
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceIntermolecular C–H silylations of arenes and heteroarenes with mono-, bis-, and tris(trimethylsiloxy)hydrosilanes: control of silane redistribution under operationally diverse approaches†
Noah
Swann
a,
Kiki
Tang‡
a,
Jihyeon
Nam‡
a,
Jooyeon
Lee
a,
Marek
Domin
b,
Thomas E.
Shaw
c,
Stosh A.
Kozimor
 c,
Salina
Som
a and
Kangsang L.
Lee
*a
c,
Salina
Som
a and
Kangsang L.
Lee
*a
aUniversity of Central Florida, Department of Chemistry, 4111 Libra Drive, PSB #255, Orlando, FL, USA 32816. E-mail: k.lee@ucf.edu
bMass Spectrometry Centre, Boston College, 245 Beacon Street, Chestnut Hill, MA 02467, USA
cLos Alamos National Laboratory, P.O. Box 1663, Los Alamos, NM 87545, USA
First published on 1st July 2024
Abstract
Efficient catalytic protocols for C–H silylations of arenes and heteroarenes with sterically and electronically different hydrosiloxysilanes are disclosed. The silylations are catalyzed by a well-defined Rh-complex (1 mol%), derived from [Rh(1,5-hexadiene)Cl]2 and a bulky BINAP type ligand. This catalyst not only promotes C–Si bond formation affording the desired products in up to 95% isolated yield, but also can suppress the silane redistribution side reactions of HSiMe2(OTMS). The protocol can also be applied for the C–H silylations of more reactive HSiMe(OTMS)2 with a much lower catalyst loading (0.25 mol%) and even with sterically demanding HSi(OTMS)3. The steric bulk of the arene substituent and hydrosiloxysilane is a major factor in determining the regioselectivity and electronic effect as secondary. The current method can be performed under operationally diverse conditions: with/without a hydrogen scavenger or solvent.
Introduction
The siloxysilane framework is widely used in the syntheses of polysiloxane-based silicone materials.1 Due to their unique properties and versatility, polysiloxanes are used in many different applications, ranging from common consumer products (baby bottles, toys, shampoo, detergents) to high-tech products (electronics, automotives, aero- or spacecraft).2 Their high biocompatibility and durability in the human body further expand the importance and utility of silicone materials in biomedical science and engineering.1a,2b,e In order to prepare such polysiloxane materials, various functional groups are required in the polymer matrix; many functional alkyl groups can be readily incorporated by other organic transformations including hydrosilylations.3 For aryl substituents, however, only the phenyl group has been heavily employed due to the lack of a reliable synthetic method. Although there are some traditional methods to form aryl–Si bonds including reactions between aryl Grignard or aryl lithium reagents and chlorosiloxysilanes, the arene scope is significantly limited due to their intrinsic incompatibility with electrophilic functional groups in the aryl groups.4 Furthermore, such methods are not welcome in industrial scale production because of the use of non-recyclable ethereal solvents and considerable amount of magnesium- or lithium salt waste at the end.5 Thus, to improve the physical and chemical properties of silicone materials, more reliable synthetic routes are needed to incorporate various aryl groups into the siloxane framework.C–H silylation is one of the most direct and atom-economical pathways to incorporate such functional aryl groups.6–9 Though there are many reports on C–H silylation reactions, most of the catalytic systems either require a pre-engineered directing group in the arenes,6 or involve a more reactive but less useful trialkyl- or phenylsilane reagent,7,8 or have limited hydrosiloxysilane scope [only highly reactive HSiMe(OTMS)2].9 Considering the dynamic structural framework of numerous silicone materials that contain simple and/or complex –(OSiMe2)n– units, structurally diverse hydrosiloxysilanes are needed for C–H silylations with various arenes and heteroarenes.
Here, we report operationally diverse synthetic methods to access functional aryl- and heteroaryl siloxysilanes by intermolecular C–H silylations of mono-, bis-, or tris(trimethylsiloxy)hydrosilanes with various arenes and heteroarenes. In the presence of 0.25–1 mol% catalyst, C–H silylations can be performed under neat conditions with low/moderate concentration of arenes, or under solvent conditions. Development of a well-defined catalyst led us to successfully control redistribution of a hydrosiloxysilane, resulting in the desired products in up to 95% isolated yield. The efficiency of the silylation can be further improved by portion-wise addition of hydrosiloxysilanes. In addition, the current protocols can be applied for double C–H silylation or carried out without alkene as a hydrogen scavenger.
