
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceQuantitative analysis of protein lipidation and acyl-CoAs reveals substrate preferences of the S-acylation machinery†
Carla
Busquets-Hernández
 a,
Silvia
Ribó‡
a,
Esther
Gratacós-Batlle§
a,
Daniel
Carbajo
a,
Alexandra
Tsiotsia
a,
Juan B.
Blanco-Canosa
a,
Luke H.
Chamberlain
b and
Gemma
Triola
*a
a,
Silvia
Ribó‡
a,
Esther
Gratacós-Batlle§
a,
Daniel
Carbajo
a,
Alexandra
Tsiotsia
a,
Juan B.
Blanco-Canosa
a,
Luke H.
Chamberlain
b and
Gemma
Triola
*a
aDepartment of Biological Chemistry, Institute for Advanced Chemistry of Catalonia (IQAC-CSIC), Barcelona, Spain. E-mail: gemma.triola@iqac.csic.es
bStrathclyde Institute of Pharmacy and Biomedical Sciences, University of Strathclyde, Glasgow, UK
First published on 9th July 2024
Abstract
Protein palmitoylation or S-acylation has emerged as a key regulator of cellular processes. Increasing evidence shows that this modification is not restricted to palmitate but it can include additional fatty acids, raising the possibility that differential S-acylation contributes to the fine-tuning of protein activity. However, methods to profile the acyl moieties attached to proteins are scarce. Herein, we report a method for the identification and quantification of lipids bound to proteins that relies on hydroxylamine treatment and mass spectrometry analysis of fatty acid hydroxamates. This method has enabled unprecedented and extensive profiling of the S-acylome in different cell lines and tissues and has shed light on the substrate specificity of some S-acylating enzymes. Moreover, we could extend it to quantify also the acyl-CoAs, which are thioesters formed between a fatty acid and a coenzyme A, overcoming many of the previously described challenges for the detection of such species. Importantly, the simultaneous analysis of the lipid fraction and the proteome allowed us to establish, for the first time, a direct correlation between the endogenous levels of acyl-CoAs and the S-acylation profile of its proteome.
Introduction
S-acylation is the reversible modification of a cysteine residue with a fatty acid via a thioester bond. This lipid modification has a key role in the regulation of protein localization, stability and trafficking and it also modulates multiple cellular processes, including signal transduction and apoptosis.1–3 It has been largely considered that palmitate is the most common lipid modification and as a result, S-acylation has been often referred to as palmitoylation. However, it is now clear that proteins can be acylated with other fatty acids.4–6 In addition, there is a growing body of evidence suggesting a correlation between heterogenous acylation and different functional outcomes.7–9 Thus, it has been reported that proteins modified with unsaturated fatty acids show higher insertion rates into membranes and an increased clustering tendency.10 In addition, modification of Fyn with unsaturated or polyunsaturated fatty acids reduced its raft localization and resulted in decreased T-cell signal transduction.7 More recently, palmitate and oleate were shown to coexist on a single residue of the GNAI protein. This different composition has functional consequences, as only oleoylation prevents EGFR signaling by removing GNAI from detergent-resistant fractions.8Nevertheless, most of these previous findings have been obtained after metabolic labeling with radioactive or azide/alkyne-tagged fatty acids. These species can be metabolized to shorter, longer or unsaturated fatty acids thereby complicating the confident identification of lipid residues in the native state and their correlation with protein function. As a result, methods allowing direct analysis of the fatty acids linked to proteins under endogenous conditions would be preferred.
Recent advances in mass spectrometry have gained interest in the characterization of proteins and their posttranslational modifications (PTM). Two complementary approaches are being explored nowadays. The most widely used approach (bottom-up) relies on the tryptic digestion of the proteins followed by analysis of the resulting peptides by mass spectrometry (MS). However, the strong hydrophobicity of the lipid moieties challenges their application to lipid-modified proteins. To overcome this limitation, a method combining solid phase enrichment coupled with an open search mode was recently reported, leading to the identification of palmitate, palmitoleate, myristate and octanoate moieties attached to peptide fragments.11 Alternatively, top-down proteomics has emerged as a promising tool to characterize intact proteins. This approach allows the identification of multiple proteoforms (different protein products derived from a single gene and combining genetic variations, alternative splicing and PTMs) and it was recently employed to detect the palmitoylated K-Ras4A.12 Despite these advances, these protein-centered methods are not optimal for proper quantification of the lipid abundance, that are better addressed using lipid-centered approaches.
