
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceUniversality of critical active site glutamate as an acid–base catalyst in serine hydroxymethyltransferase function†
Victoria N.
Drago
 a,
Robert S.
Phillips
bc and
Andrey
Kovalevsky
*a
a,
Robert S.
Phillips
bc and
Andrey
Kovalevsky
*a
aNeutron Scattering Division, Oak Ridge National Laboratory, Oak Ridge, TN 37831, USA. E-mail: kovalevskyay@ornl.gov
bDepartment of Chemistry, University of Georgia, Athens, GA 30602, USA
cDepartment of Biochemistry and Molecular Biology, University of Georgia, Athens, GA 30602, USA
First published on 3rd July 2024
Abstract
Serine hydroxymethyltransferase (SHMT) is a key enzyme in the one-carbon metabolic pathway, utilizing the vitamin B6 derivative pyridoxal 5′-phosphate (PLP) and vitamin B9 derivative tetrahydrofolate (THF) coenzymes to produce essential biomolecules. Many types of cancer utilize SHMT in metabolic reprogramming, exposing the enzyme as a compelling target for antimetabolite chemotherapies. In pursuit of elucidating the catalytic mechanism of SHMT to aid in the design of SHMT-specific inhibitors, we have used room-temperature neutron crystallography to directly determine the protonation states in a model enzyme Thermus thermophilus SHMT (TthSHMT), which exhibits a conserved active site compared to human mitochondrial SHMT2 (hSHMT2). Here we report the analysis of TthSHMT, with PLP in the internal aldimine form and bound THF-analog, folinic acid (FA), by neutron crystallography to reveal H atom positions in the active site, including PLP and FA. We observed protonated catalytic Glu53 revealing its ability to change protonation state upon FA binding. Furthermore, we obtained X-ray structures of TthSHMT-Gly/FA, TthSHMT-L-Ser/FA, and hSHMT2-Gly/FA ternary complexes with the PLP-Gly or PLP-L-Ser external aldimines to analyze the active site configuration upon PLP reaction with an amino acid substrate and FA binding. Accurate mapping of the active site protonation states together with the structural information gained from the ternary complexes allow us to suggest an essential role of the gating loop conformational changes in the SHMT function and to propose Glu53 as the universal acid-base catalyst in both THF-independent and THF-dependent activities of SHMT.
1 Introduction
Serine hydroxymethyltransferase (SHMT) is a pyridoxal 5′-phosphate (PLP) dependent metabolic enzyme vital to the one-carbon metabolism pathway in both prokaryotes and eukaryotes.1,2 The enzymatic activity of SHMT is essential for the synthesis of purines, thymine nucleotides, methionine, as well as other fundamental biomolecules.3,4 SHMT catalyzes the reversible cleavage of L-serine (L-Ser) to glycine (Gly), transferring a single carbon unit to tetrahydrofolate (THF) to produce 5,10-methylenetetrahydrofolate (5,10-MTHF) (Fig. 1a). In addition, SHMT catalyzes the reversible THF-independent cleavage of β-hydroxy amino acids, decarboxylation of aminomalonate, and racemization and transamination of L- and D-alanine.5 Prokaryotes and some eukaryotes such as Plasmodium spp. possess one isoform of SHMT, whereas in mammals two SHMT isoforms exist – the cytosolic SHMT1 and mitochondrial SHMT2 – compartmentalizing one-carbon metabolism.6,7 Both SHMT1 and SHMT2 carry out the same biochemical reaction and share a ∼66% sequence identity.8 Prokaryotic SHMTs, such as Thermus thermophilus SHMT (TthSHMT, Fig. 1b), form obligate homodimers, however, eukaryotic SHMTs are homotetrameric defined as a dimer of obligate dimers.6,9,10 Several bacterial SHMTs, as demonstrated with TthSHMT,11Bacillus stearothermophilus,12 and Enterococcus faecium13 SHMTs and a plant SHMT14 have been used as model enzymes to study human SHMTs by virtue of the high conservation of the enzyme's active site from all sources. In our structural studies, we have been using TthSHMT as the model for human mitochondrial SHMT2 (hSHMT2) because it affords high diffraction quality crystals facilitating neutron crystallographic studies and the active sites of the two enzymes are fully conserved allowing for direct comparison of the structures.11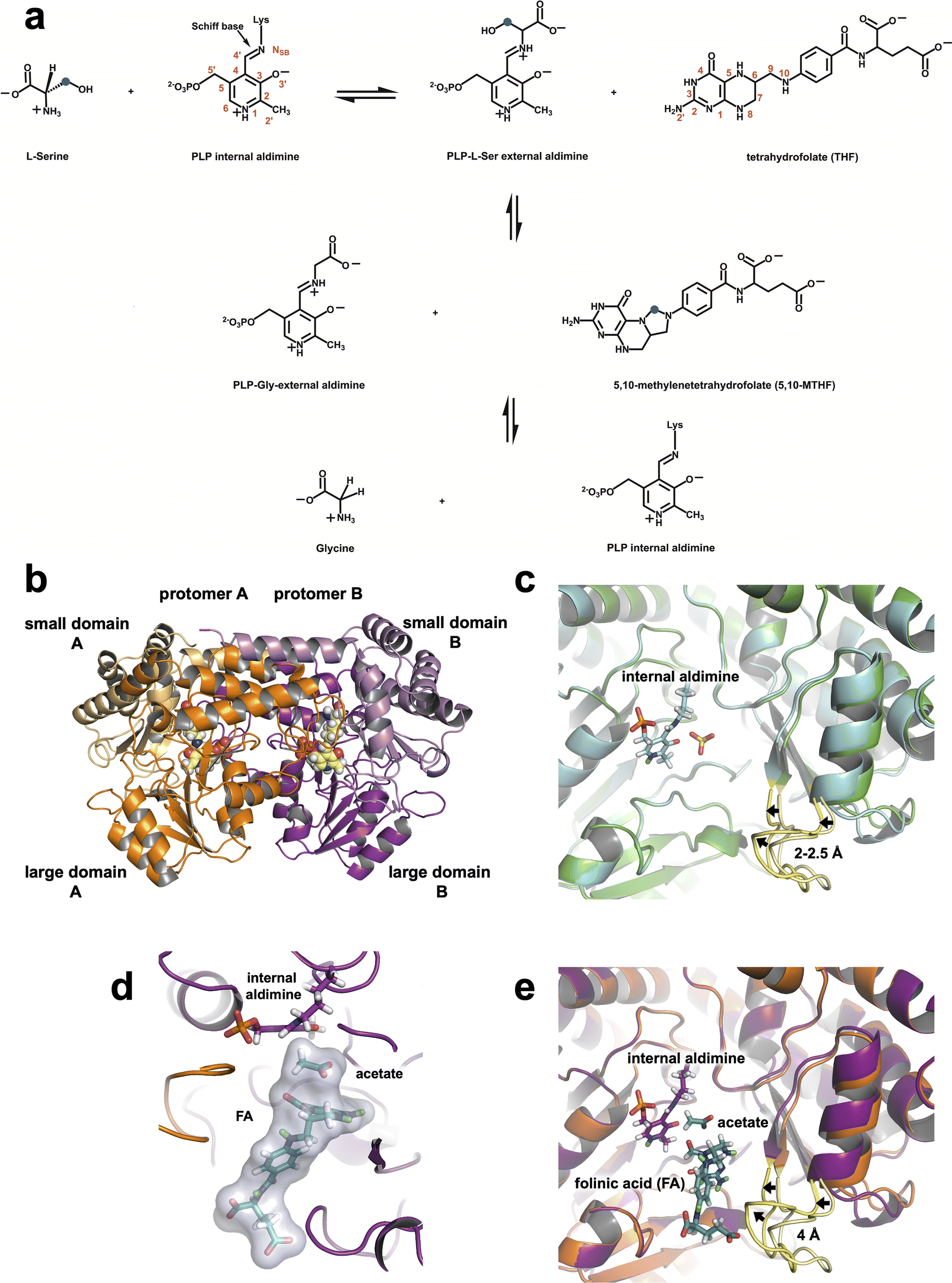 | ||
| Fig. 1 SHMT catalyzed reaction and the 3D structures of TthSHMT. (a) THF-dependent conversion of L-Ser to Gly. Carbon atom marked with blue dot is the 1C unit transferred from L-Ser to THF. (b) Overall fold of Thermus thermophilus SHMT (TthSHMT) with protomer A (orange) and protomer B (magenta) as well as their respective domains labeled. The PLP internal aldimine is shown in CPK representation (yellow carbons). (c) Zoomed-in view of the TthSHMT holoenzyme active site showing superposition of protomers A and B (PDB: 8SUJ). The gating loops are colored yellow. An open conformation is seen in protomer A (green cartoon, light yellow loop) and protomer B shows a partially closed conformation (cyan cartoon, bright yellow loop). (d) Positions of FA and the acetate molecule in the active site of the TthSHMT/FA complex neutron structure. PLP is covalently linked to Lys226 in the internal aldimine, an acetate molecule is bound in the cationic binding site, and folinic acid (FA) is located in the peripheral binding site blocking the entrance to the active site. (e) Superposition of protomer A from the TthSHMT neutron structure (PDB: 8SUJ, orange cartoon, light yellow gating loop) and protomer B from the TthSHMT/FA neutron structure (magenta cartoon, bright yellow loop) demonstrating closure of the gating loop upon FA binding. | ||
hSHMT2 plays an essential role in the metabolic reprogramming attained by many cancer types that results in the enzyme overexpression to promote cancer cell proliferation and growth.15–20 hSHMT2 overexpression has also been linked to cancer cell drug resistance.4,21,22 Consequently, hSHMT2 has emerged as a promising target for the design of antimetabolite chemotherapies.4,23–25 While some encouraging inhibitors targeting hSHMT2 have been developed in recent years, including SHIN1/2, AGF347 and AGF355 (ref. 23 and 25–30) that bind to the enzyme's active site competing with the physiological THF substrate, it is critical to improve our understanding of SHMT catalysis at the atomic level both to discern between the competing catalytic mechanisms proposed in the literature and to advance the design of hSHMT2-specific inhibitors. Identifying the amino acid residues acting as general acid and base catalysts and, thereby, discriminating between the catalytic mechanisms can only be accomplished through knowledge of the protonation states of the enzyme active site, PLP, and THF at various stages of the reaction. Because protonation states are determined by the presence or absence of hydrogen (H) atoms on specific chemical groups, the locations and movement of H atoms throughout the catalytic process need to be accurately determined and visualized. Such atomic details of protein structures can be achieved by neutron crystallography.31
Structure-based drug design relies on the three-dimensional structure of enzymes and their respective ligand binding sites to guide the design of inhibitors, with X-ray crystallography prevailing as the technique of choice for structure determination. While X-ray crystallography excels at resolving positions of nonhydrogen atoms, it usually lacks information on the positions of H atoms which account for ∼50% of all atoms in proteins.32,33 Because H atoms play essential roles in several noncovalent interactions such as H bonding, van der Waal's forces, and electrostatics, determining H atom positions and thus protonation states, H atoms provide valuable information on substrate and ligand binding, proton transfer reactions, and protein–protein interactions.32,34 Whereas the X-ray scattering magnitude of H atoms is substantially smaller compared to other atoms present in protein structures, the neutron scattering lengths of H and its isotope deuterium (D) atoms are similar to those of the “heavier elements” (C, N, O). Hence, H/D positions can be resolved at moderate resolutions with neutron crystallography.11,35–40 This unique feature of neutron crystallography renders it a powerful technique for uncovering the structural and mechanistic information arising from precise protonation states and accurate all-atom models for inhibitor design.41–44 Moreover, cold neutrons (λ = ∼2–5 Å) do not cause radiation damage to protein crystals permitting determination of radiation damage-free structures at room temperature.
In all PLP-dependent enzymes, including SHMT, the PLP coenzyme is covalently linked to a catalytic lysine (Lys) residue forming a Schiff base linkage referred to as the internal aldimine in the holoenzyme. Almost universally, the first reaction step is the formation of an external aldimine (not covalently linked to the enzyme) generated through the transaldimination reaction where the substrate amino acid α-amine displaces the Lys ε-amine. For SHMT, the catalysis starts with the production of L-Ser-bound external aldimine, PLP-L-Ser. PLP-L-Ser can subsequently react, releasing free formaldehyde and Gly in a THF-independent fashion. This THF-independent conversion of L-Ser to Gly is believed to proceed through the retro-aldol mechanism with a general base abstracting the PLP-L-Ser β-hydroxyl.9,45,46 Conversely, THF binding results in the THF-dependent conversion of L-Ser to Gly and the formation of 5,10-MTHF. There is no accepted mechanism for THF-dependent catalysis in SHMT and the proposed reaction mechanisms require up to six different general acids and bases.45,47–49 It is therefore of great importance to pinpoint protonation states of the SHMT active site amino acids, PLP, and THF to uncover the reaction mechanism of SHMT catalysis. In our previous study11 we utilized room-temperature neutron crystallography to reveal the atomic organization of the TthSHMT holoenzyme active site and of the pre-Michaelis complex where L-Ser amino acid was captured in the peripheral substrate binding site normally occupied by THF. By revealing the positions of H/D atoms, accurately assigning protonation states, and mapping H bonding networks we concluded that Glu53 (Glu98 in hSHMT2) is the best candidate for a general base catalyst, whereas all previously proposed active site His residues could not undergo protonation state changes, being constrained by H bonding networks, and hence would not be able to participate in catalysis.