Results and discussion
We initiated our study by optimizing the C–H silylation between benzene and the mono(siloxy)hydrosilane, HSiMe2(OTMS), as the initial arene and silane (Table 1). In the presence of [Rh(1,5-hexadiene)Cl]2 and a bidentate phosphine ligand (L1), the desired product P1 was not observed. Instead, a significant amount of silane redistribution byproduct was obtained (54%). With L2, we started to obtain P1 (72% conv. and 55% isolated yield, entry 2) together with the undesired redistribution byproduct (28%). When BINAP ligand (L3) was employed, the silylation was less efficient (44% conv., entry 3). With a bulkier BINAP (L4), however, the conversion was significantly increased (74% conv. and 57% yield, entry 4) without improvement in the redistribution (26%). Although a few different Rh salts were tested for the silylations, the similar silane redistribution (19–26%) and silylation efficiencies were observed (74–81% conversions, entries 5–7).10 A cationic Rh salt, on the other hand, increased the redistribution (52%, entry 8).|
|
|||||
|---|---|---|---|---|---|
| Entry | [Rh] | Ligand | [Rh] loading | Conv.; yield | Si-redistribution |
| a All reactions were performed under N2. Conversions were determined by 1H NMR analysis. | |||||
| 1 | [Rh(1,5-hexadiene)Cl]2 | L1 | 1 mol% | <5%; – | 54% |
| 2 | [Rh(1,5-hexadiene)Cl]2 | L2 | 1 mol% | 72%; 55% | 28% |
| 3 | [Rh(1,5-hexadiene)Cl]2 | L3 | 1 mol% | 44%; – | 18% |
| 4 | [Rh(1,5-hexadiene)Cl]2 | L4 | 1 mol% | 74%; 57% | 26% |
| 5 | [Rh(nbd)Cl]2 | L4 | 1 mol% | 81%; 53% | 19% |
| 6 | [Rh(coe)2Cl]2 | L4 | 1 mol% | 74%; 55% | 26% |
| 7 | [Rh(ethylene)2Cl]2 | L4 | 1 mol% | 77%; 64% | 23% |
| 8 | Rh(nbd)2BF4 | L4 | 2 mol% | 48%; – | 52% |

|
|||||
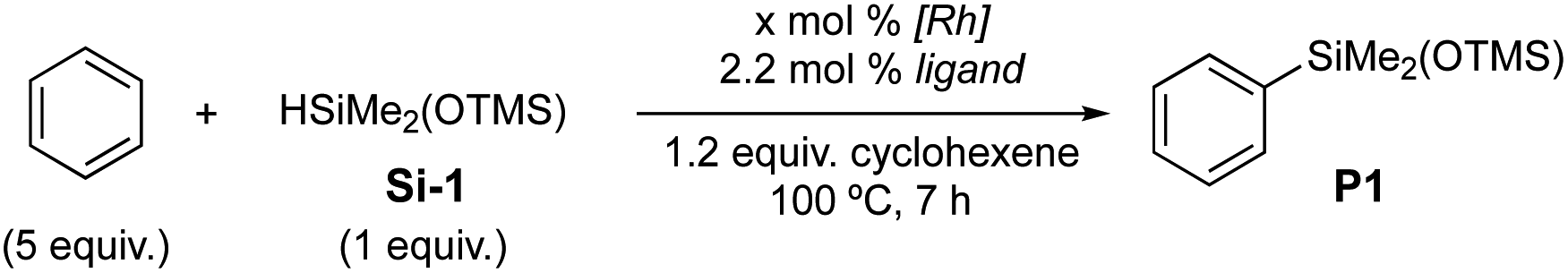
To improve the efficiency of the C–H silylations and to suppress the competing silane redistribution,11 several preformed complexes were prepared with L4. As shown in Table 2, C1 derived from [Rh(nbd)2Cl]2 slightly decreased the conversion to the desired product (79% conv., entry 1). Compared to in situ reactions (entries 6 and 7, Table 1), the corresponding preformed complexes (C2 and C3) marginally improved the efficiency of the silylation (78% and 82% conversions, entries 2 and 3, Table 2). The complex C4, however, was much more efficient in delivering the desired silylation product (93% conv., entry 4) with a significantly lower silane redistribution (7% vs. 26% in entry 4, Table 1). This increased conversion to the desired product P1 might be explained either by the improved efficiency of C4 toward the silylation, or by suppressing the silane redistribution by C4.
|
|
||||
|---|---|---|---|---|
| Entry | [Rh]/L | Preformed catalyst | Conv. | Si-redistribution |
| a All reactions were performed under N2. Conversions were determined by 1H NMR analysis. | ||||
| 1 | [Rh(nbd)Cl]2/L4 | C1 | 79% | 21% |
| 2 | [Rh(coe)2Cl]2/L4 | C2 | 78% | 22% |
| 3 | [Rh(ethylene)2Cl]2/L4 | C3 | 82% | 18% |
| 4 | [Rh(1,5-hexadiene)Cl]2/L4 | C4 | 93% | 7% |

Although hydrosiloxysilanes are generally known as a class of reactive hydrosilanes with stable Si–O–Si bonds,3 we have observed silane redistribution in our studies. In order to further understand the reactivity difference between in situ-formed catalysts and preformed complexes (Tables 1 and 2), we decided to study the redistribution tendency of each component of the catalysts. First, like other preformed complexes used in Table 2 (C1–C3),10 we have synthesized C4 from [Rh(1,5-hexadiene)Cl]2 and L4 in THF at 22 °C (Scheme 1). X-ray crystallography revealed that the complex was obtained in a dimeric form.10 Bond lengths of Rh and bridging Cl atoms are between 2.42 and 2.43 Å and the distance between two Rh centres is 3.60 Å. The bond angles of Cl–Rh–Cl and Rh–Cl–Rh are 81° and 96°, respectively.