Thioesters, but not esters, can be selectively cleaved upon treatment with hydroxylamine at neutral pH, resulting in the formation of a free sulfhydryl at the cysteine residue and the conversion of the released fatty acids into a fatty acid hydroxamate (FAH, Fig. 1A).13 First attempts to identify the resulting FAHs were based on gas chromatography-MS (GC-MS) analysis, which requires derivatization to form a methyl ester and could only be used to assess gross changes in protein acylation.4,14 Nowadays, GC-MS analysis of lipids has been mainly overtaken by MS coupled to liquid chromatography (LC). We have previously described the development of an LC-MS method to identify and quantify the fatty acids linked to cysteine residues.10 However, this method relied on the use of a fluorescently labeled hydroxylamine derivative, and the high amounts required of this synthetic compound challenged its wider application. Since we are convinced that this field of research could strongly benefit from robust, general and simple methods enabling the direct and sensitive analysis of S-linked fatty acids, we decided to explore the use of the commercially available hydroxylamine (NH2OH) for the global profiling and quantification of the S-acylome. Herein, we present a general method able to resolve and accurately identify and quantify the wide composition of the S-acylome. The generality of the method enables its use in cell lines and mice tissues giving important insights into the heterogeneous composition of the lipid modifications present in proteins. Moreover, the method could also be applied to profile not only the proteome but also the lipid fraction (acyl-CoAs) overcoming many important limitations in the detection of this highly unstable species and enabling for the first time the simultaneous identification and quantification in tissue samples of both the acyl-CoAs pool and the S-acylome in a single experiment.
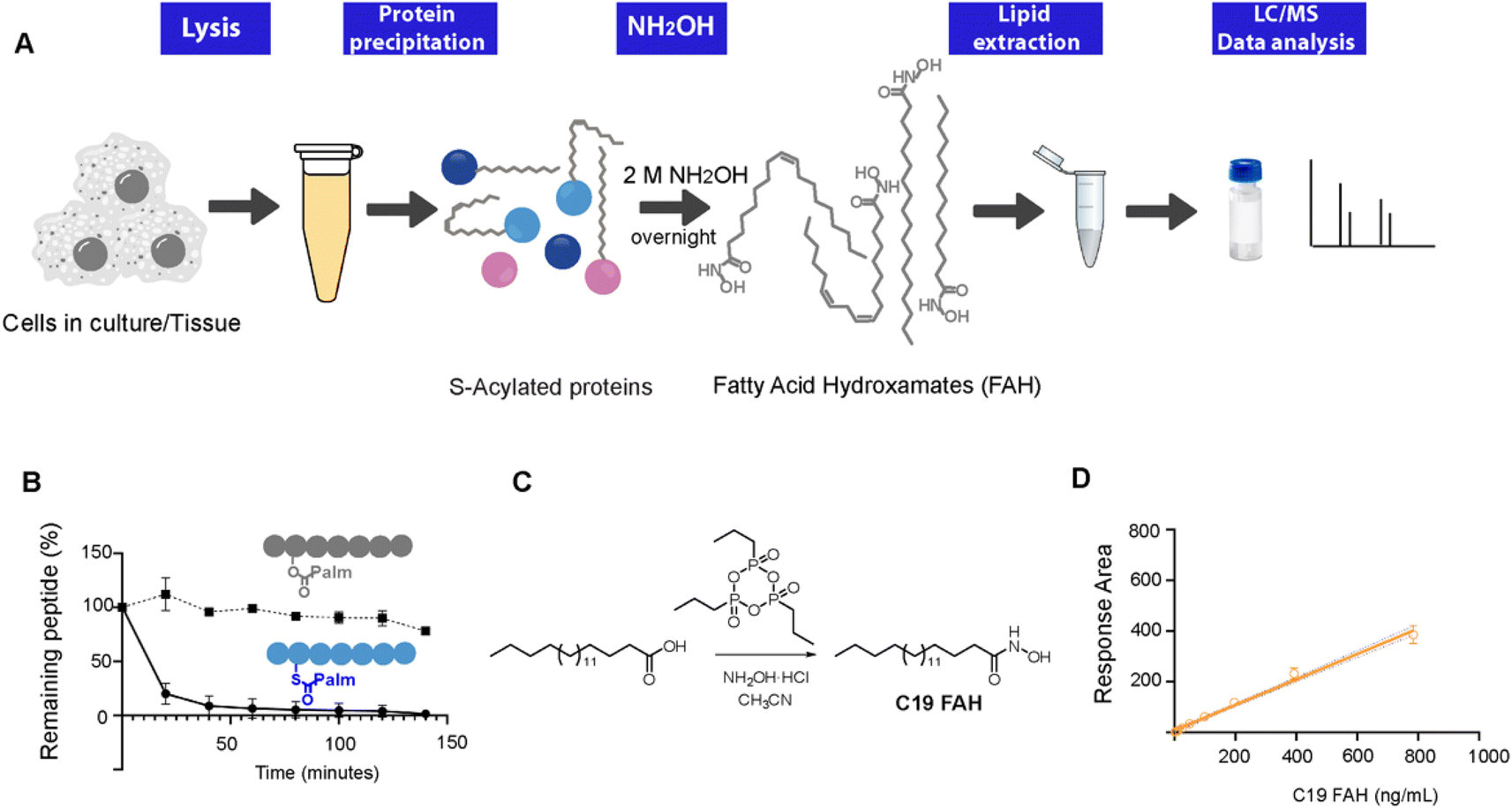 | ||
| Fig. 1 (A) Workflow of the method. NH2OH-based treatment of S-acylated proteins results in the release of the linked lipids as FAHs that can be analyzed and quantified by MS upon the addition of an internal standard. (B) HPLC monitoring of the thioester/ester bond hydrolysis of peptides 1 and 2 at room temperature under the presence of a 0.5 M solution of NH2OH. (C) Synthesis of the standard C19 FAH. (D) Standard curve corresponding to the MS-detection of C19 FAH. | ||
Results
Thioester cleavage by hydroxylamine treatment
