Acc. Chem. Res., 2009, 42, 1910–1921 CrossRef CAS PubMed.
- C. E. Ramirez, S. Chen, N. E. Powers-Riggs, I. Schlesinger, R. M. Young and M. R. Wasielewski, J. Am. Chem. Soc., 2020, 142, 18243–18250 CrossRef CAS PubMed.
- H. Shigemitsu, K. Ohkubo, K. Sato, A. Bunno, T. Mori, Y. Osakada, M. Fujitsuka and T. Kida, JACS Au, 2022, 2, 1472–1478 CrossRef CAS PubMed.
- X. Zhao, S. He, J. Wang, J. Ding, S. Zong, G. Li, W. Sun, J. Du, J. Fan and X. Peng, Adv. Mater., 2023, 2305163 CrossRef CAS PubMed.
- C. Huang, C. Liang, T. Sadhukhan, S. Banerjee, Z. Fan, T. Li, Z. Zhu and P. Zhang, Angew. Chem., Int. Ed., 2021, 60, 9474–9479 CrossRef CAS PubMed.
- S. Altenhöfer, K. A. Radermacher, P. W. M. Kleikers, K. Wingler and H. H. H. W. Schmidt, Antioxid. Redox Signaling, 2014, 23, 406–427 CrossRef PubMed.
- R. Hou, M. Li, X. Ma, H. Huang, H. Lu, Q. Jia, Y. Liu, X. Xu, H.-B. Li and Z. Bo, ACS Appl. Mater. Interfaces, 2020, 12, 46220–46230 CrossRef CAS.
- W. Xiang, X. Xu, Y. Huang, L. Yu, R. Li, Y. Jiang and Q. Peng, Chem. Eng. J., 2022, 441, 136058 CrossRef CAS.
- T.-J. Wen, Z.-X. Liu, Z. Chen, J. Zhou, Z. Shen, Y. Xiao, X. Lu, Z. Xie, H. Zhu, C.-Z. Li and H. Chen, Angew. Chem., Int. Ed., 2021, 60, 12964–12970 CrossRef CAS PubMed.
- Y.-c. Liu, G.-j. Liu, W. Zhou, G.-l. Feng, Q.-y. Ma, Y. Zhang and G.-w. Xing, Angew. Chem., Int. Ed., 2023, 62, e202309786 CrossRef CAS PubMed.
- Y. Bu, X. Zhu, H. Wang, J. Zhang, L. Wang, Z. Yu, Y. Tian, H. Zhou and Y. Xie, Anal. Chem., 2021, 93, 12059–12066 CrossRef CAS PubMed.
- N. Zhang, L. Wang, H. Wang, R. Cao, J. Wang, F. Bai and H. Fan, Nano Lett., 2018, 18, 560–566 CrossRef CAS PubMed.
- Y. Sheng, W. Li, L. Xu and Y. Zhu, Adv. Mater., 2022, 34, 2102354 CrossRef CAS PubMed.
- X. Liu, W. Zhan, G. Gao, Q. Jiang, X. Zhang, H. Zhang, X. Sun, W. Han, F.-G. Wu and G. Liang, J. Am. Chem. Soc., 2023, 145, 7918–7930 CrossRef CAS PubMed.
- Z. Zhou, K. Maxeiner, P. Moscariello, S. Xiang, Y. Wu, Y. Ren, C. J. Whitfield, L. Xu, A. Kaltbeitzel, S. Han, D. Mücke, H. Qi, M. Wagner, U. Kaiser, K. Landfester, I. Lieberwirth, D. Y. W. Ng and T. Weil, J. Am. Chem. Soc., 2022, 144, 12219–12228 CrossRef CAS.
- S. Chagri, D. Y. W. Ng and T. Weil, Nat. Rev. Chem, 2022, 6, 320–338 CrossRef PubMed.
- J. Yang, J. Jing and Y. Zhu, Adv. Mater., 2021, 33, 2101026 CrossRef CAS.
- R. Yu, H. Yao, L. Hong, Y. Xu, B. Gao, J. Zhu, Y. Zu and J. Hou, Adv. Energy Mater., 2018, 8, 1802131 CrossRef.
- S. Okazaki and K. Takeshita, Appl. Magn. Reson., 2018, 49, 881–892 CrossRef CAS.
- A. Roshanzadeh, H. Kang, S.-H. You, J. Park, N. D. Khoa, D.-H. Lee, G.-J. Kim and E.-S. Kim, Biosens. Bioelectron., 2019, 146, 111753 CrossRef CAS PubMed.
- A. Chiarugi, C. Dölle, R. Felici and M. Ziegler, Nat. Rev. Cancer, 2012, 12, 741–752 CrossRef CAS.
- S. Panchavarnam, P. Pushpanandan and M. Ravikanth, Inorg. Chem., 2022, 61, 1562–1570 CrossRef CAS PubMed.
- S. Hu, L. Hu, X. Zhu, Y. Wang and M. Liu, Angew. Chem., Int. Ed., 2021, 60, 19451–19457 CrossRef CAS.
- S. I. van Dijk, C. P. Groen, F. Hartl, A. M. Brouwer and J. W. Verhoeven, J. Am. Chem. Soc., 1996, 118, 8425–8432 CrossRef CAS.
- X. Chen, J. Wang, Y. Chai, Z. Zhang and Y. Zhu, Adv. Mater., 2021, 33, 2007479 CrossRef CAS PubMed.
- A. D. Nidhankar, Goudappagouda, D. S. Mohana Kumari, S. K. Chaubey, R. Nayak, R. G. Gonnade, G. V. P. Kumar, R. Krishnan and S. S. Babu, Angew. Chem., Int. Ed., 2020, 59, 13079–13085 CrossRef CAS PubMed.
- R. J. Hafner, L. Tian, J. C. Brauer, T. Schmaltz, A. Sienkiewicz, S. Balog, V. Flauraud, J. Brugger and H. Frauenrath, ACS Nano, 2018, 12, 9116–9125 CrossRef CAS PubMed.
- Y. Bu, M. Rong, J. Wang, X. Zhu, J. Zhang, L. Wang, Z. Yu, Y. Tian, H. Zhou and Y. Xie, Anal. Chem., 2022, 94, 8373–8381 CrossRef CAS PubMed.
- H. Yuan, Z. Han, Y. Chen, F. Qi, H. Fang, Z. Guo, S. Zhang and W. He, Angew. Chem., Int. Ed., 2021, 60, 8174–8181 CrossRef CAS PubMed.
- M. Tian, W. Chen, G. Hong, M. Chen, F. Song, W.-H. Zheng and X. Peng, et al. , ACS Mater. Lett., 2023, 5, 1922–1928 CrossRef CAS.
- K. Li, K. Xu, Y. He, Y. Yang, M. Tan, Y. Mao, Y. Zou, Q. Feng, Z. Luo and K. Cai, ACS Nano, 2023, 17, 4667–4687 CrossRef CAS PubMed.
- M. Wei, J. Bai, X. Shen, K. Lou, Y. Gao, R. Lv, P. Wang, X. Liu and G. Zhang, ACS Nano, 2023, 17, 11345–11361 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4sc02199a |
| This journal is © The Royal Society of Chemistry 2024 |