 | ||
| Scheme 1 Synthesis of a well-defined Rh-complex C4 and its X-ray structure. | ||
With C4, we investigated the redistribution tendencies of each catalytic component. As depicted in Scheme 2, the hydrosilane Si-1 alone does not undergo the redistribution at 100 °C for 7 h. When 1 mol% of [Rh(1,5-hexadiene)Cl]2 was added to Si-1, however, the redistribution byproduct started to appear in 1H NMR after 0.5 h (6%). A significant amount of the byproduct was observed in 3 h (55%), but during the next 4 hours the redistribution byproduct was slightly increased (67% after 7 h). On the other hand, the Lewis-basic phosphine L4 did not cause the side reaction. When the preformed complex C4 was subjected to Si-1, negligible amount of the redistribution byproduct was observed by 1H NMR (<5%). The similar trend of the silane redistribution was also seen in 29Si NMR spectroscopy analysis.10 This finding may partially explain the much lower redistribution with C4 (7%, entry 4 in Table 2), compared to 26% redistribution by the in situ protocol (entry 4, Table 1). Furthermore, this indicates that by using the well-defined catalyst (C4), the redistribution side reaction can be significantly suppressed. The different redistribution tendencies of [Rh(1,5-hexadiene)Cl]2 and C4 may be explained by the steric and electronic effects; the less sterically hindered and more Lewis acidic [Rh(1,5-hexadiene)Cl]2 can readily interact with Si-1, compared to C4 with the electron-rich and bulky ligand. The observed minimal redistribution (7%, entry 4 in Table 2) might result from the interaction of Si-1 with other active catalytic species in the C–H silylation process.
 | ||
| Scheme 2 Redistribution side-reactions of Si-1 by catalytic components. The intensity of peaks between 3.2 and 5.2 ppm was manually increased for clarity (vs. peaks between 0 and 1.0 ppm). For unprocessed 1H and 29Si NMR spectra, see the ESI.† | ||
With the well-defined complex C4, we set out to investigate the C–H silylation of mono(siloxy)hydrosilane (Si-1). As shown in Scheme 3, benzene and naphthalene undergo the silylations to afford the desired products P1 and P2 in 71% and 41% of isolated yield, respectively. Electron-rich anisole and sterically hindered t-butylbenzene are effective substrates for the silylation (P3, 57% conv. and P4, 59% conv.). Halogen-containing arenes are also compatible with the catalytic system (P5 and P6). For substituted arenes, o-silylation products are generally obtained as major products (P3–P6) except P4 in which m-silylation is dominant due to the steric bulk of the t-butyl group. The silylations of heteroarenes are efficient to furnish desired products in up to 71% yield (P7–P12) where α-silylation products are major regioisomers. Chelating thiofuran (P10) and benzothiophene10 were relatively less effective (41% yield and 36% yield, respectively).
 | ||
| Scheme 3 C–H silylations of arenes and heteroarenes with mono(siloxy)silane. | ||
Next, the current protocol was tested for the silylation of bis(siloxy)silane Si-2 (Scheme 4). It is noteworthy that Si-2 is generally much more reactive in the C–H silylation than Si-1 with no silane redistribution.9b–g Thus, 0.25 mol% of C4 is sufficient to catalyse the C–H silylation to afford the desired product at an even lower reaction temperature (80 °C, Scheme 4vs. 100 °C for Si-1 in Scheme 3). The silylations of benzene, polyarene, anisole, and halogen-containing arenes proceeded efficiently to afford the desired products in up to 95% isolated yield (P13–P18). For the substituted arenes, o-silylation products were still the major products (P15–P18), but the ratio of m-silylation products were slightly increased, compared to P3–P6 shown in Scheme 3. This difference may be attributed to the steric bulkiness of Si-2 (vs.Si-1). Electronically activated heteroarenes are generally more effective in this C–H silylation, furnishing the silylation products in 80–92% conversions. The high reactivity of Si-2 was also evidenced in the product distribution of heteroarene silylations; more and/or diverse double C–H silylation products are obtained (P19–P21). Fused heteroarenes were equally reactive toward the silylation, so that P22 and P23 were isolated in 66% and 88% yields, respectively. As aforementioned, sulfur-containing arenes are less reactive, delivering P24 in moderate yields (44% yield, Scheme 4).