Here, we build upon our previous study11 and present a 2.3 Å resolution neutron structure of TthSHMT complex with (6S)-5-formyltetrahydrofolate (folinic acid, FA), a substrate analog of THF and inhibitor of SHMT, and an acetate molecule found at the cationic substrate binding site mimicking an amino acid substrate. FA is substituted at the N5 position with a formyl group, resembling the carbinolamine form of THF. Therefore, the current TthSHMT/FA neutron structure bears resemblance to an intermediate in the catalytic mechanism. Moreover, we obtained X-ray structures of TthSHMT-Gly/FA, TthSHMT-L-Ser/FA, and hSHMT2-Gly/FA ternary complexes containing PLP-Gly or PLP-L-Ser external aldimines providing insights into the organization and structural rearrangements in the active site associated with the amino acid substrate and FA binding. Importantly, we observed neutral Schiff base nitrogen and neutral His residues in TthSHMT/FA in agreement with the previous neutron structure of TthSHMT holoenzyme. A D atom was located on the carboxylic moiety of Glu53 indicating its protonation in TthSHMT/FA. The protonation states observed in our neutron structure of TthSHMT/FA and structural organization and comparison of the external and internal aldimine complexes allow us to propose a central role of Glu53 acting as the universal general acid–base catalyst in both THF-independent and THF-dependent transformations catalyzed by SHMT.
2 Results
2.1 Functional role of the gating loop in TthSHMT
TthSHMT, and other prokaryotic SHMTs, exist and function as biological homodimers.12,50 Each protomer of TthSHMT can be divided into large (residues 33–284) and small (residues 7–32 and 285–407) domains (Fig. 1b). A seven-stranded mixed β-sheet surrounded by α-helices arranged in an α/β/α sandwich makes up the large domain. The small domain is composed of anti-parallel β-strands and α-helices in a two-fold α/β sandwich. The active sites are formed by the residues from both protomers as discussed further below. Homodimeric TthSHMT crystallizes as a complete dimer present in the asymmetric unit, with seemingly identical active sites in protomers A and B. However, crystal packing contacts alter the functionalities of the two active sites, as was demonstrated by our structures of L-Ser and D-Ser complexes reported previously.11L-Ser was found within the peripheral THF-binding site (henceforth referred to as the peripheral binding site) of protomer A representing a pre-Michaelis complex, whereas D-Ser bound deeper inside the active site at the cationic amino acid substrate binding site (henceforth referred to as the cationic binding site) in protomer B mimicking the Michaelis complex. The entrance to the active site is believed to be controlled by a gating loop spanning residues 342–356 that can adopt open and closed conformations before and after a THF substrate analogue, such as folinic acid, or some inhibitors would bind, respectively, according to crystal structures of SHMT from various species.12,51–53 In the crystal structure of TthSHMT holoenzyme, where PLP is connected to the catalytic Lys226 as internal aldimine (PDB ID: 8SUJ),11 we detected the gating loop adopting two conformations even before substrate binding, an open conformation in protomer A and a partially closed conformation in protomer B (Fig. 1c). In protomer B, the gating loop residues shifted by ∼2–2.5 Å compared to their positions in protomer A. The gating loop has crystal contacts in protomer A that may restrict its conformational flexibility and dynamics, but it should be able to move freely in protomer B where the closest symmetry-related molecule is over 13 Å away (Fig. S1A†). This observation may explain the differential binding of ligands to protomers A and B in TthSHMT structures and apparent lack of reactivity (or inertness) of protomer A active site towards natural substrates like L-Ser or Gly, as demonstrated previously11 and discussed below.2.2 Joint XN structure of TthSHMT/FA complex
The fundamental chemical structure framework of vitamin B9 and its derivatives, such as FA, can be broken into three components: pterin ring, 4-aminobenzoyl group, and a glutamate (mono or poly) tail. The pterin group is placed tightly at the bottom of the peripheral binding site near the acetate molecule, effectively blocking off the entrance to PLP in the TthSHMT/FA complex. The 4-aminobenzoyl group extends through a hydrophobic channel and the glutamate tail reaches the bulk solvent at the enzyme surface within the peripheral substrate binding site (Fig. 1d). Analysis of the peripheral binding site in protomer B of TthSHMT/FA complex reveals that the gating loop assumed the fully closed conformation, moving beyond the intermediate closed conformation of protomer B in the holoenzyme structure. In the TthSHMT/FA complex, the gating loop moves as much as 4 Å toward FA relative to the open conformation seen in protomer A (Fig. 1e and S1B†), leading to a cascade of additional structural rearrangements that are more pronounced than those detected between the two protomers in the holoenzyme structure.11 Specifically, the loop containing residues 318–324 moves ∼3 Å in the same direction as the gating loop. In concert with this structural shift, the nearby antiparallel β-sheet consisting of two β-strands (residues 313–317 and 356–360) and an adjacent α-helix (residues 325–335) tilt by ∼7° (Fig. S1C and S1D†). The tight fit of FA in this site suggests that the gating loop may have initially opened to allow for the substrate analogue to enter, and then closed back to keep the ligand bound. Such a mechanism of FA binding to TthSHMT is supported by the absence of the ligand in protomer A, where the gating loop remains in the open conformation as in the holoenzyme structure and cannot close presumably due to crystal contacts. The gating loop movement upon FA binding closes the active site entrance as illustrated using the protein surface representation in Fig. S1E and S1F.†
![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N double bond rotated 37° above the plane of the PLP on the si face and away from the cationic substrate binding site occupied by the acetate molecule. In our two previous neutron structures of TthSHMT, this dihedral angle was measured between 29° and 38°, and such out-of-plane geometry was also observed for the neutral Schiff base of the PLP internal aldimine in the neutron structure of AAT (PDB ID: 5VJZ).37 The phenolic oxygen O3′ in TthSHMT/FA is deprotonated and negatively charged. The O3′ charge is stabilized through the electronic resonance with the combined π-conjugated system of the pyridine ring and Schiff base. As determined in our previous neutron structures of TthSHMT, O3′ participates in an unconventional C–H⋯O bond with the Cδ2 atom of neutral His200 having the H⋯O and C⋯O distances of 2.1 and 2.9 Å, respectively (Fig. 2a). Consequently, His200 is oriented such that it uses its non-protonated Nδ1 to make an H bond with the protonated Nε2 of His312 (D⋯N distance of 2.0 Å, N⋯N distance of 3.0 Å), whereas the protonated Nε2 of His200 contacts the carboxylate group of the acetate molecule. His312, also observed as neutral, is further anchored to its own backbone amide ND by an H bond with a D⋯N distance of 2.2 Å and N⋯N distance of 2.9 Å (Fig. 2b). In addition, the PLP's pyridine ring is sandwiched between the neutral His122 and the methyl side chain of Ala199 making hydrophobic π–π stacking and C–H⋯π interactions, respectively, which together with the H bonds secure its orientation. His225 located near the phosphate group of the PLP is also observed as singly protonated. We thus note that the protonation states of the active site PLP in the internal aldimine form and His residues remain unchanged upon FA binding in the TthSHMT/FA complex. Moreover, identical protonation states of the PLP internal aldimine were also observed by neutron crystallography in aspartate aminotransferase (AAT), another enzyme in the fold-type I PLP-dependent family.37
N double bond rotated 37° above the plane of the PLP on the si face and away from the cationic substrate binding site occupied by the acetate molecule. In our two previous neutron structures of TthSHMT, this dihedral angle was measured between 29° and 38°, and such out-of-plane geometry was also observed for the neutral Schiff base of the PLP internal aldimine in the neutron structure of AAT (PDB ID: 5VJZ).37 The phenolic oxygen O3′ in TthSHMT/FA is deprotonated and negatively charged. The O3′ charge is stabilized through the electronic resonance with the combined π-conjugated system of the pyridine ring and Schiff base. As determined in our previous neutron structures of TthSHMT, O3′ participates in an unconventional C–H⋯O bond with the Cδ2 atom of neutral His200 having the H⋯O and C⋯O distances of 2.1 and 2.9 Å, respectively (Fig. 2a). Consequently, His200 is oriented such that it uses its non-protonated Nδ1 to make an H bond with the protonated Nε2 of His312 (D⋯N distance of 2.0 Å, N⋯N distance of 3.0 Å), whereas the protonated Nε2 of His200 contacts the carboxylate group of the acetate molecule. His312, also observed as neutral, is further anchored to its own backbone amide ND by an H bond with a D⋯N distance of 2.2 Å and N⋯N distance of 2.9 Å (Fig. 2b). In addition, the PLP's pyridine ring is sandwiched between the neutral His122 and the methyl side chain of Ala199 making hydrophobic π–π stacking and C–H⋯π interactions, respectively, which together with the H bonds secure its orientation. His225 located near the phosphate group of the PLP is also observed as singly protonated. We thus note that the protonation states of the active site PLP in the internal aldimine form and His residues remain unchanged upon FA binding in the TthSHMT/FA complex. Moreover, identical protonation states of the PLP internal aldimine were also observed by neutron crystallography in aspartate aminotransferase (AAT), another enzyme in the fold-type I PLP-dependent family.37
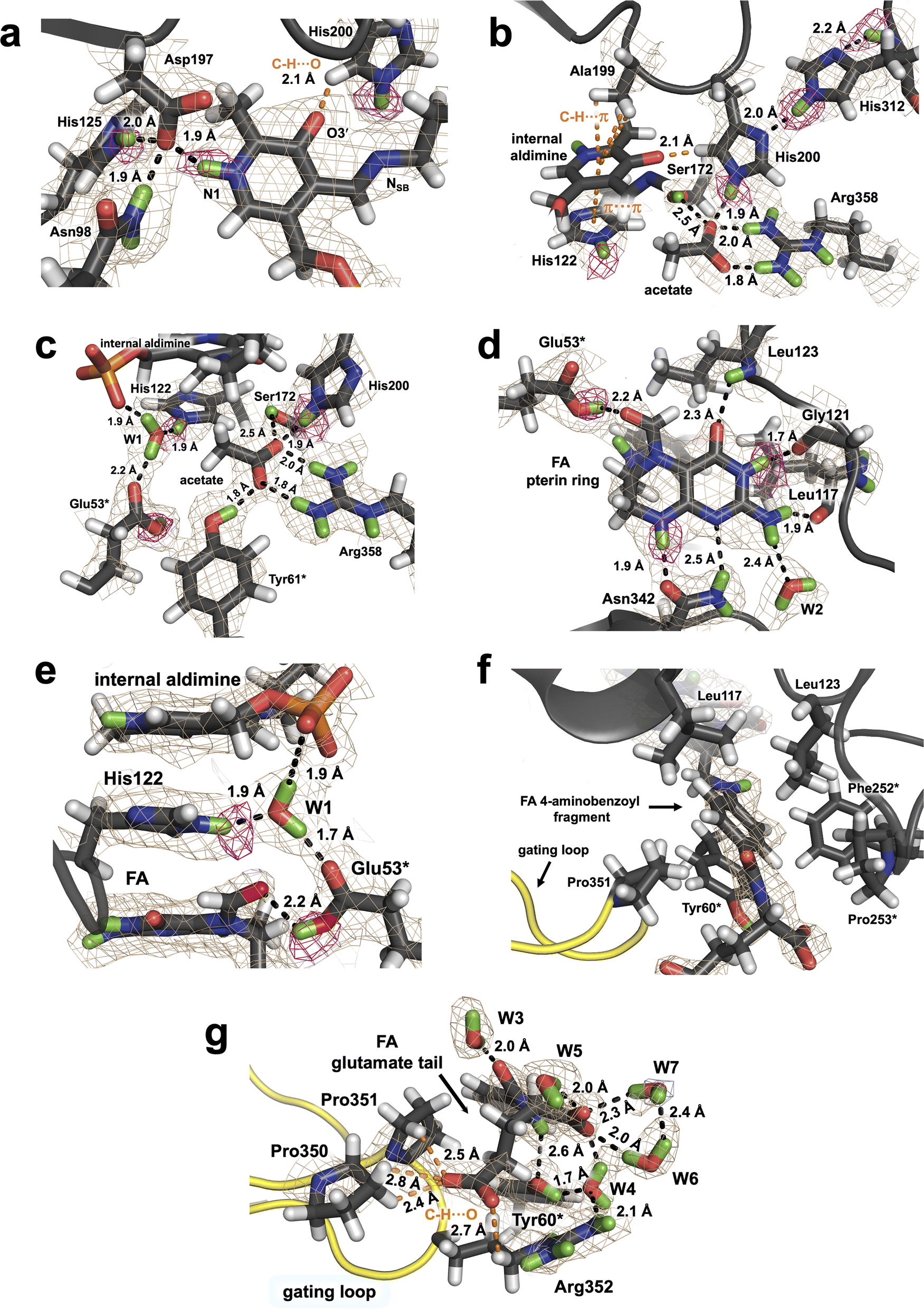 | ||
| Fig. 2 Binding and interactions of PLP and FA in the active site of TthSHMT protomer B. (a) Protonation states of the PLP internal aldimine and surrounding residues. (b) Binding of acetate molecule and the H binding network connecting PLP O3′ to His312 main chain. (c) A different view of the acetate molecule and the coordination of conserved water W1. (d) H bonding network around the FA's pterin moiety showing protonation of Glu53* (protomer A). (e) Stacking of PLP's pyridine, His122 and the FA's pterin. (f) Hydrophobic pocket binding the 4-aminobenzoyl group of FA. (g) Binding, hydration and interactions of the FA monoglutamate tail near the gating loop (yellow). The 2FO–FC neutron scattering length density map for TthSHMT/FA active site residues and bound ligands is depicted in wheat mesh and is contoured at 1σ level. The omit FO–FC difference neutron scattering length density map is shown in magenta mesh contoured at 2σ level. | ||
Unlike the THF substrate, FA is substituted at the N5 position with a formyl group and N5 adopts the trigonal planar geometry. The aldehyde carbonyl is rotated towards Glu53* carboxyl, and 2.7 Å and 3.0 Å distances separate the oxygen atoms. The difference FO–FC neutron scattering length density map revealed a D atom on Oε2 of Glu53* which is 2.2 Å away from the FA's carbonyl oxygen (Fig. 2d and S2†). Consequently, Glu53* is protonated, thus neutral, donating its D atom in an H bond with FA, which nonetheless cannot be regarded as a strong H bond because the carboxylic O–D bond is rotated out of the carboxyl moiety plane by 41°. Glu53* also accepts a D atom from a tightly bound water W1 (Fig. 2e), whose position is stabilized further by H bonds with His122 and the PLP phosphate. Notably, the FA's pterin, His122 imidazole and PLP pyridine groups are stacked against each other. It is of note that Glu53* is found to be deprotonated and negatively charged in the holoenzyme neutron structure.11 In addition, Glu53* is the only active site amino acid residue observed to undergo a protonation state change upon FA binding.