To explore the optimal conditions for the selective cleavage of thioesters mediated by NH2OH treatment, we synthesized two peptides, corresponding to the C-terminus of K-Ras4A, and bearing a palmitic acid linked via a thioester (1) or an ester bond (2) (ESI Fig. S1–S3†). The hydroxylaminolysis of the S-palmitoylated peptide (1) and the O-palmitoylated peptide (2) was then monitored by high-performance liquid chromatography (HPLC). The complete disappearance of the peptide thioester was already detected after 60 min treatment with a 0.5 M solution of NH2OH, whereas the peptide ester was not hydrolyzed under the same conditions (Fig. 1C). Similar results were obtained after an overnight treatment with a 2 M solution of NH2OH (ESI Fig. S5†).Alternatively, the formation of the resulting C16 FAH was also analyzed by LC-MS. Accurate quantification required the use of an internal standard, belonging to the same compound class and having a similar structure and MS ionizability, that will be spiked at a known concentration into the sample. The FAH derived from the odd-chain fatty acid nonadecanoic acid (C19 FAH) was chosen to avoid potential overlap with endogenous species. The C19 FAH was obtained in good yields in a two-step one-pot procedure: first, in situ activation of the fatty acid with the cyclic propane phosphonic acid anhydride (T3P®) followed by reaction with the unprotected hydroxylamine (Fig. 1C).15 To overcome the reported low solubility of this compound class,16,17 which challenges its LC-MS analysis, optimal signal detection was achieved using as a mobile phase a mixture of water and methanol containing ammonium formate and formic acid. The linearity of the response and the limit of detection (LOD) and quantification (LOQ) of the C19 FAH were next determined using the signal-to-noise method (S/N). A S/N of 3 is generally accepted for estimating the LOD, and a S/N ratio of 10 is used for estimating the LOQ.18 The working range was established as the range of concentration from the LOQ to the maximum of the calibration curve, maintaining the correlation coefficient (r2) above 0.98. The MS response was linear with concentrations up to 783 ng mL−1 and the LOD and LOQ were established as 1.5 ng mL−1 (or 38.3 fmols of injected product) and 6.12 ng mL−1 (156.3 fmols) respectively (Fig. 1D). At this point, quantification of the formed C16 FAH could be performed by LC-MS analysis after overnight treatment of the S-acylated and O-acylated peptides with NH2OH 2 M. Whereas clear formation of the C16 FAH could be observed from the S-palmitoylated peptide (1), only a negligible amount was formed in the case of the O-palmitoylated peptide (2). The efficiency of the cleavage at different pH values (7.5, 7 and 6.5) was explored, and better conversions were obtained at the higher pH 7.5 (ESI Fig. S6†). Thus, NH2OH reacts efficiently and selectively with thioesters to generate a stable product that is detectable by LC-MS with high sensitivity, and esters do not contribute to FAH formation.