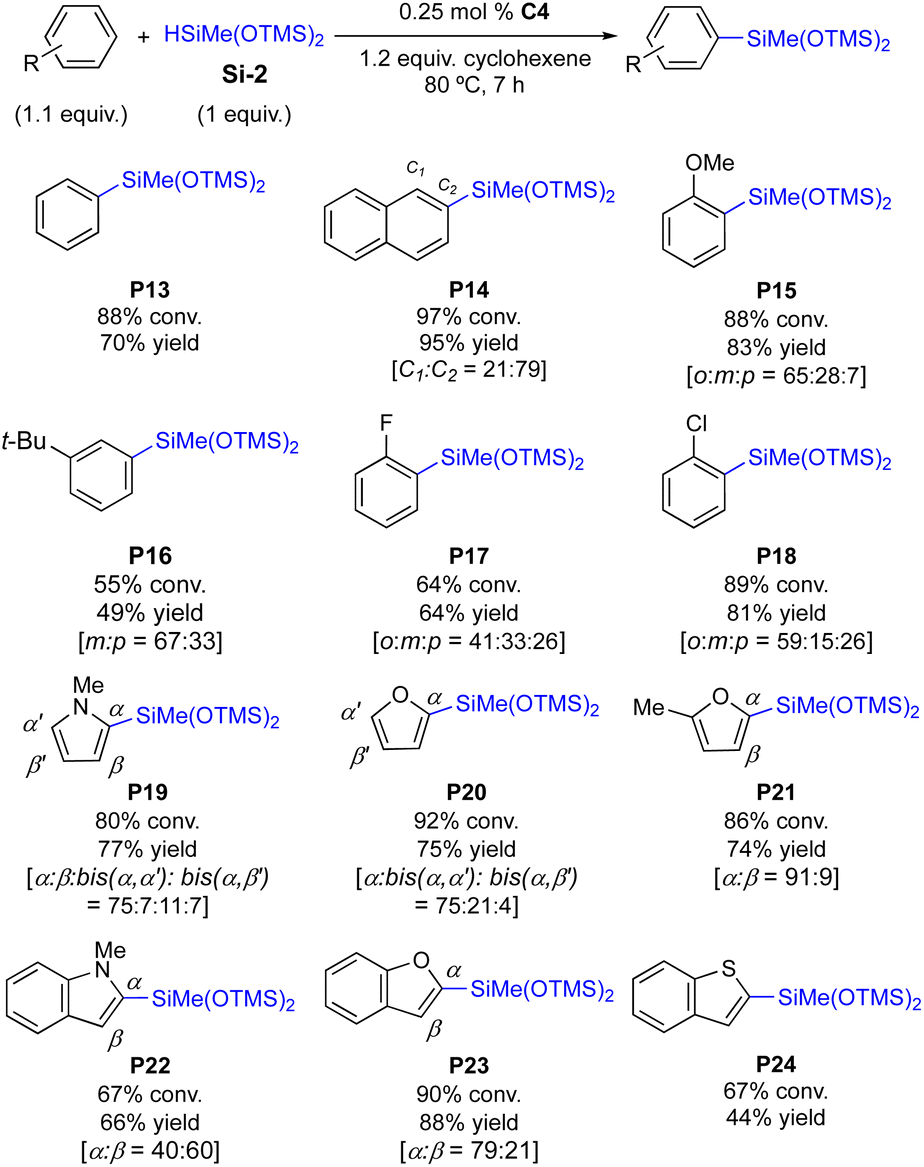 | ||
| Scheme 4 C–H silylation of arenes and heteroarenes with bis(siloxy)silane. | ||
The present protocol was further expanded toward the silylation of a sterically demanding tris(siloxy)hydrosilane Si-3. As shown in Scheme 5, the silylations of Si-3 require a relatively high catalyst loading (1 mol%) and an elevated reaction temperature (120 °C).10 With the sterically bulky Si-3, it is found that the in situ generated catalyst is slightly more efficient over the preformed C4 complex in general. This reaction condition was operable because any redistribution of Si-3 was not observed. For example, P25 is obtained in 92% yield in the presence of the in situ generated catalyst (vs. 80% yield with the preformed C4, Scheme 5). A polycyclic arene is an effective substrate to afford the desired silylation product in 66% yield (P26). The silylation of electron-rich anisole is less efficient (53% conv., P27) with an increased amount of p-silylation product (45%). Fluorobenzene is effective enough to deliver the desired product P28 (71% yield) and the m-substituted product was obtained as the major product. This substitution pattern is in accordance with the observations in the reactions of Si-1 and Si-2, in which o-silylation products are the major products with halogen substituents, but m-silylation becomes favoured as the steric bulkiness of the silane increases (P5vs.P17vs.P28). This also implies that the current protocols are more sensitive to the steric effects of both arene substituents and silanes than the electronics of arene substituents: the steric bulk is a major determining factor of the regioselectivity in this C–H silylation. Heteroarenes are generally less efficient in the silylation with Si-3, furnishing the desired products in moderate yields (37–85% conversions, P29–P33). This observation is opposite to the reactions with Si-1 and Si-2 shown in Schemes 3 and 4.
