
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Selective sodium halide over potassium halide binding and extraction by a heteroditopic halogen bonding [2]catenane†
Hui Min
Tay
 a,
Andrew
Docker
ab,
Carol
Hua
c and
Paul D.
Beer
*a
a,
Andrew
Docker
ab,
Carol
Hua
c and
Paul D.
Beer
*a
aChemistry Research Laboratory, Department of Chemistry, University of Oxford, Mansfield Road, Oxford, OX1 3TA, UK. E-mail: paul.beer@chem.ox.ac.uk
bYusuf Hamied Department of Chemistry, University of Cambridge, Lensfield Road, Cambridge CB2 1EW, UK
cSchool of Chemistry, The University of Melbourne, Parkville, Victoria 3010, Australia
First published on 18th July 2024
Abstract
The synthesis and ion-pair binding properties of a heteroditopic [2]catenane receptor exhibiting highly potent and selective recognition of sodium halide salts are described. The receptor design consists of a bidentate halogen bonding donor motif for anion binding, as well as a di(ethylene glycol)-derived cation binding pocket which dramatically enhances metal cation affinity over previously reported homo[2]catenane analogues. 1H NMR cation, anion and ion-pair binding studies reveal significant positive cooperativity between the cation and anion binding events in which cation pre-complexation to the catenane subsequently ‘switches-on’ anion binding. Notably, the heteroditopic catenane displayed impressive selectivity for sodium halide recognition over the corresponding potassium halides. We further demonstrate that the catenane is capable of extracting solid alkali metal salts into organic media. Crucially, the observed solution phase binding selectivity for sodium halides translates to superior functional extraction capabilities of these salts relative to potassium halides, overcoming the comparatively higher lattice enthalpies NaX > KX dictated by the smaller alkali metal sodium cation. This is further exemplified in competitive solid–liquid experiments which revealed the exclusive extraction of sodium halide salts from solid mixtures of sodium and potassium halide salts.
Introduction
The field of supramolecular host–guest receptors based on mechanically interlocked molecules (MIMs) has expanded dramatically over the past two decades. A distinguishing feature of MIM-based receptors is the presence of topologically unique binding environments, which through judicious design, can be tuned to achieve size and shape complementarity towards a specific target guest.1–9 The resulting improvements in binding affinity and selectivity over non-interlocked receptor analogues have motivated the construction of numerous MIM-based cation10–14 and anion15–20 receptors over the years.Heteroditopic receptors capable of the simultaneous binding of cations and anions, or ion-pair recognition, have been shown to exhibit dramatically augmented ion binding affinities over monotopic cation or anion receptors.21–26 These favourable binding properties have fuelled the rising prominence of ion-pair receptors in a myriad of applications including salt extraction/solubilisation,27–37 membrane transport38,39 and the recognition of biologically-relevant zwitterions.40,41
Despite these successes, the design of ion-pair receptors capable of distinguishing chemically-similar ions remains a persistent challenge. Of particular interest is the ability to discriminate between sodium and potassium ions, which are the primary cationic biological electrolytes and underpin many of the functions requisite for life.42–44 Their shared monocationic nature, spherical geometry and similar ionic radii make selective recognition of these ions a significant challenge in synthetic receptor systems,45 a difficulty underscored by the scarcity of heteroditopic ion-pair receptors capable of selectively binding sodium or potassium salts.35,46,47
The demonstrated efficacy of MIM-based receptors to engineer otherwise challenging ion selectivity profiles, alongside their modular nature which enables facile incorporation of multiple binding sites, makes them exquisitely suited to function as selective heteroditopic ion-pair receptors. It is therefore somewhat surprising that examples of MIM-based ion-pair receptors remain remarkably rare.48–53 Prompted by the envisaged utility of MIM-based ion-pair receptors, we previously reported a series of homo[2]catenanes capable of alkali metal halide binding via a di(ethylene glycol)-derived binding site for cation complexation alongside a bis(iodotriazole)benzene halogen bonding donor motif for anion binding.54 While this work highlighted the potential of exploiting the highly pre-organised binding cavities of catenanes to elicit the desired selectivity in sodium and potassium cation binding, the relatively moderate individual cation and anion affinities of these receptors precluded strong ion-pair binding.
Herein we report a heteroditopic hetero[2]catenane (Fig. 1) possessing a modified cation binding site serving to significantly increase its cation affinity, which in turn leads to a dramatic enhancement in the ion-pair binding affinity of the hetero[2]catenane relative to the homo[2]catenanes. Crucially, the use of di(ethylene glycol) linkers generates a cation binding pocket that exhibits a significantly closer size complementarity to the smaller sodium cation over the larger potassium cation. Notably, 1H NMR ion-pair binding studies revealed a preference for binding sodium halide salts over the corresponding potassium halide salts. Importantly, the observed preference for sodium halide binding in the solution phase manifests in superior and exclusive extraction of sodium halide salts, as demonstrated by a series of solid–liquid extraction studies.