The pterin moiety of FA, like THF and folic acid, can assume two tautomeric forms, in which either N1 or N3 (Fig. 1a) is protonated shifting the endocyclic double bond from C2N3 to C2N1, respectively.54 As a result, N1 and N3 can switch their roles between an H bond donor and H bond acceptor, although DFT quantum chemical studies of over 30 pteridines showed that N3-lactam tautomer is the most stable.55 In the TthSHMT/FA complex, the neutron scattering length density maps unequivocally indicate that N1 is not protonated and N3 is protonated (Fig. 2d). This tautomer is apparently locked in by several H bonding interactions with TthSHMT residues. Specifically, N1 accepts a D atom and N8 donates its D in an H bond bridge that connects the pterin moiety to the Asn342 side chain that is properly oriented to support these two H bonds. N3-D is H bonded with the backbone carbonyl oxygen of Gly121, whereas the exocyclic carbonyl adjacent to N3 accepts a D atom from the main chain amide of Leu123 to form a second H bond bridge. The N2′ amine found to have the trigonal planar configuration with its plane rotated by 30° relative to the pterin's plane forms H bonding interactions with the main chain carbonyl of Leu117 and a structural water molecule (W2, Fig. 2d). Remarkably, the rest of the FA structure makes no additional H bonds with the enzyme. The closest interaction is that of the amide ND of the FA glutamate tail with the phenolic oxygen of Tyr60* having the D⋯O distance of 2.6 Å (N⋯O distance of 3.5 Å). The 4-aminobenzoyl fragment thus uses predominantly hydrophobic interactions to anchor its position within the peripheral binding site and is situated in a hydrophobic cage made up by the side chains of Tyr60*, Phe252*, Pro253*, Leu117, Leu123, and Pro351 (Fig. 2f and g). Staggered π–π stacking with Tyr60* and C–H⋯π interactions with the side chain of Leu123 stabilize the 4-aminobenzoyl position. The closest C⋯C contacts with Tyr60* are 3.5 Å and those with Leu123 are 3.8 Å. The structural arrangement of the hydrophobic cage clusters Leu117, Leu123, Phe252* and Pro253* into a hydrophobic patch (Fig. 2f). As a result of being located at the dimer interface, the hydrophobic patch adopts a more rigid geometry, and the positions of these residues are almost invariant relative to the holoenzyme. Conversely, Tyr60* phenolic side chain rotates 21° to avoid clashing with FA and Pro351 from the gating loop. The main chain carboxylate of the FA glutamate moiety is hydrated by four surface water molecules that shield its negative charge. The side chain carboxylate, on the other hand, is stabilized through interactions with Pro350, Pro351 and the positively charged Arg352 located at the tip of the gating loop through the unconventional C–H⋯O H bonds (Fig. 2g).
2.3 X-ray structures of TthSHMT-Gly/FA and TthSHMT-L-Ser/FA complexes
The TthSHMT-Gly/FA and TthSHMT-L-Ser/FA ternary complexes were obtained by soaking the holoenzyme crystals with FA and either of the substrates, Gly or L-Ser. Room-temperature X-ray data were collected on the stable TthSHMT-Gly/FA complex and the structure was obtained at 2.0 Å resolution. However, the TthSHMT-L-Ser/FA complex had to be cryogenically trapped to discourage the THF-independent retro-aldol cleavage of L-Ser, requiring the X-ray diffraction data to be collected at 100 K. The TthSHMT-L-Ser/FA structure was thus obtained at 1.8 Å resolution. As anticipated, FA was observed only in protomers B in these two structures bound identically as in the TthSHMT/FA complex. Also, in both complexes, the corresponding amino acid substrate displaces Lys226 from the internal aldimine form with PLP through a transaldimination reaction to yield a substrate-bound PLP-Gly or PLP-L-Ser external aldimine states, again detected only in protomers B (Fig. 3a and b). Surprisingly, however, protomer A of TthSHMT-Gly/FA complex lost its PLP cofactor and is observed in the apo-state (Fig. 3a). Conversely, the PLP internal aldimine form remained intact in protomer A of TthSHMT-L-Ser/FA complex, and an L-Ser molecule is found at the cationic amino acid substrate binding site (Fig. 3b). We first describe the structural organization of the active sites in protomers B in these two complexes and then proceed to consider the geometries of the active sites in protomers A.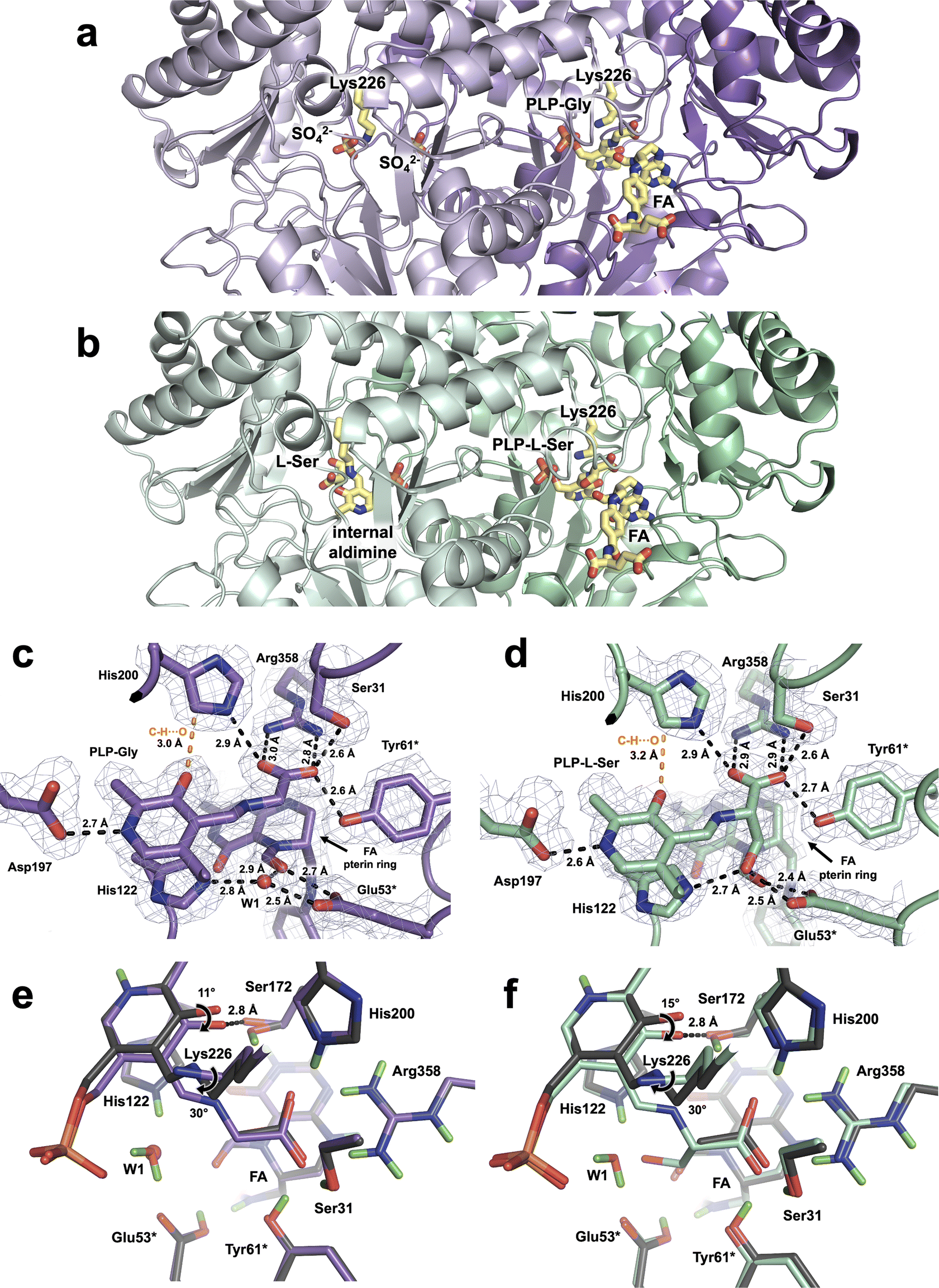 | ||
| Fig. 3 TthSHMT-Gly/FA and TthSHMT-L-Ser/FA complexes. (a) Overview of the TthSHMT-Gly/FA ternary complex. Protomer B (purple) contains the PLP-Gly external aldimine and FA. Protomer A (light purple) lost the PLP cofactor and is thus seen in the apo-form with two sulfate ions occupying the cationic binding site and the previous position of the PLP phosphate group. (b) Overview of the TthSHMT-L-Ser/FA ternary complex. Protomer B (green) contains the PLP-L-Ser external aldimine and FA. Protomer A (light green) retains PLP in the internal aldimine form and an unreacted L-Ser molecule is found in the cationic binding site. (c) The architecture of the active site in TthSHMT-Gly/FA. The 2FO–FC electron density map is shown as gray mesh contoured at 1σ level. (d) The architecture of the active site in TthSHMT-L-Ser/FA. The 2FO–FC electron density map is shown as gray mesh contoured at 2σ level. In both (c) and (d) H bonds are shown as black dashed lines, and the unconventional C–H⋯O interactions are represented by the orange dashed lines. (e) Superposition of TthSHMT-Gly/FA (purple carbons) onto the TthSHMT/FA structure (dark gray carbons) showing that conformational differences in the active sites are localized to the PLP region. (f) Superposition of TthSHMT-L-Ser/FA (green-cyan carbons) onto the TthSHMT/FA structure (dark gray carbons). | ||
In both TthSHMT-Gly/FA and TthSHMT-L-Ser/FA, as also observed in TthSHMT/FA, FA binding effectively blocks the entrance to the active site restricting bulk solvent from reaching PLP-Gly or PLP-L-Ser. The FA binding pose is identical in all three structures, with very similar interactions (Fig. S3†). FA's N5 atom keeps the trigonal planar configuration and the aldehyde group faces the Glu53* carboxylic group with the O⋯O distances of 2.7 and 2.4 Å in TthSHMT-Gly/FA and TthSHMT-L-Ser/FA, respectively, indicating formation of H bonds. Importantly, Glu53* makes an additional H bond of 2.5 Å using the other carboxylate oxygen with either a structural water molecule, W1, in TthSHMT-Gly/FA or with the PLP-L-Ser β-hydroxyl group in TthSHMT-L-Ser/FA. It appears this water templates the position of the β-hydroxyl group in the PLP-L-Ser external aldimine as it is displaced by the L-Ser side chain oxygen atom. Based on our observations in the TthSHMT/FA XN structure, we suggest that Glu53* is also protonated in TthSHMT-Gly/FA and TthSHMT-L-Ser/FA and donates its H atom in H bonds with the FA's aldehyde.