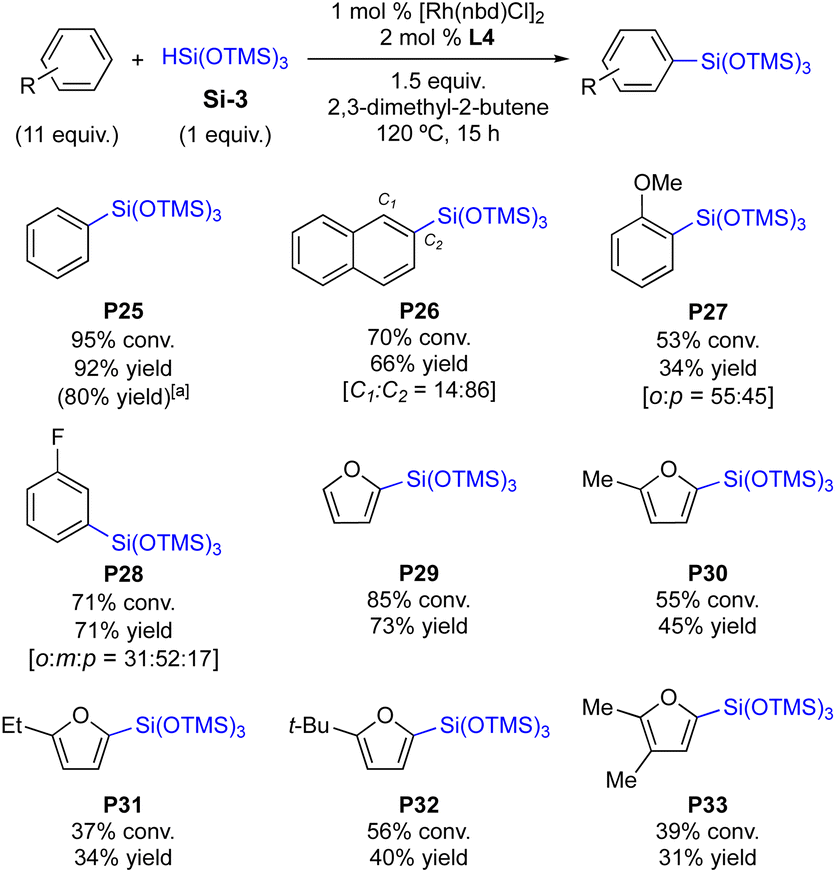 | ||
| Scheme 5 C–H silylation of arenes and heteroarenes with tris(siloxy)silane. | ||
Next, we briefly investigated the feasibility of double C–H silylations. With 2.5 equivalent of more reactive Si-2 and in the presence of 0.25 mol% C4, the double silylation of furan proceeded efficiently to afford P34 as a major product (>98% conv., 60% yield, Scheme 6). The double silylation products of N-methyl pyrrole were isolated in 86% yield with sizable amount of two different double silylation products (P36 and P37 in 67![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :33 ratio). Though less efficient, the corresponding silylation of thiofuran resulted in appreciable amount of the product P38 along with the mono-silylation product P39 (27:73 ratio).
:33 ratio). Though less efficient, the corresponding silylation of thiofuran resulted in appreciable amount of the product P38 along with the mono-silylation product P39 (27:73 ratio).
 | ||
| Scheme 6 Double C–H silylation of heteroarenes. | ||
In order to further suppress the redistribution of silanes, thereby improving overall efficacy, the silylation reactions were performed by portion-wise addition of silanes. As illustrated in Scheme 7, the silylations involving Si-1, which is prone to redistribution became more efficient by the portion-wise addition protocol (P1–P7, 58–95% conversions). With relatively stable Si-2 and Si-3, similar efficiencies were observed in the silylation reactions (P13–P20 with Si-2 and P25–P29 with Si-3). Overall, the portion-wise protocol was especially useful for unactivated arenes rather than heteroarenes. Additionally, this portion-wise silylation protocol involving various hydrosiloxysilanes was further tested in the presence of a solvent (THF) and similar results were obtained.10
 | ||
| Scheme 7 Portion-wise C–H silylation of arenes and heteroarenes with various siloxysilanes. | ||
The current protocol was further tested for hydrogen scavenger-free C–H silylations. As shown in Scheme 8, the overall efficiency of the silylations of unactivated arenes was significantly decreased without alkenes as exemplified in P1 (32% conv. vs. 93% conv. in Scheme 3). For heteroarenes, however, similar (P8: 65% vs. 71% conv. in Scheme 3) or in some cases, much higher efficiencies are achievable (P12: 81% conv. vs. 51% conv. in Scheme 3).