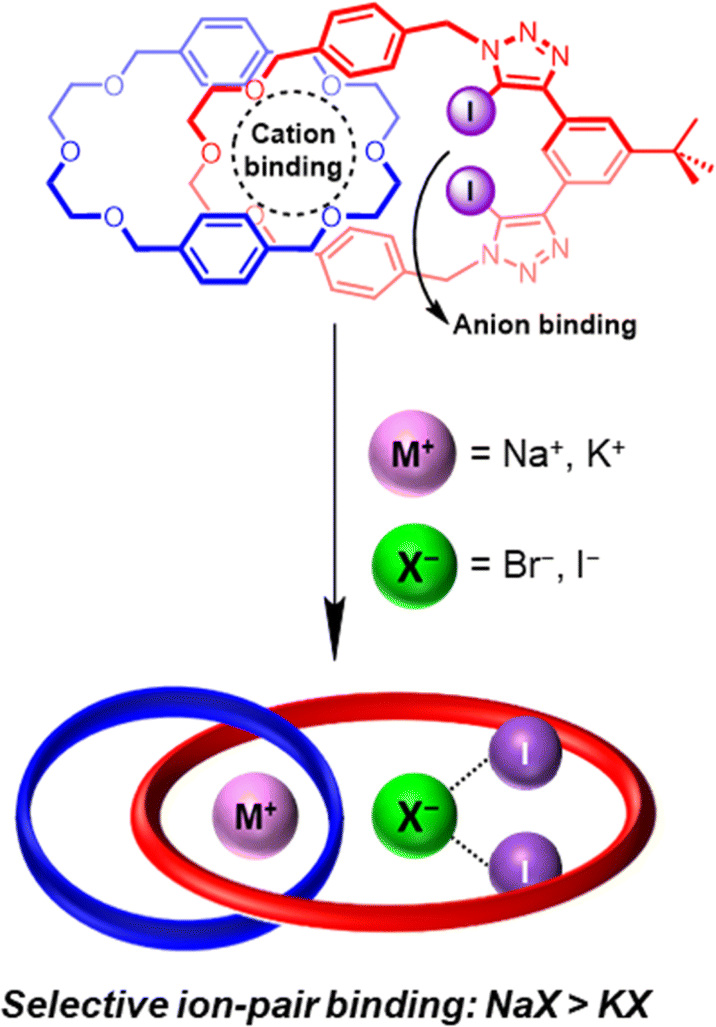 | ||
| Fig. 1 Schematic showing the chemical structure (top) and cartoon representation (bottom) of a hetero[2]catenane which functions as a heteroditopic ion-pair receptor capable of potent and selective binding of sodium halides. | ||
Results and discussion
Synthesis and characterisation
It was envisaged that the target hetero[2]catenane could be prepared via a cation template-directed strategy55–61 in which a sodium ion directs the self-assembly of a pseudo[2]rotaxane complex between the crown ether like di(ethylene)glycol-functionalised macrocycle 1 and bis(azide) 2. Subjecting the threaded bis(azide) to a copper(I)-catalysed azide–alkyne cycloaddition (CuAAC) macrocyclisation reaction with a bis(iodoalkyne) would concurrently generate the second interlocked macrocycle and the halogen bonding anion binding site. To this end, macrocycle 1, NaBArF and bis(azide) 2 were pre-complexed in CH2Cl2 for 30 minutes, after which the bis(iodoalkyne) 3 was added, followed by dropwise addition of Cu(MeCN)4PF6 and tris[(1-benzyl-1H-1,2,3-triazol-4-yl)methyl]amine (TBTA). The reaction was allowed to stir at room temperature overnight, then washed with basic EDTA solution and water. Preparative thin-layer chromatographic purification afforded the target hetero[2]catenane 4 in 11% yield, which was characterised by 1H NMR, 13C NMR and high-resolution ESI-MS. It is postulated that the low yield is attributed to the competing macrocyclisation reaction to afford non-interlocked 5, which was isolated as the major by-product of the reaction (Scheme 1).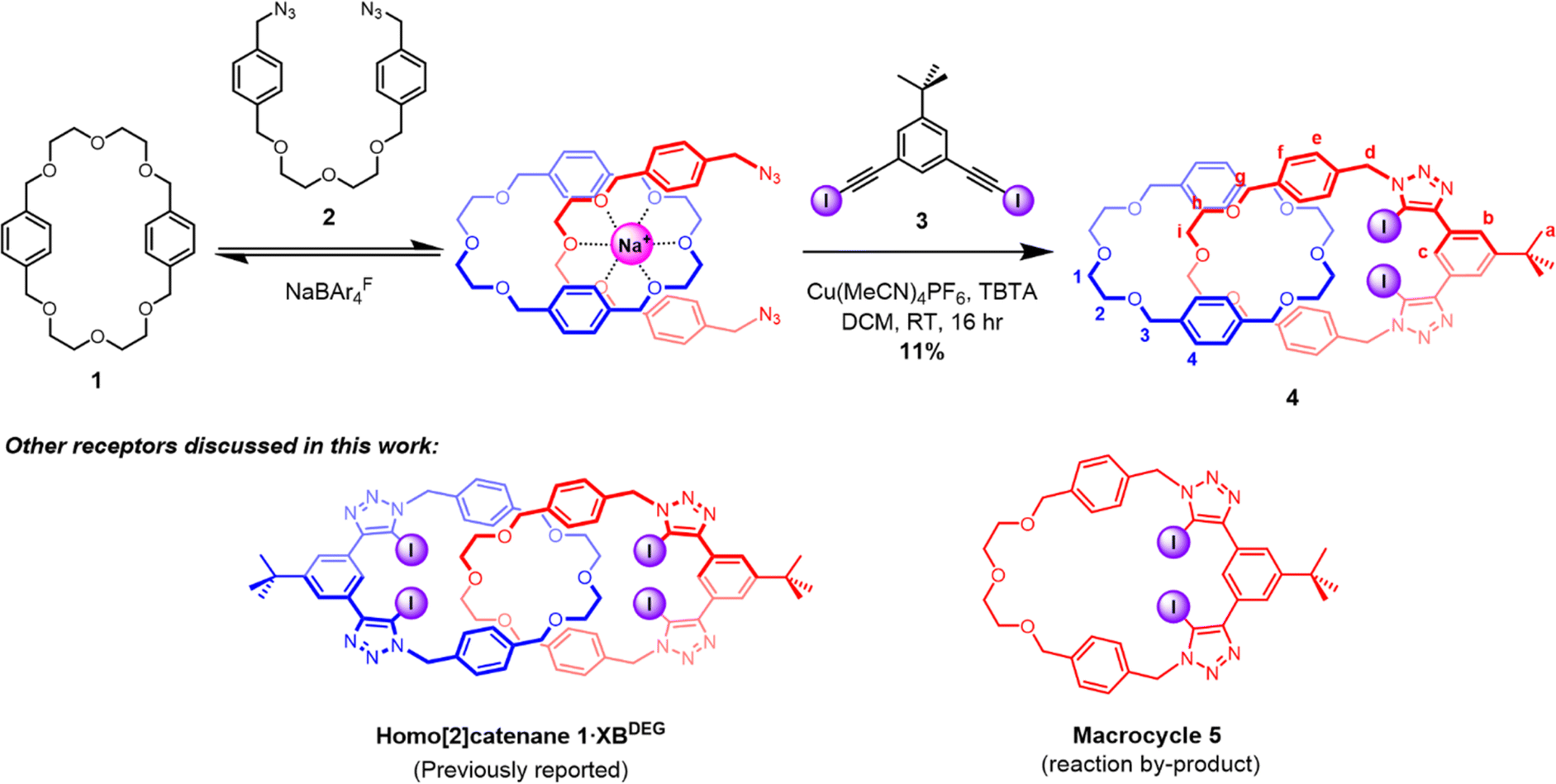 | ||
| Scheme 1 Synthesis of hetero[2]catenane 4 using a sodium cation template-directed strategy. The chemical structures of macrocycle 5 and a previously reported homo[2]catenane 1·XBDEG are shown below. | ||
Comparing the 1H NMR spectrum of [2]catenane 4 in CDCl3 solution to those of its constituent macrocycles 1 and 5 revealed diagnostic peak shifts consistent with an interlocked topology (Fig. 2). The proton resonances arising from macrocycle 1 underwent significant upfield shifts upon formation of the [2]catenane, attributed to the shielding effect associated with the enforced proximity of these environments to the shielding ring currents of the xylene spacers in 5. No proton signal desymmetrisation of the macrocycles was observed, indicating that under ambient conditions in CDCl3 the relative co-conformational circumrotation of the constituent macrocycles is facile, i.e. fast relative to the NMR timescale. Significant perturbations were also observed in the signals arising from 5, most notably upfield shifts and coalescence of protons Hh and Hi, indicating that the ethylene glycol region of 5 is threaded through the cavity of 1.
 | ||
| Fig. 2 Stacked 1H NMR spectra of macrocycle 1 (top), [2]catenane 4 (middle) and XB macrocycle 5 (bottom) (500 MHz, CDCl3, 298 K). | ||
1H NMR binding studies