The active site of protomer A in TthSHMT-L-Ser/FA complex maintains the PLP cofactor in the internal aldimine form. Unexpectedly, however, we found an L-Ser molecule bound in the cationic substrate binding site surrounded by His200*, Arg358*, Glu53 and Tyr61 residues (Fig. 3b and S4B†). L-Ser clearly did not react with the PLP to form the external aldimine as it did in protomer B and adopts a unique binding orientation not seen in our previous complex with D-Ser.11 Instead of being anchored tightly by a salt bridge to Arg358 with its carboxylate as D-Ser was, L-Ser is rotated such that the carboxylate makes a 2.7 Å H bond with His200, yet keeping one H bond with Arg358 having the distance of 2.9 Å (Fig. S4C†). The L-Ser amino group is also not oriented towards the internal aldimine but rather faces Glu53* and Tyr61* making 2.8 and 2.9 Å H bonds, respectively, with their side chains and a 2.6 Å H bond with an active site water molecule. As a result, the sidechain β-hydroxyl group of L-Ser is not interacting with Glu53*; it is shifted to form a 2.6 Å H bond with Ser31*. Consequently, L-Ser has been trapped in an intermediate orientation in the cationic binding site of protomer A in TthSHMT-L-Ser/FA and has to reorient by flipping its α-amino group towards the PLP C4′ for the transaldimination reaction to ensue.
2.4 X-ray structure of hSHMT2-Gly/FA complex
In our studies we utilize TthSHMT as a model enzyme to glean the structure–function information about hSHMT2 while attempts are under way to obtain neutron-quality crystals of the human enzyme. For this study, we were able to co-crystallize hSHMT2 with Gly and FA to result in a stable ternary complex hSHMT2-Gly/FA, which is identical to that obtained for TthSHMT but has not been previously reported. A 2.1 Å resolution X-ray diffraction dataset was collected on a hSHMT2-Gly/FA crystal at room-temperature. hSHMT2-Gly/FA crystallizes in a novel orthorhombic unit cell with space group P2221. The asymmetric unit contains two copies of hSHMT2 dimers (Fig. 4a). The expected tetrameric biological assembly of hSHMT2 is generated through crystallographic symmetry operations. Protein chains A and C make up one of the dimers, whereas chains B and D form the other dimer. The dimers are essentially equivalent, having the RMSD of 0.1 Å. All four chains contain both the PLP-Gly external aldimine and bound FA. In chains A and B, the β-hairpin flap motif (residues 293–311) is resolved but remains disordered in chains C and D. The flap motif is conserved in mammalian SHMTs but is not found in bacterial homologs. For example, in TthSHMT the flap motif is reduced to a four-residue loop (residues 238–241). Studies with the cytosolic hSHMT1 (ref. 57) suggest the flap motif is important in stabilizing the tetrameric assembly, PLP binding, and binding affinity of THF and presumably raises the pKa of the catalytic Glu residue (Glu75 in hSHMT1 and Glu98 in hSHMT2).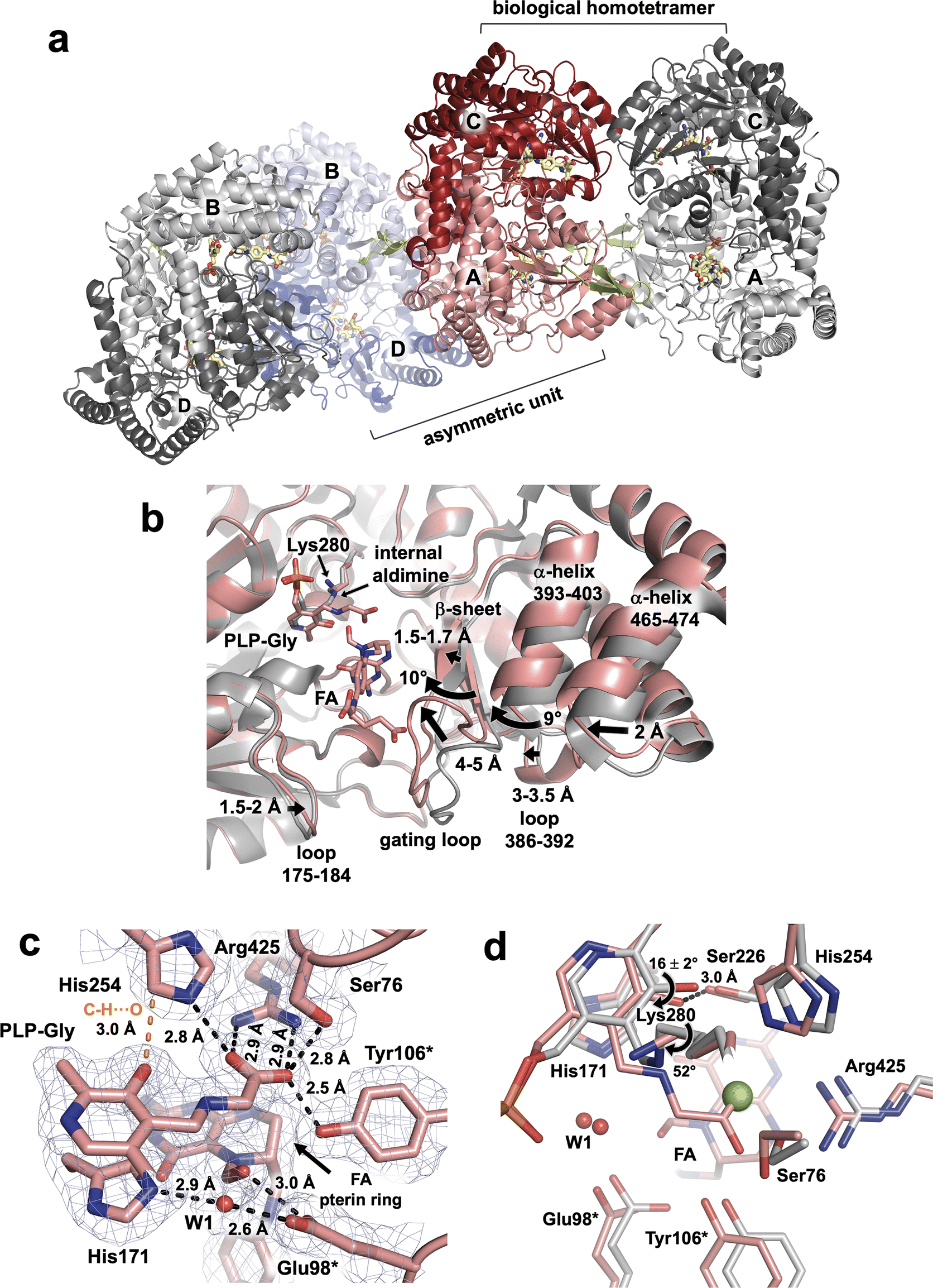 | ||
| Fig. 4 hSHMT2-Gly/FA complex and the structural details of the active site. (a) Assembly of the biological tetramer in hSHMT2-Gly/FA ternary complex. Two hSHMT2 dimers (protomers A/C: red/pink and protomers B/D: blue/light blue) occupy the asymmetric unit whereas the tetramers are generated through the crystallographic two-fold axes. The β-hairpin flap motifs (green, residues 293–311) are resolved in protomers A and B from separate dimers but are disordered in protomers C and D. (b) Superposition of the hSHMT2-Gly/FA complex (pink, protomer A) onto holoenzyme hSHMT2 structure (light grey, protomer A, PDB ID: 8SSJ) showing several structural rearrangements associated with the FA binding. (c) The architecture of the active site in hSHMT2-Gly/FA. The 2FO–FC electron density map is shown as gray mesh contoured at 1σ level. H bonds are shown as black dashed lines, and the unconventional C–H⋯O interaction is represented by the orange dashed line. (d) Superposition of hSHMT2-Gly/FA (pink carbons) onto the hSHMT2 holoenzyme structure (gray carbons) showing conformational differences in the active sites caused by the PLP-Gly external aldimine formation. Chloride ion present in hSHMT2 holoenzyme and located in the cationic binding site is represented by the green sphere. | ||
As observed in the TthSHMT structures, FA binding induces a series of structural rearrangements in hSHMT2 compared to the holoenzyme (PDB ID 8SSJ).11 The gating loop (residues 410–423), adjacent to the FA binding cleft at the entrance to the active site, moves by 4–5 Å to assume a closed conformation upon FA binding (Fig. 4b). The gating loop movement results in the adjacent loop (residues 386–392) to shift ∼3–3.5 Å in unison. In concert, a short β-strand (residues 408–410) preceding the gating loop shifts by 1.5–1.7 Å, whereas the two β-strands making a nearby antiparallel β-sheet (residues 381–385 and 423–427) and an α-helix (residues 393–403) flanking the gating loop tilt by ∼10° and ∼9°, respectively. The movement of the 393–403 α-helix triggers another α-helix (residues 465–474) to move by ∼2 Å. On the opposite side of the FA's glutamate tail, a loop containing residues 175–184 tilts toward FA by 1.5–2 Å. Interestingly, in TthSHMT this loop (residues 126–130) is truncated by five residues and undergoes significantly less pronounced shift of 0.7–0.8 Å. The cascade of motions of the secondary structure elements in the hSHMT2 peripheral binding site prompted by the FA binding fully closes the active site in a similar fashion as was also observed for TthSHMT. The active site closure is identical in all four chains of hSHTM2-Gly/FA complex as the chains A–D superimpose on each other with the RMSDs of 0.14 ± 0.02 Å (Fig. S5†).
Because the crystals of hSHMT2-Gly/FA complex were obtained by co-crystallization, unlike the TthSHMT-Gly/FA complex which formed through soaking, all four protein chains in hSHMT2-Gly/FA reacted with Gly affording the PLP-Gly external aldimine, displacing and freeing the catalytic Lys280 from its covalent linkage with PLP (Fig. 4c). The ε-amino group of the released Lys280 adopts a position on the si face of PLP 3.4–3.6 Å above the Schiff base C4′. As in TthSHMT-Gly/FA, the conformational flexibility of the Lys280 side chain is reduced and its position is stabilized by H bonds with Thr277 and Tyr96* ensuring the Lys280's ε-amino group is readily available for the reverse reaction. The pyridine ring of the PLP-Gly external aldimine is tilted towards His171 by 16 ± 2° in hSHMT2-Gly/FA compared to its position in the hSHMT2 internal aldimine (PDB ID: 8SSJ) in all protein chains (Fig. 4d). The tilt of the pyridine ring brings PLP-O3′ closer to Ser226 to form an H bond. Rotation of the Schiff base relative to the pyridine ring plane decreases dramatically in the hSHMT2 PLP-Gly external aldimine compared to that in the internal aldimine (PDB ID: 8SSJ) and the Schiff base becomes nearly co-planar with the PLP pyridine ring. However, there are slight differences between the four protein chains in hSHMT2-Gly/FA. In chains A and D, the Schiff bases are on the re face of the PLP, with the dihedral angles of −13 and −14°, respectively, as was also detected in TthSHMT-Gly/FA. Conversely, in chains B and C, the Schiff bases do not rotate as much, remaining on the si face of the PLP with the dihedral angles of 9 and 2°, respectively. The binding interactions of PLP, including the H bond between PLP-N1 and Asp251, the C–H⋯O bond between His254 and PLP-O3′, π-interactions with Ala253 and His171, and H bonds within the phosphate binding pocket, are retained by the PLP-Gly external aldimine compared to the internal aldimine and exhibit similar bond distances in all the active sites. A salt bridge with Arg425 anchors the PLP-Gly carboxylate in a rigid geometry in the cationic substrate binding site along with H bonds with the side chains of His254, Ser76, and Tyr106* (Fig. 4c).
The pterin moiety of the bound FA in hSHMT2-Gly/FA blocks the entrance to the substrate binding pocket and sequesters the active site from bulk solvent, leaving only one water molecule (W1) near the catalytic site exactly as it did in TthSHMT (Fig. 4c and S6A†). The oxygen atoms in the N5 aldehyde group and the 4′ carbonyl displace two water molecules occupying those positions in the open, internal aldimine form of hSHMT2. The formyl group on FA-N5 rotates towards Cα of PLP-Gly, positioning the N5 aldehyde O atom 3.1–3.3 Å from the external aldimine Cα and forming 2.8–3 Å H bonds with the carboxylate of the catalytic Glu98*. Glu98* participates in an H bonding network through its interaction with W1 which also connects His171, FA's aldehyde and the PLP-Gly phosphate (Fig. S6B†). W1 appears to be conserved between TthSHMT-Gly/FA and hSHMT2-Gly/FA structures, templating the location of the β-hydroxyl of PLP-L-Ser external aldimine as was demonstrated in the TthSHMT-L-Ser/FA structure.