 | ||
| Scheme 8 C–H silylation of arenes and heteroarenes without the hydrogen scavenger. | ||
Conclusions
In this report, Rh-based catalytic platforms for C–H silylations of arenes and heteroarenes were developed to prepare various arylsiloxysilanes from mono-, bis-, and tris(siloxy)hydrosilanes. The suppression of the redistribution by the well-defined catalyst C4 was an important finding, allowing us to utilize the labile Si-1 for C–Si bond formations and to improve overall catalytic efficiency. The same protocol can be further extended to the C–H silylations with much more reactive Si-2 and with the sterically demanding Si-3. The regioselectivity was mainly affected by the steric bulkiness of arene substituents or hydrosilanes and the electronic effects of the substituents were a secondary factor. The current protocols can be applied to introduce two siloxane moieties to a heteroarene and performed by a portion-wise addition of silanes, or, although limited, without a hydrogen scavenger. Thus, this study can make a positive impact on innovating numerous silicone materials and products by introducing various functional aryl groups into polysiloxane systems.Data availability
All the data supporting this article have been included in the main text and the ESI.† Crystallographic data have been deposited at the CCDC under 2351158.Author contributions
N. S., K. T., J. N., J. L., S. S. and K. L. conceptualized the research and performed the investigation. M. D. studied HRMS. T. E. S. and S. A. K. analysed X-ray crystals. K. L. L. wrote the manuscript with contributions from all authors. K. L. L. supervised this study.Conflicts of interest
There are no conflicts to declare.Acknowledgements
N. Swann and K. Tang are thankful for the UCF undergraduate research program. This work was financially supported by NSF (CHE-2246567) and UCF. T. E. Shaw acknowledges helpful discussion with Dr Brian L. Scott (Los Alamos National Laboratory) for the X-ray crystallography. We thank the U.S. Department of Energy, Office of Science, Office of Basic Energy Sciences, Heavy Element Chemistry program (2020LANLE372) for funding the X-ray diffractometer portion of this work (S. A. K. and T. E. S.). LANL is an affirmative action/equal opportunity employer managed by Triad National Security, LLC, for the National Nuclear Security Administration of the U.S. DOE.Notes and references
- For selected recent reviews, see: (a) M. A. Brook, Silicon in Organic, Organometallic and Polymer Chemistry, Wile, New York, 2000 Search PubMed; (b) The Chemistry of Organic Silicon Compounds, ed. Z. Rappoport and Y. Apeloig, Wiley, Chichester, 2003 Search PubMed; (c) Y. Abe and T. Gunji, Prog. Polym. Sci., 2004, 29, 149–182 CrossRef CAS; (d) P. Lucas and J. Robin, Adv. Polym. Sci., 2007, 209, 111–147 CAS; (e) Silicon Polymers, vol. 235, ed. A. M. Muzafarov, Adv. Polym. Sci., 2011, pp. 1–228 Search PubMed; (f) E. Yilgor and I. Yilgor, Prog. Polym. Sci., 2014, 39, 1165–1195 CrossRef CAS and references therein..
- For selected reviews, see: (a) J. Mark, Acc. Chem. Res., 2004, 37, 946–953 CrossRef CAS PubMed; (b) G. Giordano and M. F. Refojo, Prog. Polym. Sci., 1998, 23, 509–532 CrossRef CAS; (c) J. C. McDonald and G. M. Whitesides, Acc. Chem. Res., 2002, 35, 491–499 CrossRef CAS PubMed; (d) J. G. C. Veinot and T. J. Marks, Acc. Chem. Res., 2005, 38, 632–643 CrossRef CAS PubMed; (e) J. M. Lambert and J. Biomed, Mater. Res. B, 2006, 78, 167–180 Search PubMed; (f) D. R. Paul and J. E. Mark, Prog. Polym. Sci., 2010, 35, 893–901 CrossRef CAS; (g) B. A. Kamino and T. P. Bender, Chem. Soc. Rev., 2013, 42, 5119–5130 RSC; (h) J. J. Chrusciel and E. Lesniak, Prog. Polym. Sci., 2015, 41, 67–121 CrossRef CAS; (i) I. You, M. Kong and U. Jeong, Acc. Chem. Res., 2019, 52, 63–72 CrossRef CAS PubMed; (j) R. Baier, M. Ricotta, V. Andolina, F. Siraj, R. Forsberg and A. Meyer, Adv. Polym. Sci., 2019, 284, 367–376 CrossRef; (k) A. Roy and M. Oestreich, Angew. Chem., Int. Ed., 2021, 60, 4408–4410 CrossRef CAS PubMed; (l) H. Wang, X. Zhang, Y. Li and L. Xu, Angew. Chem., Int. Ed., 2022, 61, e202210851 CrossRef CAS PubMed.
- (a) B. Marciniec, Comprehensive Handbook on Hydrosilylation, Pergamon, 1992 Search PubMed; (b) S. Putzien, O. Nuyken and F. E. Kuhn, Prog. Polym. Sci., 2010, 35, 687–713 CrossRef CAS.
- (a) R. L. Merker and M. J. Scott, J. Am. Chem. Soc., 1963, 85, 2243–2244 CrossRef CAS; (b) L. S. Luh, Y. S. Wen, H. Tobita and H. Ogino, Bull. Chem. Soc. Jpn., 1997, 70, 2193–2200 CrossRef CAS.