To investigate the potential of the hetero[2]catenane 4 to function as an ion-pair receptor for alkali metal halide salts, 1H NMR binding studies with sodium and potassium tetrakis[3,5-bis(trifluoromethyl)phenyl]borate (MIBArF4, where MI = Na+, K+) were initially undertaken to establish the cation binding ability of 4 in 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 CD3CN/CDCl3.
:1 CD3CN/CDCl3.
Upon addition of NaBArF4 and KBArF4 to 4, large perturbations were observed in the ethylene glycol proton resonances of the two constituent macrocycles, as well as the proximal benzylic singlets Hg and H3, consistent with convergent coordination of the alkali metal cation by the polyether oxygen atoms of both macrocycles (Fig. S7–S8†). A substantial downfield shift of the xylene protons H4 from macrocycle 1 was also observed, possibly indicative of a co-conformational rearrangement that rotates these aromatic groups away from the macrocyclic cavity of 5 to accommodate the cation in the binding site, a postulated structure of which is shown in Fig. 3a.
 | ||
| Fig. 3 Ion-pair binding studies of hetero[2]catenane 4, including: (a) proposed ion-pair binding mode, showing the pre-complexation of the cation in the di(ethylene glycol)-derived binding cavity, followed by anion binding at the XB donor group. (b) Truncated stacked 1H NMR spectra of 4 upon progressive addition of TBABr in the presence of 1 eq. NaBArF4. (c) Binding isotherms of 4, showing changes in the chemical shift of Hc with increasing equivalents of TBAX salts (X = Br, I) in the presence of 1 eq. MBArF4 (M = Na, K). For (b) and (c), [receptor] = 1.0 mM. Solvent = 3:1 CD3CN/CDCl3 (v/v). T = 298 K. | ||
The binding isotherms generated by monitoring the upfield perturbations of ethylene glycol proton Hi rapidly approached saturation upon the addition of 1 equiv. of the cation (Fig. S36†). Fitting the isotherms to a 1:1 host–guest stoichiometric binding model using Bindfit analysis62 gave cation association constants of approximately 58000 M−1 and 28000 M−1 for Na+ and K+ respectively, indicating preferential sodium over potassium binding, although the high fitting errors (10–20%) precluded an accurate determination of the Na+ > K+ binding selectivity (Table 1). Importantly, this represents a significant enhancement (>10-fold) in cation affinity relative to previously reported homo[2]catenane 1·XBDEG (Ka (Na+) = 756(11) M−1, Ka (K+) = 284(2) M−1), which possesses a cation binding site comprised of two ethylene glycol linkers from the corresponding non-interlocked constituent macrocycles 5. The symmetric nature of macrocycle 1 would conceivably allow [2]catenane 4 to adopt a larger number of co-conformations in which convergent cation coordination by the polyether regions of its two constituent macrocycles can be achieved. Crucially, analogous cation binding studies conducted on the non-interlocked macrocyclic components 1 and 5 in 1:1 CD3CN/CDCl3 (Fig. S32–S36†) revealed no significant cation affinity, highlighting the necessity of the interlocked binding cavity for cation binding.