Profiling the S-acylome in cells
Next, the method was applied to profile the lipid moieties bound to proteins in different cultured cell lines. First, to optimize their LC separation and establish their retention times, 5 additional reference standards (C14, C16, C18:2, C20:4, C22:1), with different chain lengths and double bond numbers, were prepared from the corresponding fatty acids following a procedure analogous to the one applied for the synthesis of C19 FAH. For the characterization of the S-acylome, cells were lysed in the presence of the thioesterase inhibitor palmostatin B to prevent cleavage of the S-acyl groups,19 and the cell lysates were treated with a mixture of chloroform/methanol to precipitate and delipidate the proteome in a single step.20 Optimal results for a complete cleavage of the S-acylome were obtained when treating the proteome overnight with a 2 M solution of NH2OH. Shorter times or lower concentrations resulted in the detection of lower amounts of FAHs. In addition, complete cleavage was confirmed by the disappearance of peptide 1, when it was spiked into the sample before NH2OH treatment. Moreover, to assess the complete cleavage of the S-acylome, samples were also immunoblotted for the cysteine string protein (CSP), a molecular chaperone of the Hsp40 protein family which is extensively palmitoylated on a central string domain that contains 14 cysteine residues. As the removal of its palmitate groups gives a clear detectable shift in molecular weight, the band-shift induced by the NH2OH treatment serves as a robust assessment of complete depalmitoylation (ESI Fig. S7†).21,22After treatment of the proteome with NH2OH, the released FAHs were then extracted with a chloroform/methanol/water mixture at room temperature. Before lipid extraction, 100 pmols of C19 FAH were spiked into the samples for proper relative quantification. Collected lipid extracts were dried and stored until LC-MS-based analysis. The identity of the formed FAHs were unambiguously confirmed by accurate mass and elemental composition and by comparing their retention time with the ones observed for synthesized reference compounds. Each peak was quantified relative to the amount of the added internal standard. The extraction method is based on the Bligh and Dyer protocol, extensively used for lipid sample preparation.23 However, to explore the optimal recovery and extraction of the FAHs, different control experiments were performed. First, to quantify the loss of analyte during the extraction, the recovery of the internal standard was measured by comparing the peak areas obtained when the C19 FAH was spiked in a sample before the extraction with the one where the standard was directly added to a matrix blank (C19 FAH in methanol) (ESI Fig. S8†). This measurement established a percentage of recovery of 80%. Then, the amounts of FAHs extracted from a cell lysate were measured after two sequential extraction steps. The second extraction cycle did not substantially increase the extraction efficiency of the investigated FAHs since only a residual 10% was recovered in this step, thus providing evidence that the FAHs were extracted to a remarkable degree already during the first extraction step. Moreover, when extraction was performed at higher temperatures or longer times did not result in significant efficiency improvement.
This approach was then employed to profile the S-acylome of different cell lines, the human lung adenocarcinoma cell line A549, the human colon adenocarcinoma cell line HT29, the hepatocarcinoma cells HepG2, the melanoma cells A375, and the human embryonic kidney cells HEK293 (ESI Fig. S9† shows representative examples of the obtained chromatograms). The level of FAHs formed ranged from the higher levels detected in HT29 cells (2200 pmols per mg of protein) to the lowest detected in HepG2 and HEK293 (around 1600 pmols per mg of protein) (Fig. 2A). In general, all cell lines showed high levels of proteins modified with saturated FAH, comprised between the 69% detected in HT29 cells and the 58% observed HepG2 cells.