The pterin ring orientation is determined by H bonds with the active site residues that are conserved between hSHMT2 and TthSHMT. Hence, these H bonds are conserved between TthSHMT-Gly/FA and hSHMT2-Gly/FA complexes. Briefly, in hSHMT2-Gly/FA, FA-N1 and -N8 have H bonds with Asn410, and FA-N2′ amine makes H bonds with Leu166 and a water molecule (W2). The backbone carbonyl of Gly170 interacts with the N3 cyclic secondary amine. The exocyclic carbonyl accepts an H bond from the backbone amide of Leu172 (Fig. S6A†). The FA 4-aminobenzoyl group docks in a peripheral binding site made up of hydrophobic residues from both protomers. The side chains Tyr105*, Phe320*, Pro321*, Leu166, Leu172, and Ala418 make up the hydrophobic cage surrounding the 4-aminobenzoyl group (Fig. S6C†). The glutamate tail extends towards the surface of hSHMT2, reaching the bulk solvent. The structure of the glutamate tail is flexible and interacts with hSHMT2 through the gating loop, consisting of H bonds with Ser417, Ala418 and Ile419 (Fig. S6D†). Taken together, the modes of PLP-Gly and FA binding to the active sites of TthSHMT and hSHMT2 are very similar because the active site residues are conserved in these two proteins.
3 Discussion
The two major chemical transformations catalyzed by the SHMT enzyme are the THF-independent cleavage of β-hydroxy amino acids, such as L-Ser, to generate corresponding free aldehydes58,59 and THF-dependent synthesis of 5,10-MTHF.2,47,60 Within the SHMT active site these reactions proceed through the general acid-base catalysis governed by finely tuned proton transfer events between a substrate, PLP, THF and active site amino acid side chains. To determine the mechanism(s) by which SHMT catalyzes transformations of L-Ser into Gly and L-Ser/THF into Gly/5,10-MTHF positions and movement of H atoms along the reaction pathways have to be directly identified with high accuracy and confidence. In this study we have built upon our previous work using neutrons to pinpoint H atoms in the active site of TthSHMT with PLP in the internal aldimine form and bound to a THF mimic, FA. We also captured TthSHMT in ternary complexes containing PLP-Gly or PLP-L-Ser external aldimine forms and FA, and hSHMT2 in complex with PLP-Gly and FA. Moreover, the crystal packing effects permitted observation of the TthSHMT apo-form with no PLP present in the active site and a Michaelis complex mimic with L-Ser bound in the cationic binding site.Detailed structural analyses and comparisons lead us to suggest an essential role of the gating loop (residues 342–356 in TthSHMT and residues 410–423 in hSHMT2) in the SHMT enzyme function controlling the open-to-closed conformational change during THF binding and restricting bulk solvent access to the active site interior. A functional role of the open-to-closed conformational change for the SHMT active site was proposed in a seminal paper by Schirch et al.61 (1991), although due to the lack of an available crystal structure specific residues could not be pinpointed. Later, Trivedi et al.12 (2002) suggested based on the crystal structures of binary and ternary complexes of Bacillus stearothermophilus SHMT that amino acid substrate alone did not induce sizable conformational changes of the active site whereas binding of Gly and FA showed appreciable movement of the loop region spanning residues 342–352. Comparison of the gating loop conformational flexibility in protomers A and B in our structures suggests that the gating loop has two functional roles in SHMT: (1) its ability to close the active site is required for the substrate THF to bind, as FA binding was not detected in protomers A of any TthSHMT structures, but was observed in protomers B and all four hSHMT2 protomers; and (2) its conformational flexibility is instrumental in the active site reactivity towards the amino acid substrates as the internal aldimine PLP was unreactive towards L-Ser in protomers A but proceeded to PLP-L-Ser external aldimine in protomers B and to PLP-Gly in all four hSHMT2 protomers.
Several mechanisms for THF-independent and THF-dependent SHMT catalysis have been proposed. The dominant view for the THF-independent conversion of L-Ser, and other β-hydroxy amino acids, into the corresponding aldehydes is the retro-aldol mechanism, in which a general base located near the PLP-L-Ser external aldimine abstracts the proton of the β-hydroxyl group resulting in Cα–Cβ bond cleavage.9,45,46 Using neutron crystallography, we have previously accurately mapped the H atom positions within TthSHMT active site in the internal aldimine form.11 Glu53 was found to be deprotonated and all His residues were observed as neutral. Glu53 (Glu98 in hSHMT2) was proposed as the best candidate for the general base because none of the other residues could undergo protonation/deprotonation processes. Hence, we confirmed the retro-aldol mechanism as the most probable route for the THF-independent reaction. Conversely, there is no accepted mechanism for the THF-dependent SHMT catalysis.12,45,47,48,60,62,63 In one hypothesis, the first step is identical to the THF-independent reaction, with a formaldehyde molecule formed transiently before it is attacked by N5 of THF to give a THF N5-carbinolamine (N5–CH2–OH) intermediate. A different mechanism proposes a direct nucleophilic attack by the THF N5 amine on Cβ of PLP-L-Ser external aldimine producing the same carbinolamine intermediate. This attack by THF N5 could also proceed with the simultaneous dehydration of the Cβ hydroxyl to generate a covalent intermediate having a direct bond between THF N5 and PLP Cβ, as is suggested by yet another mechanism. Remarkably, up to six chemical groups were suggested to participate in the THF-dependent reaction as general acid-base catalysts in these mechanisms.
In this study, unlike in our previous neutron structure of TthSHMT,11 we observed protonation of Glu53 in the TthSHMT/FA complex on the carboxylic oxygen that would face the THF N5 in the native ternary complex containing THF and PLP-L-Ser. Strategically positioned within the active site in the near proximity to the β-hydroxyl of PLP-L-Ser, and N5 and N10 of THF, capable of undergoing protonation/deprotonation events and conserved in all SHMT enzymes, Glu53 could be considered the universal general acid-base catalyst facilitating both THF-independent and THF-dependent transformations by SHMT enzyme (Fig. 5). It is important to note that previous mutagenesis studies revealed that Glu-to-Gln substitution of Glu53, or the equivalent Glu residue in SHMT enzymes from other species, nearly abolishes the THF-dependent catalysis, pointing to the critical functional role of this residue.45,63,64 Building on our experimental observations, we now propose the following steps in the THF-dependent mechanism for SHMT catalysis. (1) In the ternary complex containing the PLP-L-Ser external aldimine and bound THF, the negatively charged (deprotonated) Glu53 can either abstract a proton from the β-hydroxyl of PLP-L-Ser or the THF N5. The latter would be the only viable proton transfer, whereas the former would be a futile route because the protonated trigonal planar THF N5 is too weak of a nucleophile to react with formaldehyde. (2) Deprotonated, negatively charged, THF N5 is a strong nucleophile that can directly attack Cβ to result in THF N5-carbinolamine intermediate. Note that at this stage Glu53 is protonated. (3) In the subsequent step, Glu53 donates its proton to the hydroxyl group of the THF N5-carbinolamine, returning to the deprotonated state, leading to the release of a water molecule and production of the THF N5-methyleneimine intermediate. Importantly, N5 in the THF N5-methyleneimine intermediate is positively charged but also the methylene carbon is activated through resonance. (4) In the last step of the transformation, Glu53 deprotonates THF N10 increasing its nucleophilicity to facilitate the N10 attack on the N5 methylene carbon that results in the production of the final product 5,10-MTHF.
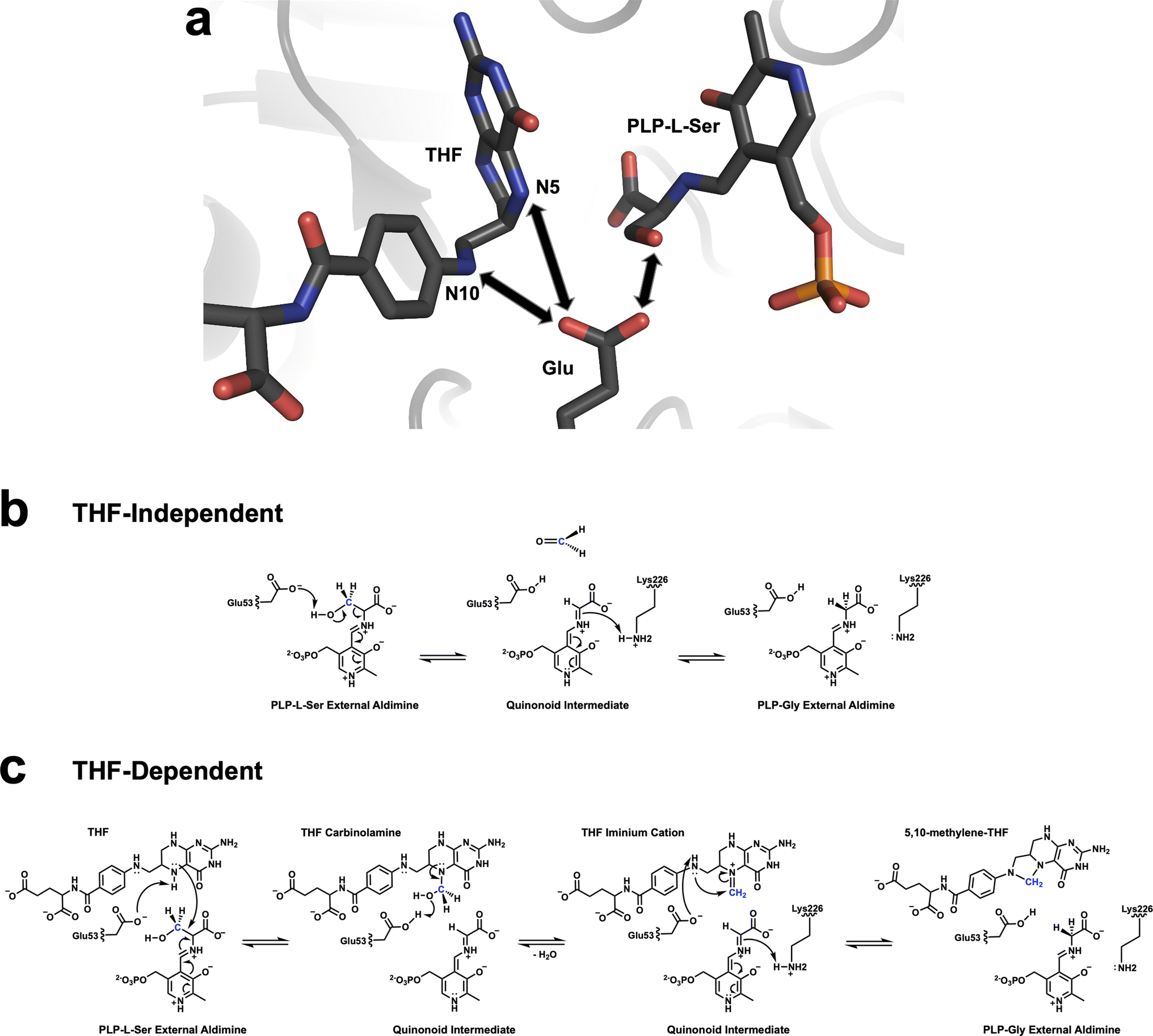 | ||
| Fig. 5 Positioning of the catalytic Glu53 and the proposed enzyme mechanisms for SHMT. (a) The strategic positioning of Glu53 in close proximity to the β-hydroxyl group of the PLP-L-Ser external aldimine, as well as FA N5 and N10 in the TthSHMT active site suggests Glu53 carries out protonation/deprotonation as a universal general acid-base catalyst in the SHMT THF-independent and THF-dependent catalyses. (b) The proposed retro-aldol mechanism for the THF-independent reaction catalyzed by the SHMT active site glutamate. (c) The proposed direct displacement mechanism for THF-dependent reaction catalyzed by the SHMT active site glutamate. In both (b) and (c) the catalytic Lys carries out transformation of the PLP quinonoid intermediate into the PLP-Gly external aldimine. | ||
4 Conclusion
Using neutron crystallography, we have located the positions of H atoms and thereby determined the protonation states in the active site and FA binding pocket in the TthSHMT/FA complex, where PLP is in the internal aldimine form and an acetate molecule occupies the cationic binding site. The organization of this complex resembles the carbinolamine form of the native THF coenzyme. In agreement with our previous neutron structure of TthSHMT holoenzyme, the Schiff base nitrogen is neutral and all the active site His residues were found in a neutral state. Whereas in our previous neutron structure, Glu53 was determined to be deprotonated and negatively charged, in the neutron structure of TthSHMT/FA Glu53 is protonated and the carboxylic oxygen participates in an H bond with the FA carbonyl oxygen. The X-ray structures of the TthSHMT-Gly/FA, TthSHMT-L-Ser/FA, and hSHMT2-Gly/FA ternary complexes revealed the architecture of the active site with both the amino acid external aldimine and FA bound. Similarities with the TthSHMT/FA neutron structure suggest Glu53 is also protonated in the ternary complexes. Based on the active site protonation state observations and our structural analyses, we propose key roles of the gating loop conformational flexibility and Glu53 protonation changes in the function of SHMT. Consequently, Glu53 is suggested to perform the general acid–base catalysis in both THF-independent and THF-dependent activities of SHMT.5 Methods
5.1 TthSHMT expression and purification
A detailed description of the expression and purification of TthSHMT was previously published.11 The glyA gene encoding TthSHMT was codon-optimized, synthesized, and cloned into the kanamycin-resistant pJ411 plasmid (ATUM, Newark, CA) with a DNA sequence encoding for an N-terminal poly-histidine-(His6)-tag with a 34 amino acid long linker and a TEV protease cleavage tag. The TthSHMT containing plasmid was transformed into the E. coli BL21(DE3) expression vector. The transformed cells were grown in Luria–Bertani (LB) media with 50 μg mL−1 kanamycin. Cell cultures were grown at 37 °C to an optical density of 0.8–1.0 and induced overnight with 1 mM isopropyl β-D-1-thiogalactopyranoside (IPTG) at 22 °C. The induced cells were centrifuged at 5660 rpm at 4 °C to harvest the cells and were subsequently resuspended in 50 mM sodium phosphate pH 7.5, 500 mM NaCl, and 10 mM imidazole at a ratio of 5 mL of lysis buffer per gram of wet cell paste. Lysozyme was added to the resuspended cells at a concentration of 0.1 mg mL−1 and stirred on ice for 30 min. Sonication was used to disrupt the cells and the lysate was clarified by centrifugation at 17![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 rpm (∼30000g). Affinity chromatography with a HisTrap FF (5 mL) nickel column equilibrated with 20 mM HEPES pH 7.5, 100 mM NaCl, and 10 mM imidazole was performed on the supernatant and pure TthSHMT was eluted from the column with a linear gradient of 20 mM HEPES pH 7.5, 100 mM NaCl, and 500 mM imidazole. TEV protease (1 mg TEV protease/100 mg of tagged protein) was added to purified TthSHMT and dialyzed against 20 mM HEPES pH 7.5, 100 mM NaCl, and 1 mM EDTA overnight to remove the poly-histidine tag at room temperature. The TEV protease-treated TthSHMT was loaded onto a HisTrap FF (5 mL) nickel column equilibrated with 20 mM HEPES pH 7.5, 100 mM NaCl, and 10 mM imidazole and pure, untagged TthSHMT was eluted in the flow-through. TthSHMT was dialyzed against 40 mM NaOAc pH 5.4 and 1 mM PLP, concentrated to 19 mg mL−1 and stored in 20% (v/v) glycerol at −30 °C.