|
a
K
a values calculated using Bindfit with a 1:1 host–guest binding model. Errors are shown in parantheses. All cations added as BAr4F− salts. T = 298 K. [Receptor] = 1.0 mM.
b Reported in ref. 54.
|
|||
|---|---|---|---|
| Solvent | 1:1 CD3CN/CDCl3 |
3:1 CD3CN/CDCl3 |
|
| Receptor | Homo[2]catenane 1·XBDEG | Hetero[2]catenane 4 | |
| Na+ | 756(11)b | 58000 (6000) |
20000(750) |
| K+ | 284(2)b | 27000 (5000) |
3737(91) |
To further investigate the relative affinities of [2]catenane 4 for sodium and potassium ions, the 1H NMR cation binding studies were repeated in the more competitive solvent mixture 3:1 CD3CN/CDCl3 (Fig. S9–S10 and S37†), wherein cation binding affinities should be attenuated relative to the 1:1 CD3CN/CDCl3 solvent system. In this solvent system the Ka(Na+) value was determined to be approximately 20000 M−1, which is significantly higher (>5-fold) than the corresponding Ka(K+) value (Table 1). This indicates that [2]catenane 4 preferentially binds sodium over potassium, presumably due to more optimal size complementarity between the di(ethylene glycol)-derived binding site and the smaller sodium cation.57
The anion binding affinity of [2]catenane 4 was investigated via analogous 1H NMR titrations with tetrabutylammonium halide salts (TBAX, where X = Cl, Br, I). In 1:1 CD3CN/CDCl3, the addition of aliquots of TBAX surprisingly resulted in significant peak broadening and merging (Fig. S11–S13†). Consequently, the halide association constants of 4 could not be reliably determined in this solvent system. In contrast, the 1H NMR signals of the [2]catenane host remained sharp in 3:1 CD3CN/CDCl3, which is attributed to the greater solubility of the [2]catenane–anion complex in this solvent system. A significant downfield perturbation of the internal benzene signal Hc was observed, suggesting that halide anion binding occurs via XB interactions with the bis(iodotriazole)benzene donor motif (Fig. S14–S16†). Bindfit62 analysis of the resulting binding isotherms (Fig. S38†) unexpectedly revealed that 4 exhibits approximately 2-fold higher halide association constants than its constituent XB macrocycle 5 (Table 2). Whilst initially surprising, since the mechanical bond between crown ether-like macrocycle 1 to macrocycle 5 to form [2]catenane 4 ostensibly introduces no additional anion binding groups, we tentatively postulate this enhanced halide affinity is attributable to the formation of ancillary CH⋯X− interactions involving the aromatic spacers of macrocycle 1, which underwent downfield perturbations upon addition of TBA halide salts, and/or co-conformational restraints imposed on the XB anion binding motif imposed by the mechanical bond.63
| Receptor | Hetero[2]catenane 4 | Macrocycle 5 |
|---|---|---|
|
a
K
a values calculated using Bindfit with a 1:1 host–guest binding model. Errors are shown in parantheses. All anions added as TBA+ salts. Solvent = 3:1 CD3CN/CDCl3. T = 298 K. [Receptor] = 1.0 mM.
|
||
| Cl− | 419(34) | 212(2) |
| Br− | 506(44) | 261(2) |
| I− | 437(29) | 277(2) |
Having established that the binding affinity of 4 for alkali metal cations is sufficient to ensure a high degree of saturation of the receptor in the presence of 1 equiv. of the cation in 1:1 CD3CN/CDCl3, 1H NMR ion-pair binding studies were conducted by adding aliquots of TBAX salts (X = Cl, Br, I) to a 1 mM solution of 4 containing an equimolar amount of MBArF4 (M = Na, K) (Fig. 3a).
Upon addition of Br− and I−, prominent downfield shifts of the internal benzene proton Hc were observed, which suggest that anion binding remains primarily mediated by XB interactions (Fig. S18, S19, S21 and S22†). Importantly, the addition of bromide and iodide causes little to no perturbation of the ethylene glycol proton signals, indicating that the cation binding sites of the catenane remain saturated throughout the titration. In contrast, addition of chloride to the receptor–cation complex induces chemical shift perturbations consistent with external ion-pairing and salt recombination (Fig. S17 and S20†), indicating that despite the high cation affinities of 4, its ion-pair binding ability remains insufficient to overcome the high lattice enthalpies of NaCl and KCl.