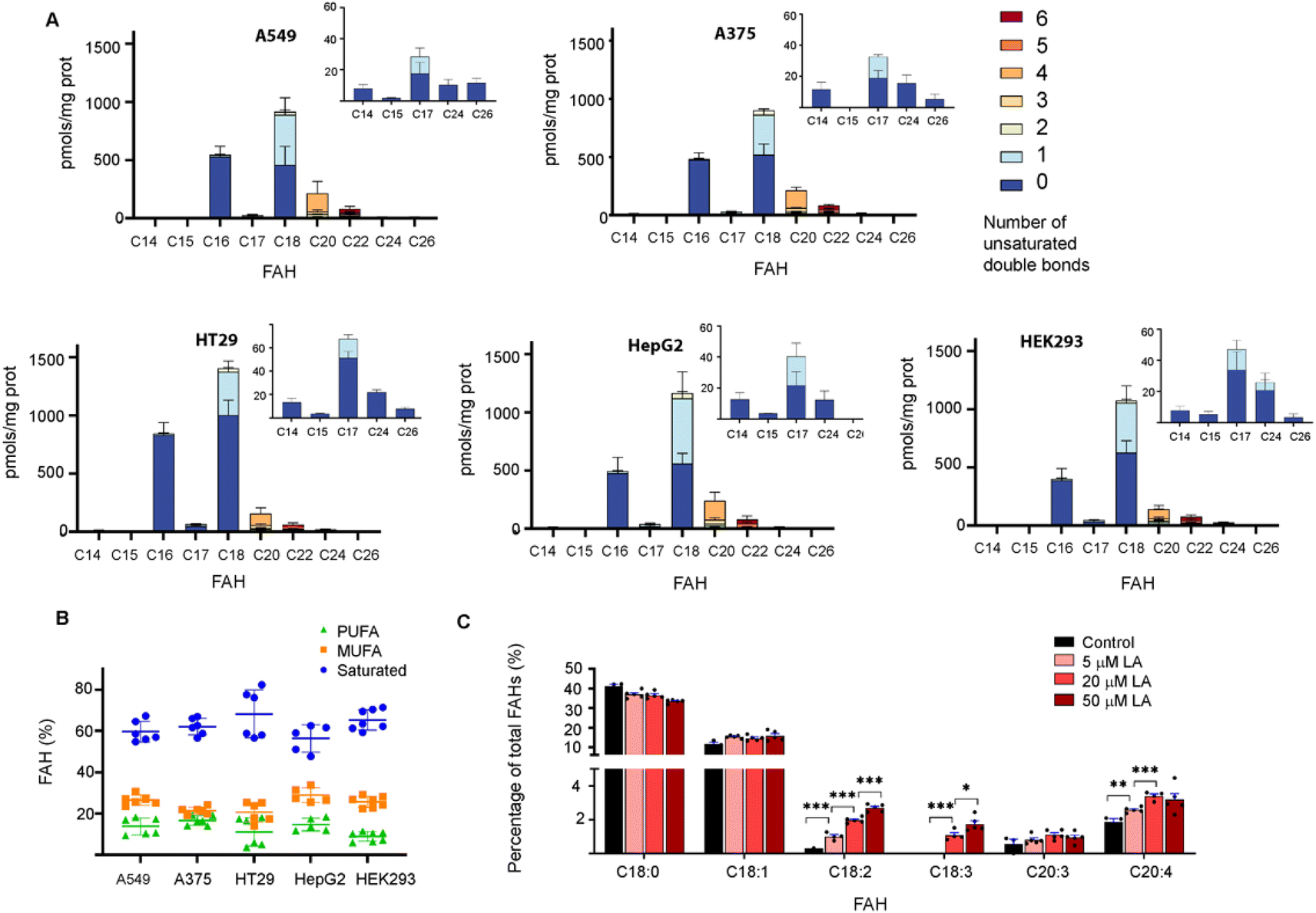 | ||
| Fig. 2 (A) Abundance of FAHs detected in the proteome of A549, A375, HT29, HepG2 and HEK293 cells. Amounts are calculated in relation to C19 FAH (100 pmols) used as internal standard and for 0.6 mg of protein sample. Colors indicate the number of unsaturations for a specific chain length. Data are from 2 independent experiments (n = 3 replicates per experiment). All panels display mean with error bars representing standard deviation. Data for all the FAHs species that were quantified are displayed. Inserts show the amount of the less abundant FAHs. Those that were not quantified showed insufficient signal intensity for the analyte. (B) Relative percentage of saturated, monounsaturated (MUFA) and polyunsaturated (PUFA) fatty acids in the cell lines tested. The horizontal line represents mean values. (C) The abundance of FAHs (in %) was detected in the proteome of HT29 cells treated with different concentrations of Linoleic Acid (LA) for 4 h (n = 5). Comparison between two means has been carried out with the unpaired two-tailed t-test and statistical differences are marked with asterisks (P values: *P < 0.05, **P < 0.01, ***P < 0.001). | ||
The second largest section was the monounsaturated fatty acids (MUFA, 21–29%) with the higher levels observed in HepG2 cells, while polyunsaturated fatty acids (PUFA) comprised only 8–17% of all the FAH detected (lowest levels in HEK293 and higher in HepG2 and A375) (Fig. 2B). These results are similar to the free fatty acid profile observed in cells in culture (45% saturated FA, 39% MUFA, 14% PUFA).24 Strikingly, when checking the individual levels of FAHs, the non-cancer cells HEK293 cells contain the higher fraction of proteins modified with C18 (37%) and the lowest fraction modified with C16 (22%). As a result, modified proteins in HEK293 cells displayed a C18/C16 ratio of >1.5, in contrast to the cancer cells that showed in most of the cases an equimolar amount of both saturated fatty acids. Moreover, HEK293 cells display the lower fraction of proteins modified with PUFA, and the ones identified contain the highest fraction of C22:6 linked fatty acids (Fig. 2A).