- (a) A. Kadam, M. Nguyen, M. Kopach, P. Richardson, F. Gallou, Z. Wan and W. Zhang, Green Chem., 2013, 15, 1880–1888 RSC; (b) S. G. Koenig, D. K. Leashy and A. S. Wells, Org. Process Res. Dev., 2018, 22, 1344–1359 CrossRef CAS.
- For recent reviews on catalytic C–H silylations, see: (a) J. F. Hartwig, Acc. Chem. Res., 2012, 45, 864–873 CrossRef CAS PubMed; (b) C. Cheng and J. F. Hartwig, Chem. Rev., 2015, 115, 8946–8975 CrossRef CAS PubMed; (c) R. Sharma, R. Kumar, I. Kumar, B. Singh and U. Sharma, Synthesis, 2015, 47, 2347–2366 CrossRef CAS; (d) Z. Xu and L.-W. Xu, ChemSusChem, 2015, 8, 2176–2179 CrossRef CAS PubMed; (e) J. F. Hartwig and E. A. Romero, Tetrahedron, 2019, 75, 4059–4070 CrossRef CAS PubMed; (f) S. C. Richter and M. Oestreich, Trends Chem., 2020, 2, 13–27 CrossRef CAS; (g) Y. Ge, X. Huang, J. Ke and C. He, Chem Catal., 2022, 2, 2898–2928 CrossRef CAS; (h) H. Khatua, S. Das, S. Patra and B. Chattopadhyay, Synthesis, 2023, 55, 3434–3453 CrossRef CAS.
- For reports on intermolecular C–H silylations of arenes without directing groups, see: (a) T. Sakakura, Y. Tokunaga, T. Sodeyama and M. Tanaka, Chem. Lett., 1987, 16, 2375–2378 CrossRef; (b) M. Ishikawa, S. Okazaki, A. Naka and H. Sakamoto, Organometallics, 1992, 11, 4135–4139 CrossRef CAS; (c) M. Ishikawa, H. Sakamoto, S. Okazaki and A. Naka, J. Organomet. Chem., 1992, 439, 19–21 CrossRef CAS; (d) Y. Uchimaru, A. M. M. Elsayed and M. Tanaka, Organometallics, 1993, 12, 2065–2069 CrossRef CAS; (e) K. Ezbiansky, P. I. Djurovich, M. LaForest, D. J. Sinning, R. Zayes and D. H. Berry, Organometallics, 1998, 17, 1455–1457 CrossRef CAS; (f) A. Naka, K. K. Lee, K. Yoshizawa, T. Yamabe and M. Ishikawa, Organometallics, 1999, 18, 4524–4529 CrossRef CAS; (g) T. Ishiyama, K. Sato, Y. Nishio and N. Miyaura, Angew. Chem., Int. Ed., 2003, 42, 5346–5348 CrossRef CAS PubMed; (h) T. Saiki, Y. Nishio, T. Ishiyama and N. Miyaura, Organometallics, 2006, 25, 6068–6073 CrossRef CAS; (i) M. Murata, N. Fukuyama, J.-i. Wada, S. Watanabe and Y. Masuda, Chem. Lett., 2007, 36, 910–911 CrossRef CAS; (j) T. Ishiyama, T. Saiki, E. Kishida, I. Sasaki, H. Ito and N. Miyaura, Org. Biomol. Chem., 2013, 11, 8162–8165 RSC; (k) M. Murai, K. Takami and K. Takai, Chem.–Eur. J., 2015, 21, 4566–4570 CrossRef CAS PubMed; (l) K. Lee, D. Katsoulis and J. Choi, ACS Catal., 2016, 6, 1493–1496 CrossRef CAS; (m) L. Rubio-Perez, M. Iglesias, J. Munarriz, V. Polo, V. Passarelli, J. J. Perez-Torrente and L. A. Oro, Chem. Sci., 2017, 8, 4811–4822 RSC; (n) B. Neil, F. Lucien, L. Fensterbank and C. Chauvier, ACS Catal., 2021, 11, 13085–13090 CrossRef CAS; (o) S. Som, J. Choi, D. Katsoulis and K. L. Lee, Chem. Sci., 2022, 13, 10759–10764 RSC.