In an analogous manner to the anion binding studies, binding isotherms for Br− and I− were constructed by monitoring the downfield shift of Hc and subsequently fitted to a 1:1 host–guest binding model (Fig. S39 and S40†) to determine apparent anion association constants (Kapp) of the monocationic catenane–Na+ complexes, assuming near-quantitative pre-complexation of the metal cation to the catenane host. The calculated Kapp values were in range 10000–15000 M−1 in 1:1 CD3CN/CDCl3, demonstrating a dramatic enhancement in the anion binding affinity of [2]catenane 4 in the presence of a co-bound sodium cation. The corresponding Kapp values of the catenane–K+ complex were found to be significantly lower (Table 3), suggesting a binding preference for sodium halide over potassium halide salts by the [2]catenane receptor. Importantly, the qualitatively similar chemical shift perturbations observed in the NaX and KX titrations, namely a downfield shift of Hc and H4 upon addition of Br− and I− with no significant changes in the ethylene glycol proton signals, indicate that ion-pair binding of KBr and KI by 4 nonetheless remains the dominant process, outcompeting the salt recombination equilibrium. These studies crucially demonstrate strong positive cooperativity between the cation and anion binding events which enables the heteroditopic [2]catenane to act as a potent ion-pair receptor.
| Solvent | Cation | Anion | K app (M−1) |
|---|---|---|---|
|
a
K
app values calculated using Bindfit with a 1:1 host–guest binding model. Errors are shown in parantheses. All cations added as BAr4F− salts and anions as TBA+ salts. T = 298 K. [Receptor] = [MBArF4] = 1.0 mM.
|
|||
| 1:1 CD3CN/CDCl3 |
Na+ | Cl− | Salt recombination |
| Br− | 10300(1700) |
||
| I− | 15200(3000) |
||
| K+ | Cl− | Salt recombination | |
| Br− | 3178(269) | ||
| I− | 2357(268) | ||
| 3:1 CD3CN/CDCl3 |
Na+ | Cl− | Salt recombination |
| Br− | 4179(204) | ||
| I− | 5392(522) | ||
| K+ | Cl− | Salt recombination | |
| Br− | 1480(69) | ||
| I− | 1243(96) | ||
In order to quantitatively measure the apparent anion binding constants of the catenane–cation complexes, the ion-pair binding studies were repeated in the more competitive solvent system 3:1 CD3CN/CDCl3 (Fig. 3b and c). Crucially, the Kapp values determined for the sodium halide salts were 2 to 5-fold higher than the analogous potassium salts (Table 3), indicating that the size-driven selectivity of the [2]catenane 4 for sodium over potassium cations translates to its ion-pair recognition properties.
Solid-state X-ray crystallographic studies
The proposed ion-pair binding mode of 4 was further corroborated by solid-state structural analysis.‡ Slow evaporation of a 1:1 CD3CN/CDCl3 solution containing hetero[2]catenane 4 in the presence of NaBArF4 and excess TBAI afforded single crystals suitable for analysis by single crystal X-ray diffraction. The solid-state crystal structure revealed a 1:1 complex of catenane 4 bound to an NaI ion-pair, in which the guest ions are bound in a host-separated ion-pair binding mode consistent with solution-phase NMR studies (Fig. 4). Notably, the polyether regions of the two interlocked macrocyclic components 1 and 5 are arranged in an orthogonal geometry to form a cation binding pocket. The sodium cation resides within this binding pocket where it is convergently bound by three oxygen atoms each from 1 and 5 to form a six-coordinate distorted octahedral metal centre. In contrast, the iodide anion is located on the periphery of the macrocycle 5 where it forms XB interactions with the bis(iodotriazole)benzene motif, as shown by the I⋯I− distances (3.4597(7)–3.4820(6) Å, which is 87–98% of the sum of the van der Waals radii of the two atoms). The absence of ancillary CH–X− interactions between macrocycle 1 and the iodide guest in 4·NaI may be due to steric constraints imposed by the formation of a cation binding pocket, which presumably restricts the inter-ring circum-rotation of the catenane.
 | ||
| Fig. 4 Solid-state structure of 4 bound to an NaI ion-pair. Hydrogen atoms have been omitted for clarity. Atom colours are as follows: black (carbon), blue (nitrogen), red (oxygen), purple (iodine), pink (sodium). | ||
Solid–liquid extraction studies
The promising ion-pair binding properties of hetero[2]catenane 4 prompted an investigation into its ability to act as an alkali halide salt extractant. To this end, solid–liquid extraction (SLE) studies were undertaken by sonicating a 1 mM CDCl3 solution of 4 in the presence of a fixed excess mass quantity of solid MX (M = Na, K; X = Cl−, Br−, I−).Large perturbations were observed in the 1H NMR spectra of 4 post-treatment with NaBr and NaI (Fig. 5c and S45c†). In particular, a dramatic upfield shift of internal benzene proton Hc was observed as well as desymmetrisation of the ethylene glycol protons Hh/I, which are respectively consistent with anion and cation binding by the [2]catenane. These results indicate complete stoichiometric extraction of both NaX salts, with the post-extraction solution containing only the [2]catenane as the ion-pair bound complex.