The presence of circulating odd-chain fatty acids (OCFA) has gained increasing interest in the last decade because their levels are correlated with a lower risk of some metabolic diseases.25
Thus, we decided to investigate if OCFA were also attached to proteins. Interestingly, certain amounts of C15 FAH and mainly C17 FAH could be detected in all the cases while no traces of other OCFA could be observed (Fig. 2A, Inserts). The absence of endogenous C19 was confirmed after profiling the S-acylome without the spiked internal standard C19 FAH (ESI Fig. S10†).
Recent data indicate that cells can sense the dietary levels of fatty acids and couple them to protein modification with lipids with consequences in cell signaling processes. Thus, competitive acylation can occur in single residues of proteins depending on the relative abundance of the fatty acids present in the cell media and these changes may have functional consequences. This is the case for example for GNAI, whose acylation with C16 leads to localization into detergent-resistant membranes, whereas acylation with C18:1, formed upon desaturation of C18, localizes the protein to non-detergent-resistant fractions resulting in reduced AKT activation and proliferation.8 Thus, we next asked whether the exposure of cells to linoleic acid (LA, C18:2), an essential fatty acid that needs to be obtained from the diet, could modulate the observed S-acylome. This fatty acid is essential because it is a precursor of the n-6 series of long-chain fatty acids. Thus, C18:2 is desaturated to form C18:3, which in turn is elongated to C20:3, and further desaturated to form arachidonic acid (C20:4), one of the most prominent PUFA and the precursor of prostaglandins or leukotrienes. With this aim, HT29 cells were treated with a 5, 20 or 50 μM concentration of the linoleic acid, that was delivered to the cells as a BSA conjugate. After 4 h incubation, the proteome was isolated and treated with NH2OH, and the released FAHs were extracted and analyzed following the established protocol. Indeed, treatment of cells with linoleic acid resulted in a substantial increase of the proteins modified with C18:2. Slight increases were also observed for the desaturation products C18:3 and C20:4, whereas the elongation product C20:3 was minimally impacted (Fig. 2C).
zDHHC enzymes display substrate preferences
Protein S-acylation is a dynamic and reversible modification leading to an acylation/deacylation cycle that regulates the localization and activity of proteins. The transfer of the acyl moiety is catalyzed by the 23-member family of zinc finger aspartate–histidine–histidine–cysteine motif (zDHHC) protein acyltransferases, while the reverse reaction is performed by a set of acyl-protein thioesterases.2 Acyl chains are transferred to proteins in a two-step process. Thus, zDHHCs become first autoacylated using acyl-CoA donors and then transfer those fatty acids onto cysteine residues from the substrate proteins. The zDHHC proteins have the ability to transfer acyl-CoAs of different chain lengths and degrees of unsaturation. Some individual substrate preferences have been described, although the mechanisms which drive this selectivity are not completely clear, yet.Initial studies performed with purified proteins and radioactive fatty acids showed that while zDHHC2 could use fatty acids with up to at least 20 carbons, zDHHC3 showed preferences for shorter chains up to C16 carbons.26 The autoacylation activity of zDHHC20 was also measured, in this case using a coupled-enzymatic assay and showed that the enzyme accepts C12, C14, C16 and C18, although it shows a preference for C16.27 We previously measured also protein S-acylation using fatty acid azides and HEK293T cells cotransfected with synaptosomal-associated protein (SNAP25) and different zDHHC enzymes. The results indicated that zDHHC3 and zDHHC7 show a preference for C14 and C16, zDHHC17 showed a preference for C16 and C18, whereas no significant preferences could be observed for zDHHC2.6
As the herein described method enables direct detection of the attached fatty acids without the use of radioactive or tagged fatty acids, to further validate the method we next profiled the cellular S-acylome in cells overexpressing different zDHHC enzymes, to explore if this approach can be used to gain novel insights into the substrate preferences of zDHHC enzymes. To this end, HEK293T cells were transfected with hemagglutinin (HA)-tagged zDHHC2, zDHHC3, zDHHC6 or zDHHC7, and the S-acylome was then extracted and analyzed using the standard procedure (ESI Fig. S11†). Fig. 3 shows that the levels of C18, C20 or C22 incorporation by zDHHC3 and zDHHC7 were markedly reduced, whereas the incorporation of C14 or C16 was substantially increased, especially in zDHHC3 overexpressing cells. No significant changes were detected in cells overexpressing the zDHHC2 enzyme and the low levels of overexpression of zDHHC6 precluded its further analysis.