000 rpm (∼30000g). Affinity chromatography with a HisTrap FF (5 mL) nickel column equilibrated with 20 mM HEPES pH 7.5, 100 mM NaCl, and 10 mM imidazole was performed on the supernatant and pure TthSHMT was eluted from the column with a linear gradient of 20 mM HEPES pH 7.5, 100 mM NaCl, and 500 mM imidazole. TEV protease (1 mg TEV protease/100 mg of tagged protein) was added to purified TthSHMT and dialyzed against 20 mM HEPES pH 7.5, 100 mM NaCl, and 1 mM EDTA overnight to remove the poly-histidine tag at room temperature. The TEV protease-treated TthSHMT was loaded onto a HisTrap FF (5 mL) nickel column equilibrated with 20 mM HEPES pH 7.5, 100 mM NaCl, and 10 mM imidazole and pure, untagged TthSHMT was eluted in the flow-through. TthSHMT was dialyzed against 40 mM NaOAc pH 5.4 and 1 mM PLP, concentrated to 19 mg mL−1 and stored in 20% (v/v) glycerol at −30 °C.
5.2 hSHMT2 expression, purification, and crystallization
A detailed description of the expression and purification of hSHMT2 was previously published.11 The SHMT2 gene encoding hSHMT2 was codon-optimized, synthesized and cloned into the kanamycin-resistant pJ411 plasmid (ATUM, Newark, CA) with a DNA sequence encoding for an N-terminal poly-histidine-(His6)-tag with a 34 amino acid long linker and a TEV protease cleavage tag. The plasmid containing hSHMT2 was transformed into BL21(DE3) competent E. coli cells. The transformed cells were grown in LB media containing 50 μg mL−1 kanamycin at 37 °C. Once the culture reached an optical density of 0.8–1.0, it was induced with 1 mM IPTG overnight at 22 °C. The cells were harvested by centrifugation at 5660 rpm at 4 °C and resuspended in 20 mM HEPES pH 7.5, 500 mM NaCl, and 10 mM imidazole at a ratio of 5 mL g−1 of wet cell paste. Lysozyme was added to the resuspended cells at a concentration of 0.1 mg mL−1 and stirred on ice for 30 min before manual sonication. The cell lysates were clarified by centrifugation at 30000g for 30 min. The crude lysate was loaded onto a HisTrap FF (5 mL) nickel column equilibrated with 20 mM HEPES pH 7.5, 500 mM NaCl, and 10 mM imidazole and eluted with of 20 mM HEPES pH 7.5, 500 mM NaCl, and 500 mM imidazole in a linear gradient. The poly-histidine tag was removed by adding TEV protease (1 mg TEV protease/100 mg of tagged protein) to the purified pooled fractions of hSHMT2 and dialyzing the sample overnight against 20 mM HEPES pH 7.5, 250 mM NaCl, and 1 mM EDTA. The TEV protease-treated protein was loaded onto a HisTrap FF (5 mL) nickel column equilibrated with 20 mM HEPES pH 7.5, 500 mM NaCl, and 10 mM imidazole and pure hSHMT2 was eluted in the flow-through. After verifying the purity by SDS-PAGE, hSHMT2 was dialyzed against 20 mM HEPES pH 7.5, 300 mM NaCl, and 1 mM PLP overnight. hSHMT2 was concentrated to 18 mg mL−1 for crystallization experiments. Fresh, never-frozen, hSHMT2 was co-crystallized with 10 mM FA in 0.5 M Gly pH 8.6 and 18% PEG 3350 at 20 °C in sitting-drop vapor diffusion setups to generate square plate-like crystals of the hSHMT2-Gly/FA complex amendable for room-temperature X-ray diffraction.
5.3 TthSHMT crystallization, soaking, and H/D-exchange
Purified aliquots of TthSHMT stored at −30 °C were thawed and dialyzed overnight against 40 mM NaOAc pH 5.4 and 1 mM PLP to remove the glycerol cryoprotectant. Crystallization of TthSHMT (∼19 mg mL−1) in 40 mM NaOAc pH 5.5, 1 M (NH4)2SO4, and 0.5 M Li2SO4 was carried out by sitting drop vapor diffusion at 16 °C, producing showers of crystals and crystal aggregates. The crystal aggregates were crushed and a seeding tool from Hampton Research was used to transfer microcrystals to large volume sitting drops by streak seeding in 9-well glass plates and sandwich box setups. This method produced large, single crystals of TthSHMT necessary for neutron diffraction. Because FA did not bind to TthSHMT in high concentrations of salt, a crystal was transferred to a fresh drop containing 40 mM NaOAc pH 5.5 and 15% PEG 4000. The crystal was left in this PEG-containing soaking condition overnight to remove the bound sulfate and, the following day, the crystal was moved into another drop containing a soaking solution with 40 mM NaOAc pH 5.5, 15% PEG 4000, and 10 mM FA. The crystal was left in the former soaking solution overnight before being mounted in a 2 mm-inner diameter quartz capillary containing a liquid plug made of 40 mM NaOAc pH 5.5, and 15% PEG 4000 in 99.8% D2O to perform H/D-vapor exchange. Crystal soaks to obtain the TthSHMT-Gly/FA and TthSHMT-L-Ser/FA ternary complexes were carried out in a similar manner, first being transferred to a drop containing 40 mM NaOAc pH 5.5, and 15% PEG 4000 overnight and then moved to a drop containing 40 mM NaOAc pH 5.5, 15% PEG 4000, 0.5 M Gly or L-Ser, and 10 mM FA.5.4 TthSHMT X-ray diffraction data collection and structure refinement
Room temperature X-ray diffraction data collection on TthSHMT/FA and TthSHMT-Gly/FA crystals and low temperature (100 K) X-ray diffraction data collection on TthSHMT-L-Ser/FA were performed on a Rigaku HighFlux HomeLab instrument equipped with a MicroMax-007 HF X-ray generator, Osmic VariMax optics, and a DECTRIS Eiger R 4M detector at ORNL. The data were indexed and integrated using the CrysAlis Pro software package (Rigaku, The Woodlands, TX), and the data were reduced and scaled in the AIMLESS program in the CCP4 software suite.65,66 The X-ray structures were solved by molecular replacement in PHASER67 using phases from PDB code 8SUJ and refined with phenix.refine in the PHENIX suite.68,69 The room temperature X-ray structure of TthSHMT/FA was subsequently used in joint XN refinement with the room temperature neutron diffraction data collected on the same crystal. Ligand restraints for FA, and PLP-Gly and PLP-L-Ser external aldimines were generated with eLBOW70 using geometry optimized by quantum chemical calculations in Gaussian 16 (ref. 71) at B3LYP/6-31 g(d,p) level of theory. The X-ray diffraction data collection and refinement statistics are presented in Table S1.†5.5 Neutron diffraction data collection
A complete room temperature neutron diffraction dataset was collected on a TthSHMT/FA crystal on the instrument MaNDi72,73 at the Spallation Neutron Source (SNS) at ORNL. The crystal was held stationary for 20 h exposures and rotated 10° around the φ-axis before collecting the next image. All neutrons between 2 and 4.16 Å were used to collect the frames, with a total of 25 images collected. The neutron diffraction data were processed and integrated with 3D time-of-flight profile fitting in Mantid.74,75 LAUENORM76,77 was used to carry out wavelength normalization and the data were then merged in SCALA.78 Neutron diffraction data collection statistics are presented in Table S1.†5.6 Joint X-ray/neutron (XN) refinement
Joint XN-refinement of the TthSHMT/FA complex was carried out using nCNS,79,80 a patch of the Crystallography & NMR Systems (CNS)79,81 software suite in the same manner as for our previous TthSHMT joint XN structures.11 A single rigid body refinement was the first step in the refinement procedure. Subsequently, several rounds of atomic position, atomic displacement parameter, and D atom occupancy refinements were performed. In between the rounds of the joint XN refinement, the structure was visualized in the molecular graphics program COOT82–84 to confirm correct side chain modeling and direct the rotation of side chain hydroxyl, thiol, and ammonium groups as well as water molecules to construct accurate H bonding networks. Water molecules were modeled and refined as D2O due to H/D-vapor exchange. TthSHMT/FA was modeled with H atoms at non-exchangeable positions because protiated protein was used in the experiment, while labile positions were initially modeled as D atoms. After D atom occupancy refinement, the exchangeable sites were modeled based on individual site occupancies, where an occupancy of −0.56 is indicative of a pure H atom and an occupancy of 1.00 reflects a pure D atom. Sites partially occupied by both H and D atoms were given two atom records with the partial occupancies adding up to 1.00. The percentage of D atom occupancy at a specific site is calculated according to the following formula: % D = (occupancy(D) + 0.56)/1.56. Joint XN refinement statistics can be found in Table S1.†5.7 hSHMT2 room temperature X-ray diffraction data collection and structure refinement
Room temperature X-ray diffraction data were collected on hSHMT2-Gly/FA on the Structural Biology Center (SBC) beamline 19-ID using a Pilatus3 X 6M detector at the Advanced Photon Source (APS). The data were integrated and scaled using the HKL3000 software suite.85 The hSHMT2-Gly/FA structure was solved by molecular replacement with PDB ID 8SSJ and the AlphaFold86 model of hSHMT2 to include the predicted structure of the flap domain which was not resolved in our previous hSHMT2 internal aldimine X-ray structure (PDB ID 8SSJ). The structure was refined against the room temperature X-ray diffraction data with phenix.refine from the PHENIX suite.68,69 The room temperature X-ray diffraction data collection and refinement statistics are presented in Table S1.†Data availability
The structures and corresponding structure factors have been deposited into the protein data bank with the PDB accession codes 9BPE for TthSHMT/FA, 9BOH for TthSHMT-Gly/FA, 9BOW for TthSHMT-L-Ser/FA, and 9BOX for hSHMT2-Gly/FA. ESI† is available online.Author contributions
V. N. D., R. S. P. and A. K. designed the study. V. N. D. expressed, purified and crystallized the proteins. V. N. D. and A. K. collected and reduced X-ray diffraction data, and refined the structures. V. N. D. and A. K. collected and reduced neutron diffraction data, and refined the joint XN structure. V. N. D., R. S. P., and A. K. wrote the paper.Conflicts of interest
There are no conflicts of interest to declare.Acknowledgements
A portion of this research used resources at Spallation Neutron Source, a DOE Office of Science User Facility operated by the Oak Ridge National Laboratory (ORNL). The beam time was allocated to MaNDi instrument on proposal number IPTS-30675. The Office of Biological and Environmental Research supported research at ORNL's Center for Structural Molecular Biology (CSMB), a DOE Office of Science User Facility. ORNL is managed by UT-Battelle LLC for DOE's Office of Science, the single largest supporter of basic research in the physical sciences in the United States. X-ray crystallographic data were in part collected at Argonne National Laboratory using Structural Biology Center (SBC) beamline ID19 at the Advanced Photon Source. Use of the Advanced Photon Source, an Office of Science User Facility operated for the U.S. Department of Energy (DOE) Office of Science by Argonne National Laboratory, was supported by the U.S. DOE under Contract No. DE-AC02-06CH11357. The authors thank Dr Changsoo Chang from the SBC for processing crystallographic data for hSHMT2-Gly/FA complex. This research was supported by a grant from NIH-GMS (R01GM137008) to R. S. P. and A. K.References
- L. Schirch, Serine hydroxymethyltransferase, Adv. Enzymol. Relat. Areas Mol. Biol., 1982, 53, 83–112 CAS.