In contrast, treatment of 4 with KBr and KI resulted in more complex 1H NMR spectra which upon closer inspection revealed two sets of proton signals corresponding to two distinct catenane species. In the case of KBr, shown in Fig. 5b, the major set exhibits chemical shifts similar to those of the unbound receptor while the minor set resembles the spectrum of the NaBr-bound catenane complexes. These observations suggest incomplete KBr extraction with the bound and unbound catenane complexes in slow exchange. Integration of these signals estimated that 43% of the hetero[2]catenane in solution was present as the ion-pair bound complex.
 | ||
| Fig. 5 Solid–liquid extraction studies of alkali metal bromide salts, showing truncated 1H NMR spectra of: (a) free catenane receptor 4, and post-extraction spectra of 4 following treatment with (b) 10 mg KBr; (c) 10 mg NaBr; (d) 5 mg NaBr and 5 mg KBr (CDCl3, 298 K, 500 MHz). The inset on the right shows the structure of 4 bound to an MX ion-pair (top) and a schematic illustrating the SLE experiment (bottom). | ||
No significant signal perturbations were observed upon treatment of 4 with NaCl or KCl (Fig. S47†), which was attributed to the high lattice enthalpy of alkali metal chloride salts outcompeting ion-pair binding to 4, and is consistent with the observation of salt recombination during titration studies of the catenane–metal complexes with TBACl.
Motivated by the observed superior performance of 4 for sodium bromide/iodide extraction over the corresponding potassium salts, we sought to further investigate the extraction preferences of the catenane receptor via a competitive NaX/KX solid–liquid extraction experiment. To this end, in an analogous procedure to the above-described experiments, a 1 mM CDCl3 solution of 4 was exposed to a mixture of NaBr/KBr or NaI/KI present in excess. In both cases, this resulted in post-extraction spectra identical to those obtained from treatment of the receptor solely with the corresponding sodium halide salt (Fig. 5d and S45d†). Crucially, this provides promising evidence that 4 is capable of preferential sodium halide extraction over the corresponding potassium halide salts. Importantly, this highlights that despite the higher lattice enthalpies of sodium halide over potassium halide salts, dictated by the smaller alkali metal sodium cation, the mechanical bond effect enables size-based discrimination and thereby achieves selective ion-pair recognition and extraction.
Conclusions
In summary, a heteroditopic [2]catenane ion-pair host system containing an interchain component di(ethylene glycol)-derived cation binding site and a bidentate halogen bonding anion binding site was synthesised via a sodium cation template-directed approach. 1H NMR cation binding studies revealed a dramatic enhancement in sodium and potassium alkali metal cation affinity relative to a previously reported series of homo[2]catenanes, as well as considerable sodium over potassium selectivity. The alkali metal halide (MX, X = Br, I) ion-pair binding properties of the catenane were probed by 1H NMR studies and X-ray crystallography, both of which were consistent with the proposed separated ion-pair binding mode. Ion-pair titrations showed strong binding of alkali metal halide salts with significant cooperativity between the cation and anion binding events, reflected in a significant ‘switch-on’ of the catenane's anion affinity upon pre-complexation with a metal cation. Importantly, the catenane was found to bind sodium halide salts 3–5 times more strongly than the corresponding potassium halide salts. Solid–liquid extraction studies highlighted the capability of the catenane to function as an efficient alkali metal halide salt extractant, in which the observed selectivity preference for sodium halide over potassium halide ion-pair binding in solution-state NMR studies impressively translated to superior and exclusive extraction of sodium halide salts from a competitive solid mixture of NaX/KX salts. The potent ion-pair binding capabilities of this heterocatenane receptor, and importantly its ability to discriminate between sodium halide and potassium halide salts, demonstrates the efficacy of exploiting the mechanical bond effect to elicit improved binding affinity and selectivity, which will serve to inform the design of future heteroditopic ion-pair receptors.Data availability
The data supporting this article have been included as part of the ESI.† Crystallographic data for compound 4·NaI has been deposited at the CCDC under 2348225 and can be obtained from https://www.ccdc.cam.ac.uk/structures.Author contributions
P. D. B. conceived the project. H. M. T. conducted the experimental studies. C. H. conducted the X-ray crystallographic analysis. All authors contributed to the writing and editing of the manuscript and agreed on the final version.Conflicts of interest
There are no conflicts to declare.Acknowledgements
H. M. T. acknowledges the Clarendon Fund and the Oxford Australia Scholarships Fund for a postgraduate research scholarship. A. D. thanks the EPSRC for funding (grant reference number: EP/N509711/1).Notes and references
- K. M. Bąk, K. Porfyrakis, J. J. Davis and P. D. Beer, Mater. Chem. Front., 2020, 4, 1052–1073 RSC.
- J. F. Stoddart, Chem. Soc. Rev., 2009, 38, 1802–1820 RSC.
- E. A. Neal and S. M. Goldup, Chem. Commun., 2014, 50, 5128–5142 RSC.
- M. Xue, Y. Yang, X. Chi, X. Yan and F. Huang, Chem. Rev., 2015, 115, 7398–7501 CrossRef CAS.
- G. Gil-Ramírez, D. A. Leigh and A. J. Stephens, Angew. Chem., Int. Ed., 2015, 54, 6110–6150 CrossRef.
- J. E. M. Lewis, M. Galli and S. M. Goldup, Chem. Commun., 2017, 53, 298–312 RSC.
- W.-X. Gao, H.-J. Feng, B.-B. Guo, Y. Lu and G.-X. Jin, Chem. Rev., 2020, 120, 6288–6325 CrossRef CAS.
- H.-N. Zhang, Y.-J. Lin and G.-X. Jin, J. Am. Chem. Soc., 2021, 143, 1119–1125 CrossRef CAS PubMed.