- For examples on intermolecular silylations of C–H bonds of heteroarenes without directing groups, see: (a) T. Ishiyama, K. Sato, Y. Nishio, T. Saiki and N. Miyaura, Chem. Commun., 2005, 5065–5067 RSC; (b) N. Tsukada and J. F. Hartwig, J. Am. Chem. Soc., 2005, 127, 5022–5023 CrossRef CAS; (c) B. Lu and J. R. Falck, Angew. Chem., Int. Ed., 2008, 47, 7508–7510 CrossRef CAS PubMed; (d) Y. Sunada, H. Soejima and H. Nagashima, Organometallics, 2014, 33, 5936–5939 CrossRef CAS; (e) Y. Minami, T. Komiyama and T. Hiyama, Chem. Lett., 2015, 44, 1065–1067 CrossRef CAS; (f) H. F. T. Klare, M. Oestreich, J.-i. Ito, H. Nishiyama, Y. Ohki and K. Tatsumi, J. Am. Chem. Soc., 2011, 133, 3312–3315 CrossRef CAS PubMed; (g) A. Fedorov, A. A. Tourtov, N. A. Swisher and R. H. Grubbs, Chem. Sci., 2013, 4, 1640–1645 RSC; (h) M. Sasaki and Y. Kondo, Org. Lett., 2015, 17, 848–851 CrossRef CAS PubMed; (i) A. A. Tourtov, W.-B. Liu, K. N. Betz, A. Fedorov, B. M. Stoltz and R. H. Grubbs, Nature, 2015, 518, 80–84 CrossRef PubMed; (j) Q.-A. Chen, H. F. T. Klare and M. Oestreich, J. Am. Chem. Soc., 2016, 138, 7868–7871 CrossRef CAS PubMed; (k) H. Fang, L. Guo, Y. Zhang, W. Yao and Z. Huang, Org. Lett., 2016, 18, 5624–5627 CrossRef CAS PubMed; (l) K. Yonekura, Y. Iketani, M. Sekine, T. Tani, F. Matsui, D. Kamakura and T. Tsuchimoto, Organometallics, 2017, 36, 3234–3249 CrossRef CAS; (m) R. Sakamoto, B.-N. Nguyen and K. Maruoka, Asian J. Org. Chem., 2018, 7, 1085–1088 CrossRef CAS; (n) Y. Gu, Y. Shen, C. Zarate and R. Martin, J. Am. Chem. Soc., 2019, 141, 127–132 CrossRef CAS PubMed; (o) W. Xu, H. Teng, Y. Luo, S. Lou, M. Nishiura and Z. Hou, Chem.–Asian J., 2020, 15, 753–756 CrossRef CAS PubMed; (p) N. Hara, N. Uemura and Y. Nakao, Chem. Commun., 2021, 57, 5957–5960 RSC; (q) K. An, W. Ma, L.-C. Liu, T. He, G. Guan, Q.-W. Zhang and W. He, Nat. Commun., 2022, 13, 847 CrossRef CAS PubMed; (r) S. Chen, J. Zhu, J. Ke, Y. Li and C. He, Angew. Chem., Int. Ed., 2022, 61, e202117820 CrossRef CAS PubMed; (s) D. Mu, S. Pan, X. Wang, X. Liao, Y. Huang and J. Chen, Chem. Commun., 2022, 58, 7388–7391 RSC; (t) Q. Wan, Z.-W. Hou, X.-R. Zhao, X. Xie and L. Wang, Org. Lett., 2023, 25, 1008–1013 CrossRef CAS PubMed.
- (a) W. A. Gustavson, P. S. Epstein and M. D. Curtis, Organometallics, 1982, 1, 884–885 CrossRef CAS ; For the silylations of HSiMe(OSiMe3)2 with Rh- and Ir-catalyst system, see ; (b) C. Cheng and J. F. Hartwig, Science, 2014, 343, 853–857 CrossRef CAS PubMed; (c) C. Cheng and J. F. Hartwig, J. Am. Chem. Soc., 2015, 137, 592–595 CrossRef CAS PubMed ; For the mechanistic study for their Rh-catalysed silylations, see ; (d) C. Cheng and J. F. Hartwig, J. Am. Chem. Soc., 2014, 136, 12064–12072 CrossRef CAS PubMed; (e) C. Karmel, Z. Chen and J. F. Hartwig, J. Am. Chem. Soc., 2019, 141, 7063–7072 CrossRef CAS PubMed; (f) C. Karmel and J. F. Hartwig, J. Am. Chem. Soc., 2020, 142, 10494–10505 CrossRef CAS PubMed; (g) C. Karmel, C. Z. Rubel, E. V. Kharitonova and J. F. Hartwig, Angew. Chem., Int. Ed., 2020, 59, 6074–6081 CrossRef CAS PubMed.
- For details, see the ESI.†.
- (a) J. W. Ryan, J. Am. Chem. Soc., 1962, 84, 4730–4734 CrossRef CAS; (b) B. Marciniec, in Comprehensive Handbook on Hydrosilylation, Pergamon, Oxford, 1992, pp.38–39 and references therein Search PubMed; (c) B. Marciniec, H. Maciejewski and U. Rosenthal, J. Organomet. Chem., 1994, 484, 147–151 CrossRef CAS; (d) H. Maciejewski, B. Marciniec and I. Kownacki, J. Organomet. Chem., 2000, 597, 175–181 CrossRef CAS; (e) V. Srinivas, Y. Nakajima, W. Ando, K. Sato and S. Shimada, J. Organomet. Chem., 2016, 809, 57–62 CrossRef CAS; (f) K. L. Lee, Angew. Chem., Int. Ed., 2017, 56, 3665–3669 CrossRef CAS PubMed; (g) Ref. 7o..
Footnotes |
| † Electronic supplementary information (ESI) available. CCDC 2351158. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d4sc03394a |
| ‡ These authors contributed equally. |
| This journal is © The Royal Society of Chemistry 2024 |