- R. Percudani and A. Peracchi, A genomic overview of pyridoxal-phosphate-dependent enzymes, EMBO Rep., 2003, 4(9), 850–854 CrossRef CAS PubMed.
- K. Herbig, E. P. Chiang, L. R. Lee, J. Hills, B. Shane and P. J. Stover, Cytoplasmic serine hydroxymethyltransferase mediates competition between folate-dependent deoxyribonucleotide and S-adenosylmethionine biosyntheses, J. Biol. Chem., 2002, 277(41), 38381–38389 CrossRef CAS.
- M. Xie and D. S. Pei, Serine hydroxymethyltransferase 2: a novel target for human cancer therapy, Invest. New Drugs, 2021, 39(6), 1671–1681 CrossRef CAS.
- K. Shostak and V. Schirch, Serine hydroxymethyltransferase: mechanism of the racemization and transamination of D- and L-alanine, Biochemistry, 1988, 27(21), 8007–8014 CrossRef CAS PubMed.
- G. Giardina, P. Brunotti, A. Fiascarelli, A. Cicalini, M. G. Costa and A. M. Buckle, et al., How pyridoxal 5'-phosphate differentially regulates human cytosolic and mitochondrial serine hydroxymethyltransferase oligomeric state, FEBS J., 2015, 282(7), 1225–1241 CrossRef CAS PubMed.
- A. Tramonti, C. Nardella, M. L. di Salvo, A. Barile, F. Cutruzzola and R. Contestabile, Human Cytosolic and Mitochondrial Serine Hydroxymethyltransferase Isoforms in Comparison: Full Kinetic Characterization and Substrate Inhibition Properties, Biochemistry, 2018, 57(51), 6984–6996 CrossRef CAS.
- T. A. Garrow, A. A. Brenner, V. M. Whitehead, X. N. Chen, R. G. Duncan and J. R. Korenberg, et al., Cloning of Human Cdnas Encoding Mitochondrial and Cytosolic Serine Hydroxymethyltransferases and Chromosomal Localization, J. Biol. Chem., 1993, 268(16), 11910–11916 CrossRef CAS PubMed.
- R. Florio, M. L. di Salvo, M. Vivoli and R. Contestabile, Serine hydroxymethyltransferase: a model enzyme for mechanistic, structural, and evolutionary studies, Biochim. Biophys. Acta, 2011, 1814(11), 1489–1496 CrossRef CAS.
- K. Snell, U. Baumann, P. C. Byrne, K. J. Chave, S. B. Renwick and P. G. Sanders, et al., The genetic organization and protein crystallographic structure of human serine hydroxymethyltransferase, Adv. Enzyme Regul., 2000, 40, 353–403 CrossRef CAS PubMed.
- V. N. Drago, C. Campos, M. Hooper, A. Collins, O. Gerlits and K. L. Weiss, et al., Revealing protonation states and tracking substrate in serine hydroxymethyltransferase with room-temperature X-ray and neutron crystallography, Commun. Chem., 2023, 6(1), 162 CrossRef CAS.
- V. Trivedi, A. Gupta, V. R. Jala, P. Saravanan, G. S. J. Rao and N. A. Rao, et al., Crystal structure of binary and ternary complexes of serine hydroxymethyltransferase from Bacillus stearothermophilus - Insights into the catalytic mechanism, J. Biol. Chem., 2002, 277(19), 17161–17169 CrossRef CAS.
- Y. Makino, C. Oe, K. Iwama, S. Suzuki, A. Nishiyama and K. Hasegawa, et al., Serine hydroxymethyltransferase as a potential target of antibacterial agents acting synergistically with one-carbon metabolism-related inhibitors, Commun. Biol., 2022, 5(1), 619 CrossRef CAS.
- M. Ruszkowski, B. Sekula, A. Ruszkowska, R. Contestabile, I. Nogues and S. Angelaccio, et al., Structural basis of methotrexate and pemetrexed action on serine hydroxymethyltransferases revealed using plant models, Sci. Rep., 2019, 9(1), 19614 CrossRef CAS.
- G. Y. Lee, P. M. Haverty, L. Li, N. M. Kljavin, R. Bourgon and J. Lee, et al., Comparative oncogenomics identifies PSMB4 and SHMT2 as potential cancer driver genes, Cancer Res., 2014, 74(11), 3114–3126 CrossRef CAS PubMed.
- G. Giardina, A. Paone, A. Tramonti, R. Lucchi, M. Marani and M. C. Magnifico, et al., The catalytic activity of serine hydroxymethyltransferase is essential for de novo nuclear dTMP synthesis in lung cancer cells, FEBS J., 2018, 285(17), 3238–3253 CrossRef CAS.
- A. M. Li, G. S. Ducker, Y. Li, J. A. Seoane, Y. R. Xiao and S. Melemenidis, et al., Metabolic Profiling Reveals a Dependency of Human Metastatic Breast Cancer on Mitochondrial Serine and One-Carbon Unit Metabolism, Mol. Cancer Res., 2020, 18(4), 599–611 CrossRef CAS PubMed.
- Z. Z. Wu, S. Wang, Q. C. Yang, X. L. Wang, L. L. Yang and B. Liu, et al., Increased Expression of SHMT2 Is Associated With Poor Prognosis and Advanced Pathological Grade in Oral Squamous Cell Carcinoma, Front. Oncol., 2020, 10, 588530 CrossRef PubMed.
- C. Yang, J. Zhang, M. Liao, Y. Yang, Y. Wang and Y. Yuan, et al., Folate-mediated one-carbon metabolism: a targeting strategy in cancer therapy, Drug Discovery Today, 2021, 26(3), 817–825 CrossRef CAS.
- X. Cui, Y. Cui, T. Du, X. Jiang, C. Song and S. Zhang, et al., SHMT2 Drives the Progression of Colorectal Cancer by Regulating UHRF1 Expression, Can J. Gastroenterol. Hepatol., 2022, 2022, 3758697 Search PubMed.
- Y. Zeng, J. Zhang, M. Xu, F. Chen, R. Zi and J. Yue, et al., Roles of Mitochondrial Serine Hydroxymethyltransferase 2 (SHMT2) in Human Carcinogenesis, J. Cancer, 2021, 12(19), 5888–5894 CrossRef CAS PubMed.
- E. Pranzini, E. Pardella, L. Muccillo, A. Leo, I. Nesi and A. Santi, et al., SHMT2-mediated mitochondrial serine metabolism drives 5-FU resistance by fueling nucleotide biosynthesis, Cell Rep., 2022, 40(7), 111233 CrossRef CAS PubMed.
- C. R. Cuthbertson, Z. Arabzada, A. Bankhead 3rd, A. Kyani and N. Neamati, A Review of Small-Molecule Inhibitors of One-Carbon Enzymes: SHMT2 and MTHFD2 in the Spotlight, ACS Pharmacol. Transl. Sci., 2021, 4(2), 624–646 CrossRef CAS.
- Z. E. Stine, Z. T. Schug, J. M. Salvino and C. V. Dang, Targeting cancer metabolism in the era of precision oncology, Nat. Rev. Drug Discov., 2022, 21(2), 141–162 CrossRef CAS.
- J. M. Katinas, M. J. Nayeen, M. Schneider, K. Shah, A. N. Fifer and L. M. Klapper, et al., Structural Characterization of 5-Substituted Pyrrolo[3,2-d]pyrimidine Antifolate Inhibitors in Complex with Human Serine Hydroxymethyl Transferase 2, Biochemistry, 2024, 63, 545–562 CAS.
- Y. Han, L. He, Y. Qi, Y. Zhao, Y. Pan and B. Fang, et al., Identification of three new compounds that directly target human serine hydroxymethyltransferase 2, Chem. Biol. Drug Des., 2021, 97(2), 221–230 CrossRef CAS.
- J. C. García-Cañaveras, O. Lancho, G. S. Ducker, J. M. Ghergurovich, X. Xu and V. da Silva-Diz, et al., SHMT inhibition is effective and synergizes with methotrexate in T-cell acute lymphoblastic leukemia, Leukemia, 2021, 35(2), 377–388 CrossRef PubMed.
- Y. Pikman, N. Ocasio-Martinez, G. Alexe, B. Dimitrov, S. Kitara and F. F. Diehl, et al., Targeting serine hydroxymethyltransferases 1 and 2 for T-cell acute lymphoblastic leukemia therapy, Leukemia, 2022, 36(2), 348–360 CrossRef CAS PubMed.
- G. S. Ducker and J. D. Rabinowitz, One-Carbon Metabolism in Health and Disease, Cell Metab., 2017, 25(1), 27–42 CrossRef CAS PubMed.
- M. J. Nayeen, J. M. Katinas, T. Magdum, K. Shah, J. E. Wong and C. E. O'Connor, et al., Structure-Based Design of Transport-Specific Multitargeted One-Carbon Metabolism Inhibitors in Cytosol and Mitochondria, J. Med. Chem., 2023, 66(16), 11294–11323 CrossRef CAS.
- M. P. Blakeley and A. D. Podjarny, Neutron macromolecular crystallography, Emerging Top. Life Sci., 2018, 2(1), 39–55 CrossRef CAS.
- N. Engler, A. Ostermann, N. Niimura and F. G. Parak, Hydrogen atoms in proteins: positions and dynamics, Proc. Natl. Acad. Sci. U. S. A., 2003, 100(18), 10243–10248 CrossRef CAS PubMed.
- A. S. Gardberg, A. R. Del Castillo, K. L. Weiss, F. Meilleur, M. P. Blakeley and D. A. Myles, Unambiguous determination of H-atom positions: comparing results from neutron and high-resolution X-ray crystallography, Acta Crystallogr., Sect. D: Biol. Crystallogr., 2010, 66(5), 558–567 CrossRef CAS PubMed.
- B. Bax, C. W. Chung and C. Edge, Getting the chemistry right: protonation, tautomers and the importance of H atoms in biological chemistry, Acta Crystallogr., Sect. D: Struct. Biol., 2017, 73(2), 131–140 CrossRef CAS.
- N. Niimura and A. D. Podjarny, Neutron protein crystallography : hydrogen, protons, and hydration in bio-macromolecules, Oxford University Press, Oxford, 2011 Search PubMed.
- O. Gerlits, T. Wymore, A. Das, C. H. Shen, J. M. Parks and J. C. Smith, et al., Long-Range Electrostatics-Induced Two-Proton Transfer Captured by Neutron Crystallography in an Enzyme Catalytic Site, Angew Chem. Int. Ed. Engl., 2016, 55(16), 4924–4927 CrossRef CAS.
- S. Dajnowicz, R. C. Johnston, J. M. Parks, M. P. Blakeley, D. A. Keen and K. L. Weiss, et al., Direct visualization of critical hydrogen atoms in a pyridoxal 5'-phosphate enzyme, Nat. Commun., 2017, 8(1), 955 CrossRef.
- V. N. Drago, S. Dajnowicz, J. M. Parks, M. P. Blakeley, D. A. Keen and N. Coquelle, et al., An N⋯H⋯N low-barrier hydrogen bond preorganizes the catalytic site of aspartate aminotransferase to facilitate the second half-reaction, Chem. Sci., 2022, 13(34), 10057–10065 RSC.
- S. Shukla, D. A. Myles and M. J. Cuneo, Mapping periplasmic binding protein oligosaccharide recognition with neutron crystallography, Sci. Rep., 2022, 12(1), 17647 CrossRef CAS PubMed.
- V. N. Drago, J. M. Devos, M. P. Blakeley, V. T. Forsyth, J. M. Parks and A. Kovalevsky, et al., Neutron diffraction from a microgravity-grown crystal reveals the active site hydrogens of the internal aldimine form of tryptophan synthase, Cell Rep. Phys. Sci., 2024, 5(2), 101827 CrossRef CAS.
- M. Kumar, K. Mandal, M. P. Blakeley, T. Wymore, S. B. H. Kent and J. M. Louis, et al., Visualizing Tetrahedral Oxyanion Bound in HIV-1 Protease Using Neutrons: Implications for the Catalytic Mechanism and Drug Design, ACS Omega, 2020, 5(20), 11605–11617 CrossRef CAS.