- J. T. Wilmore and P. D. Beer, Adv. Mater., 2024, 2309098 CrossRef CAS.
- G. Baggi and S. J. Loeb, Chem.–Eur. J., 2017, 23, 14163–14166 CrossRef CAS.
- M. Nandi, S. Bej, T. K. Ghosh and P. Ghosh, Chem. Commun., 2019, 55, 3085–3088 RSC.
- S.-M. Chan, F.-K. Tang, C.-S. Kwan, C.-Y. Lam, S. C. K. Hau and K. C.-F. Leung, Mater. Chem. Front., 2019, 3, 2388–2396 RSC.
- T. Shukla, A. K. Dwivedi, R. Arumugaperumal, C.-M. Lin, S.-Y. Chen and H.-C. Lin, Dyes Pigm., 2016, 131, 49–59 CrossRef CAS.
- M. Denis, J. Pancholi, K. Jobe, M. Watkinson and S. M. Goldup, Angew. Chem., Int. Ed., 2018, 57, 5310–5314 CrossRef CAS PubMed.
- A. Caballero, F. Zapata and P. D. Beer, Coord. Chem. Rev., 2013, 257, 2434–2455 CrossRef CAS.
- R. J. Goodwin, A. Docker, H. I. MacDermott-Opeskin, H. M. Aitken, M. L. O'Mara, P. D. Beer and N. G. White, Chem.–Eur. J., 2022, 28, e202200389 CrossRef CAS PubMed.
- J. M. Mercurio, F. Tyrrell, J. Cookson and P. D. Beer, Chem. Commun., 2013, 49, 10793–10795 RSC.
- M. J. Langton, O. A. Blackburn, T. Lang, S. Faulkner and P. D. Beer, Angew. Chem., Int. Ed., 2014, 53, 11463–11466 CrossRef CAS PubMed.
- N. G. White and P. D. Beer, Org. Biomol. Chem., 2013, 11, 1326–1333 RSC.
- Y. Cheong Tse, R. Hein, E. J. Mitchell, Z. Zhang and P. D. Beer, Chem.–Eur. J., 2021, 27, 14550–14559 CrossRef CAS.
- A. J. McConnell and P. D. Beer, Angew. Chem., Int. Ed., 2012, 51, 5052–5061 CrossRef CAS PubMed.
- J. L. Sessler, S. K. Kim, D. E. Gross, C.-H. Lee, J. S. Kim and V. M. Lynch, J. Am. Chem. Soc., 2008, 130, 13162–13166 CrossRef CAS.
- Q. He, G. I. Vargas-Zúñiga, S. H. Kim, S. K. Kim and J. L. Sessler, Chem. Rev., 2019, 119, 9753–9835 CrossRef CAS.
- B. Qiao, A. Sengupta, Y. Liu, K. P. McDonald, M. Pink, J. R. Anderson, K. Raghavachari and A. H. Flood, J. Am. Chem. Soc., 2015, 137, 9746–9757 CrossRef CAS.
- A. J. McConnell, A. Docker and P. D. Beer, ChemPlusChem, 2020, 85, 1824–1841 CrossRef CAS.
- M. M. Naseer and K. Jurkschat, Chem. Commun., 2017, 53, 8122–8135 RSC.
- P. D. Beer, P. K. Hopkins and J. D. McKinney, Chem. Commun., 1999, 1253–1254, 10.1039/A903440D.
- M. Zakrzewski, D. Załubiniak and P. Piątek, Dalton Trans., 2018, 47, 323–330 RSC.
- Q. He, G. M. Peters, V. M. Lynch and J. L. Sessler, Angew. Chem., Int. Ed., 2017, 56, 13396–13400 CrossRef CAS.
- S. G. Galbraith, P. G. Plieger and P. A. Tasker, Chem. Commun., 2002, 2662–2663, 10.1039/B208457K.
- T. Bunchuay, A. Docker, U. Eiamprasert, P. Surawatanawong, A. Brown and P. D. Beer, Angew. Chem., Int. Ed., 2020, 59, 12007–12012 CrossRef CAS.
- Y. C. Tse, A. Docker, Z. Zhang and P. D. Beer, Chem. Commun., 2021, 57, 4950–4953 RSC.
- A. Docker, J. G. Stevens and P. D. Beer, Chem.–Eur. J., 2021, 27, 14600–14604 CrossRef CAS PubMed.
- A. Docker, T. Bunchuay, M. Ahrens, A. J. Martinez-Martinez and P. D. Beer, Chem.–Eur. J., 2021, 27, 7837–7841 CrossRef CAS PubMed.
- A. Docker, I. Marques, H. Kuhn, Z. Zhang, V. Félix and P. D. Beer, J. Am. Chem. Soc., 2022, 144, 14778–14789 CrossRef CAS.
- Q. He, N. J. Williams, J. H. Oh, V. M. Lynch, S. K. Kim, B. A. Moyer and J. L. Sessler, Angew. Chem., Int. Ed., 2018, 57, 11924–11928 CrossRef CAS PubMed.
- Q. He, Z. Zhang, J. T. Brewster, V. M. Lynch, S. K. Kim and J. L. Sessler, J. Am. Chem. Soc., 2016, 138, 9779–9782 CrossRef CAS.
- A. V. Koulov, J. M. Mahoney and B. D. Smith, Org. Biomol. Chem., 2003, 1, 27–29 RSC.
- J. M. Mahoney, G. U. Nawaratna, A. M. Beatty, P. J. Duggan and B. D. Smith, Inorg. Chem., 2004, 43, 5902–5907 CrossRef CAS PubMed.
- A. Galan, D. Andreu, A. M. Echavarren, P. Prados and J. De Mendoza, J. Am. Chem. Soc., 1992, 114, 1511–1512 CrossRef CAS.
- W. Walczak, M. Zakrzewski, G. Cichowicz and P. Piątek, Org. Biomol. Chem., 2020, 18, 694–699 RSC.
- I. Mano and M. Driscoll, BioEssays, 1999, 21, 568–578 CrossRef CAS.
- P. M. Snyder, Endocr. Rev., 2002, 23, 258–275 CrossRef CAS PubMed.
- B. Hille, Biophys. J., 1978, 22, 283–294 CrossRef CAS PubMed.
- T. Dudev and C. Lim, J. Am. Chem. Soc., 2010, 132, 2321–2332 CrossRef CAS.
- D. Jagleniec, Ł. Dobrzycki, M. Karbarz and J. Romański, Chem. Sci., 2019, 10, 9542–9547 RSC.
- D. Jagleniec, S. Siennicka, Ł. Dobrzycki, M. Karbarz and J. Romański, Inorg. Chem., 2018, 57, 12941–12952 CrossRef CAS.
- R. C. Knighton and P. D. Beer, Chem. Commun., 2014, 50, 1540–1542 RSC.
- A. Brown, K. M. Mennie, O. Mason, N. G. White and P. D. Beer, Dalton Trans., 2017, 46, 13376–13385 RSC.
- M. J. Deetz, R. Shukla and B. D. Smith, Tetrahedron, 2002, 58, 799–805 CrossRef CAS.
- J. R. Romero, G. Aragay and P. Ballester, Chem. Sci., 2017, 8, 491–498 RSC.
- M. Denis, L. Qin, P. Turner, K. A. Jolliffe and S. M. Goldup, Angew. Chem., Int. Ed., 2018, 57, 5315–5319 CrossRef CAS.
- D.-H. Li and B. D. Smith, J. Org. Chem., 2019, 84, 2808–2816 CrossRef CAS PubMed.
- H. M. Tay, Y. C. Tse, A. Docker, C. Gateley, A. L. Thompson, H. Kuhn, Z. Zhang and P. D. Beer, Angew. Chem., Int. Ed., 2023, 62, e202214785 CrossRef CAS PubMed.
- T.-H. Ho, C.-C. Lai, Y.-H. Liu, S.-M. Peng and S.-H. Chiu, Chem.–Eur. J., 2014, 20, 4563–4567 CrossRef CAS PubMed.
- A. Inthasot, S.-T. Tung and S.-H. Chiu, Acc. Chem. Res., 2018, 51, 1324–1337 CrossRef CAS.
- Y.-J. Lee, T.-H. Ho, C.-C. Lai and S.-H. Chiu, Org. Biomol. Chem., 2016, 14, 1153–1160 RSC.
- Y.-H. Lin, C.-C. Lai and S.-H. Chiu, Org. Biomol. Chem., 2014, 12, 2907–2917 RSC.
- S.-T. Tung, C.-C. Lai, Y.-H. Liu, S.-M. Peng and S.-H. Chiu, Angew. Chem., Int. Ed., 2013, 52, 13269–13272 CrossRef CAS PubMed.
- Y.-W. Wu, P.-N. Chen, C.-F. Chang, C.-C. Lai and S.-H. Chiu, Org. Lett., 2015, 17, 2158–2161 CrossRef CAS PubMed.
- Y.-W. Wu, S.-T. Tung, C.-C. Lai, Y.-H. Liu, S.-M. Peng and S.-H. Chiu, Angew. Chem., Int. Ed., 2015, 54, 11745–11749 CrossRef CAS PubMed.
- D. Brynn Hibbert and P. Thordarson, Chem. Commun., 2016, 52, 12792–12805 RSC.
- A. Docker, Y. C. Tse, H. M. Tay, A. J. Taylor, Z. Zhang and P. D. Beer, Angew. Chem., Int. Ed., 2022, 61, e202214523 CrossRef CAS PubMed , A detailed discussion of factors governing the magnitude and direction of chemical shift perturbations observed during 1H NMR titrations can be found in the ESI† of this reference..
Footnotes |
| † Electronic supplementary information (ESI) available. CCDC 2348225. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d4sc03381g |
| ‡ Low temperature single crystals X-ray diffraction data were collected using an Oxford Diffraction Supernova X-ray diffractometer and reduced using CrysAlisPro. The structures were solved using SHELXS [G. Sheldrick, Acta Crystallogr., Sect. A: Found. Adv., 2015, 71, 3–8] and refined using CRYSTALS [P. W. Betteridge, J. R. Carruthers, R. I. Cooper, K. Prout and D. J. Watkin, J. Appl. Crystallogr., 2003, 36, 1487; R. I. Cooper, A. L. Thompson and D. J. Watkin, J. Appl. Crystallogr., 2010, 43, 1100–1107] and SHELXL [G. Sheldrick, Acta Crystallogr., Sect. C: Struct. Chem., 2015, 71, 3–8]. Full details are included in the accompanying ESI (CIF). The data can be obtained from the joint Cambridge Crystallographic Data Centre and Fachinformationszentrum Karlsruhe Access Structures service https://www.ccdc.cam.ac.uk/structures with deposition number 2348225. |
| This journal is © The Royal Society of Chemistry 2024 |