- L. Gajdos, M. P. Blakeley, M. Haertlein, V. T. Forsyth, J. M. Devos and A. Imberty, Neutron crystallography reveals mechanisms used by Pseudomonas aeruginosa for host-cell binding, Nat. Commun., 2022, 13(1), 194 CrossRef CAS PubMed.
- D. W. Kneller, H. Li, S. Galanie, G. Phillips, A. Labbe and K. L. Weiss, et al., Structural, Electronic, and Electrostatic Determinants for Inhibitor Binding to Subsites S1 and S2 in SARS-CoV-2 Main Protease, J. Med. Chem., 2021, 64(23), 17366–17383 CrossRef CAS PubMed.
- D. W. Kneller, H. Li, G. Phillips, K. L. Weiss, Q. Zhang and M. A. Arnould, et al., Covalent narlaprevir- and boceprevir-derived hybrid inhibitors of SARS-CoV-2 main protease, Nat. Commun., 2022, 13(1), 2268 CrossRef CAS PubMed.
- D. M. E. Szebenyi, F. N. Musayev, M. L. di Salvo, M. K. Safo and V. Schirch, Serine hydroxymethyltransferase: Role of Glu75 and evidence that serine is cleaved by a retroaldol mechanism, Biochemistry, 2004, 43(22), 6865–6876 CrossRef CAS PubMed.
- H. Nonaka, Y. Nakanishi, S. Kuno, T. Ota, K. Mochidome and Y. Saito, et al., Design strategy for serine hydroxymethyltransferase probes based on retro-aldol-type reaction, Nat. Commun., 2019, 10(1), 876 CrossRef.
- V. Schirch and D. M. Szebenyi, Serine hydroxymethyltransferase revisited, Curr. Opin. Chem. Biol., 2005, 9(5), 482–487 CrossRef CAS PubMed.
- R. G. Matthews and J. T. Drummond, Providing One-Carbon Units for Biological Methylations - Mechanistic Studies on Serine Hydroxymethyltransferase, Methylenetetrahydrofolate Reductase, and Methyltetrahydrofolate-Homocysteine Methyltransferase, Chem. Rev., 1990, 90(7), 1275–1290 CrossRef CAS.
- H. S. Fernandes, M. J. Ramos and N. M. F. S. A. Cerqueira, Catalytic Mechanism of the Serine Hydroxymethyltransferase: A Computational ONIOM QM/MM Study, ACS Catal., 2018, 8(11), 10096–10110 CrossRef CAS.
- A. Sodolescu, C. Dian, L. Terradot, L. Bouzhir-Sima, R. Lestini and H. Myllykallio, et al., Structural and functional insight into serine hydroxymethyltransferase from Helicobacter pylori, PLoS One, 2018, 13(12), e0208850 CrossRef CAS.
- J. N. Scarsdale, S. Radaev, G. Kazanina, V. Schirch and H. T. Wright, Crystal structure at 2.4 Å resolution of E. coli serine hydroxymethyltransferase in complex with glycine substrate and 5-formyl tetrahydrofolate11Edited by I. A. Wilson, J. Mol. Biol., 2000, 296(1), 155–168 CrossRef CAS PubMed.
- R. Contestabile, S. Angelaccio, F. Bossa, H. T. Wright, N. Scarsdale and G. Kazanina, et al., Role of tyrosine 65 in the mechanism of serine hydroxymethyltransferase, Biochemistry, 2000, 39(25), 7492–7500 CrossRef CAS.
- G. Schwertz, M. C. Witschel, M. Rottmann, R. Bonnert, U. Leartsakulpanich and P. Chitnumsub, et al., Antimalarial Inhibitors Targeting Serine Hydroxymethyltransferase (SHMT) with in Vivo Efficacy and Analysis of their Binding Mode Based on X-ray Cocrystal Structures, J. Med. Chem., 2017, 60(12), 4840–4860 CrossRef CAS PubMed.
- G. Gocheva, N. Petkov, A. Garcia Luri, S. Iliev, N. Ivanova and J. Petrova, et al., Tautomerism in folic acid: Combined molecular modelling and NMR study, J. Mol. Liq., 2019, 292, 111392 CrossRef.
- J. E. Gready, Theoretical studies on the activation of the pterin cofactor in the catalytic mechanism of dihydrofolate reductase, Biochemistry, 1985, 24(18), 4761–4766 CrossRef CAS.
- S. Angelaccio, F. Dworkowski, A. Di Bello, T. Milano, G. Capitani and S. Pascarella, Conformational transitions driven by pyridoxal-5'-phosphate uptake in the psychrophilic serine hydroxymethyltransferase from Psychromonas ingrahamii, Proteins, 2014, 82(10), 2831–2841 CrossRef CAS.
- S. Ubonprasert, J. Jaroensuk, W. Pornthanakasem, N. Kamonsutthipaijit, P. Wongpituk and P. Mee-Udorn, et al., A flap motif in human serine hydroxymethyltransferase is important for structural stabilization, ligand binding, and control of product release, J. Biol. Chem., 2019, 294(27), 10490–10502 CrossRef CAS PubMed.
- R. J. Ulevitch and R. G. Kallen, Studies of the reactions of lamb liver serine hydroxymethylase with L-phenylalanine: kinetic isotope effects upon quinonoid intermediate formation, Biochemistry, 1977, 16(24), 5350–5354 CrossRef CAS PubMed.
- B. Bhavani, V. Rajaram, S. Bisht, P. Kaul, V. Prakash and M. Murthy, et al., Importance of tyrosine residues of Bacillus stearothermophilus serine hydroxymethyltransferase in cofactor binding and l-allo-Thr cleavage: Crystal structure and biochemical studies, FEBS J., 2008, 275(18), 4606–4619 CrossRef CAS PubMed.
- N. A. Rao, M. Ambili, V. R. Jala, H. S. Subramanya and H. S. Savithri, Structure-function relationship in serine hydroxymethyltransferase, Biochim. Biophys. Acta, 2003, 1647(1–2), 24–29 Search PubMed.
- V. Schirch, K. Shostak, M. Zamora and M. Guatam-Basak, The origin of reaction specificity in serine hydroxymethyltransferase, J. Biol. Chem., 1991, 266(2), 759–764 CrossRef CAS PubMed.
- S. B. Renwick, K. Snell and U. Baumann, The crystal structure of human cytosolic serine hydroxymethyltransferase: a target for cancer chemotherapy, Structure, 1998, 6(9), 1105–1116 CrossRef CAS.
- V. Rajaram, B. S. Bhavani, P. Kaul, V. Prakash, N. Appaji Rao and H. S. Savithri, et al., Structure determination and biochemical studies on Bacillus stearothermophilus E53Q serine hydroxymethyltransferase and its complexes provide insights on function and enzyme memory, FEBS J., 2007, 274(16), 4148–4160 CrossRef CAS.
- J. V. K. Rao, V. Prakash, N. A. Rao and H. S. Savithri, The role of Glu74 and Tyr82 in the reaction catalyzed by sheep liver cytosolic serine hydroxymethyltransferase, Eur. J. Biochem., 2000, 267(19), 5967–5976 CrossRef CAS PubMed.
- P. R. Evans and G. N. Murshudov, How good are my data and what is the resolution?, Acta Crystallogr., Sect. D: Biol. Crystallogr., 2013, 69(7), 1204–1214 CrossRef CAS.
- M. D. Winn, C. C. Ballard, K. D. Cowtan, E. J. Dodson, P. Emsley and P. R. Evans, et al., Overview of the CCP4 suite and current developments, Acta Crystallogr. D, 2011, 67, 235–242 CrossRef CAS PubMed.
- A. J. McCoy, R. W. Grosse-Kunstleve, P. D. Adams, M. D. Winn, L. C. Storoni and R. J. Read, Phaser crystallographic software, J. Appl. Crystallogr., 2007, 40, 658–674 CrossRef CAS PubMed.
- P. D. Adams, P. V. Afonine, G. Bunkoczi, V. B. Chen, I. W. Davis and N. Echols, et al., PHENIX: a comprehensive Python-based system for macromolecular structure solution, Acta Crystallogr., Sect. D: Biol. Crystallogr., 2010, 66(Pt 2), 213–221 CrossRef CAS PubMed.
- D. Liebschner, P. V. Afonine, M. L. Baker, G. Bunkoczi, V. B. Chen and T. I. Croll, et al., Macromolecular structure determination using X-rays, neutrons and electrons: recent developments in Phenix, Acta Crystallogr. D, 2019, 75, 861–877 CrossRef CAS.
- N. W. Moriarty, R. W. Grosse-Kunstleve and P. D. Adams, electronic Ligand Builder and Optimization Workbench (eLBOW): a tool for ligand coordinate and restraint generation, Acta Crystallogr., Sect. D: Biol. Crystallogr., 2009, 65(10), 1074–1080 CrossRef CAS PubMed.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb and J. R. Cheeseman, et al., Gaussian 16 Rev. A.03, Gaussian Inc., Wallingford, CT, 2016 Search PubMed.
- L. Coates, M. J. Cuneo, M. J. Frost, J. H. He, K. L. Weiss and S. J. Tomanicek, et al., The Macromolecular Neutron Diffractometer MaNDi at the Spallation Neutron Source, J. Appl. Crystallogr., 2015, 48, 1302–1306 CrossRef CAS.
- L. Coates and B. Sullivan, The macromolecular neutron diffractometer at the spallation neutron source, Methods Enzymol., 2020, 634, 87–99 CAS.
- O. Arnold, J. C. Bilheux, J. M. Borreguero, A. Buts, S. I. Campbell and L. Chapon, et al., Mantid—Data analysis and visualization package for neutron scattering and μ SR experiments, Nucl. Instrum. Methods Phys. Res., Sect. A, 2014, 764, 156–166 CrossRef CAS.
- B. Sullivan, R. Archibald, P. S. Langan, H. Dobbek, M. Bommer and R. L. McFeeters, et al., Improving the accuracy and resolution of neutron crystallographic data by three-dimensional profile fitting of Bragg peaks in reciprocal space, Acta Crystallogr., Sect. D: Struct. Biol., 2018, 74(11), 1085–1095 CrossRef CAS.
- J. W. Campbell, Q. Hao, M. M. Harding, N. D. Nguti and C. Wilkinson, LAUEGEN version 6.0 and INTLDM, J. Appl. Crystallogr., 1998, 31, 496–502 CrossRef CAS.
- J. Helliwell, J. Habash, D. Cruickshank, M. Harding, T. Greenhough and J. Campbell, et al., The recording and analysis of synchrotron X-radiation Laue diffraction photographs, J. Appl. Crystallogr., 1989, 22(5), 483–497 CrossRef CAS.
- M. Weiss, Global indicators of X-ray data quality, J. Appl. Crystallogr., 2001, 34(2), 130–135 CrossRef CAS.
- M. Mustyakimov and P. Langan, nCNS: an open source distribution patch for CNS for macromolecular structure refinement, Los Alamos National Security, Los Alamos, NM, USA, 2007 Search PubMed.
- P. D. Adams, M. Mustyakimov, P. V. Afonine and P. Langan, Generalized X-ray and neutron crystallographic analysis: more accurate and complete structures for biological macromolecules, Acta Crystallogr., Sect. D: Biol. Crystallogr., 2009, 65(Pt 6), 567–573 CrossRef CAS PubMed.
- A. T. Brunger, P. D. Adams, G. M. Clore, W. L. DeLano, P. Gros and R. W. Grosse-Kunstleve, et al., Crystallography & NMR system: A new software suite for macromolecular structure determination, Acta Crystallogr. D, 1998, 54, 905–921 CrossRef CAS PubMed.
- P. Emsley and K. Cowtan, Coot: model-building tools for molecular graphics, Acta Crystallogr. D, 2004, 60, 2126–2132 CrossRef.
- P. Emsley, B. Lohkamp, W. G. Scott and K. Cowtan, Features and development of Coot, Acta Crystallogr., Sect. D: Biol. Crystallogr., 2010, 66(4), 486–501 CrossRef CAS PubMed.
- A. Casanal, B. Lohkamp and P. Emsley, Current developments in Coot for macromolecular model building of Electron Cryo-microscopy and Crystallographic Data, Protein Sci., 2020, 29(4), 1069–1078 CrossRef CAS PubMed.
- W. Minor, M. Cymborowski, Z. Otwinowski and M. Chruszcz, HKL-3000: the integration of data reduction and structure solution-from diffraction images to an initial model in minutes, Acta Crystallogr., Sect. D: Biol. Crystallogr., 2006, 62(8), 859–866 CrossRef PubMed.
- J. Jumper, R. Evans, A. Pritzel, T. Green, M. Figurnov and O. Ronneberger, et al., Highly accurate protein structure prediction with AlphaFold, Nature, 2021, 596(7873), 583–589 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4sc03187c |
| This journal is © The Royal Society of Chemistry 2024 |