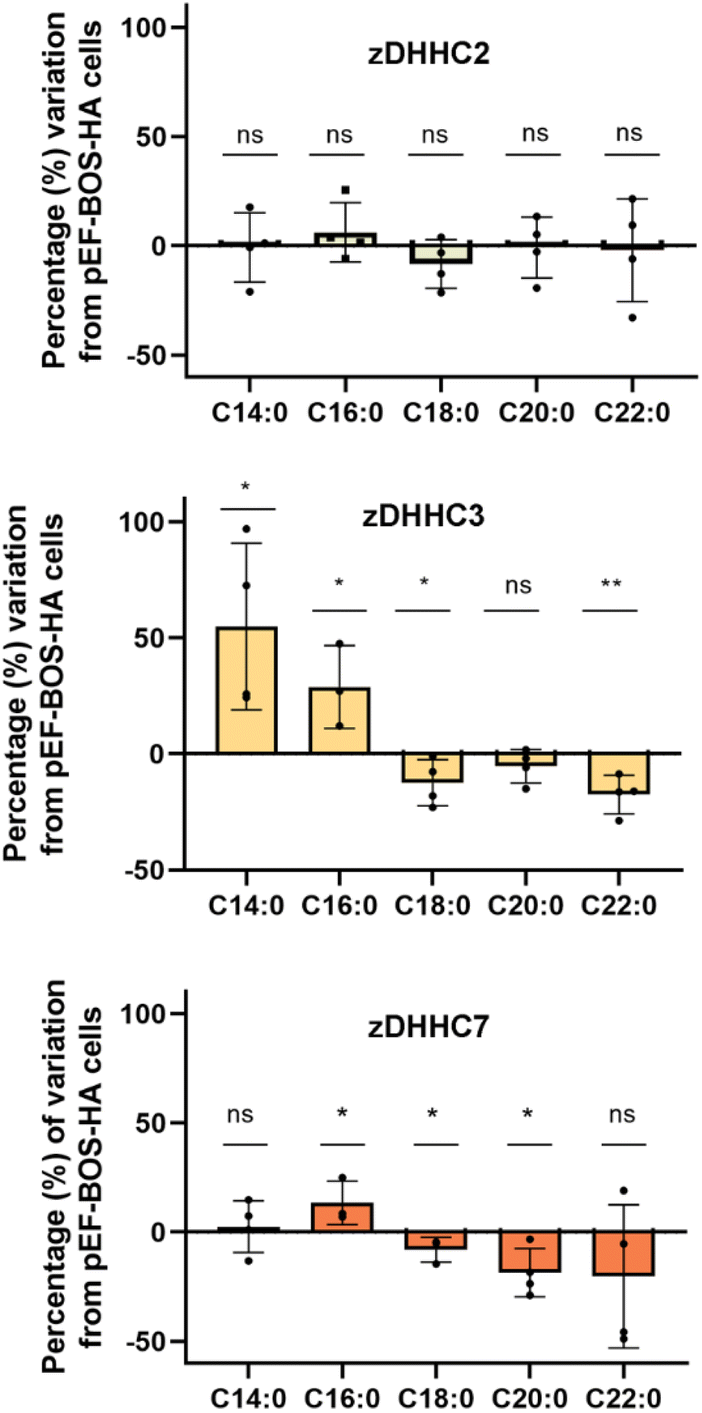 | ||
| Fig. 3 The graph displays the positive or negative percentage change in the incorporation of C14, C16, C18, C20 and C22 acyl moieties into proteins in cells overexpressing either zDHHC2, zDHHC3 or zDHHC7 enzymes compared to cells transfected with pEF-BOS-HA (negative control, empty plasmid, n = 4). Comparison between two means has been carried out with the unpaired two-tailed t-test and statistical differences are marked with asterisks (P values: *P < 0.05, **P < 0.01, ***P < 0.001, ns: not significant). | ||
NH2OH can be used to measure the S-acylome in tissue extracts
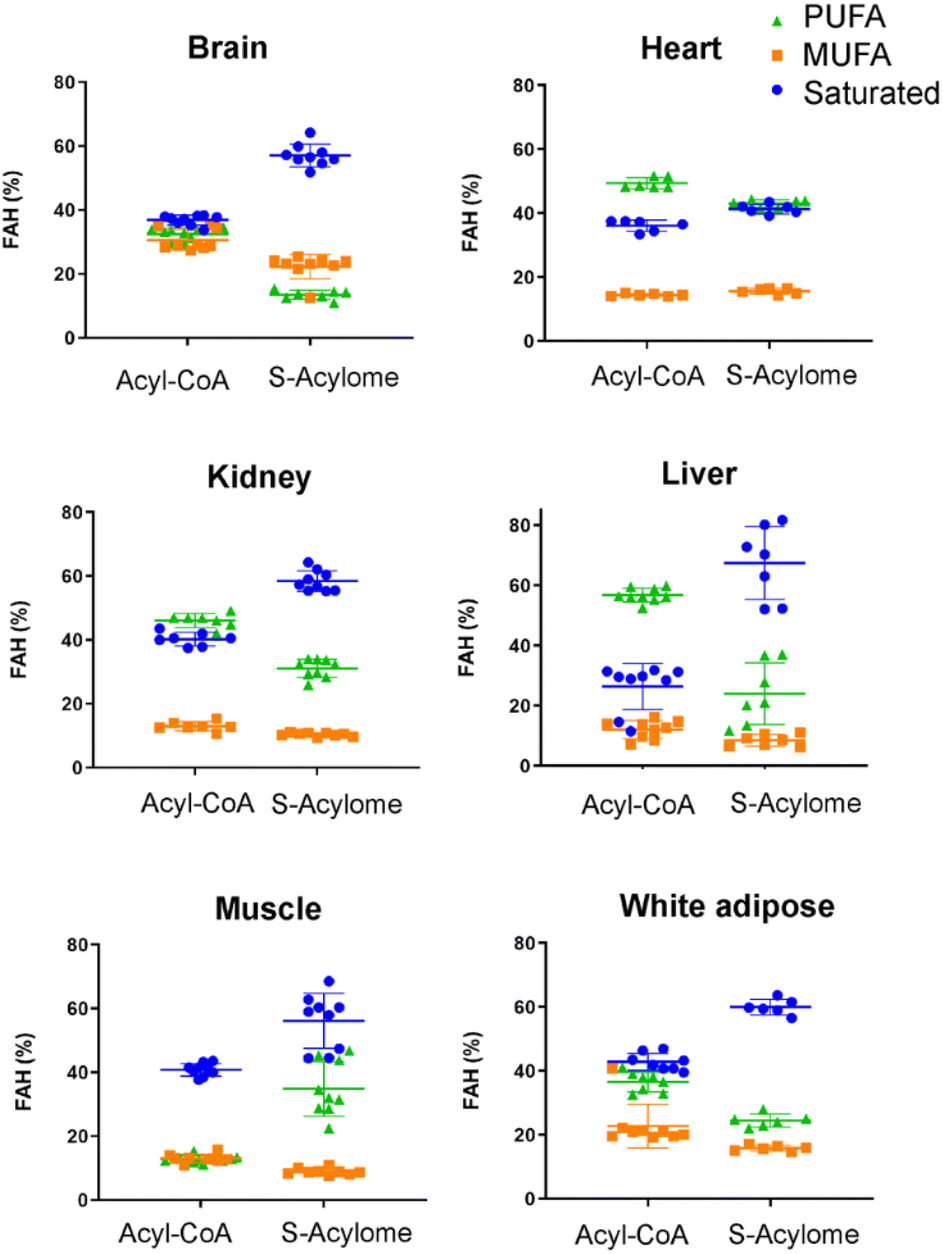
Cells in culture have limited access to lipids. This factor influences their fatty acid profile (very low in PUFA and high in MUFA) and probably also their S-acylome.24 In contrast, natural tissues have a more diverse and natural composition of fatty acids. As a result, we next decided to explore the S-acylome in different relevant mice tissues. To this end, mouse heart, liver, kidney, brain, white adipose and muscle tissues were snap frozen and after ultrasonic homogenization, the proteome was precipitated and subjected to hydroxylamine treatment. Released FAHs were then extracted and analyzed by LC-MS following the usual protocol. As depicted in Fig. 4, the composition of the S-acylome differs substantially between tissues. Shorter saturated C12 and C14 are mainly detected in significant amounts in the brain and heart, while the longer C16 and C18 are present in all tissues in considerable quantities being C18 significantly important in the liver. The presence of monounsaturated species, C16:1 and especially C18:1, is quite important in the brain, whereas the heart has considerable amounts of C18:2 and docosahexaenoic acid (C22:6), and the kidney presents also important levels of proteins modified with arachidonic acid (C20:4).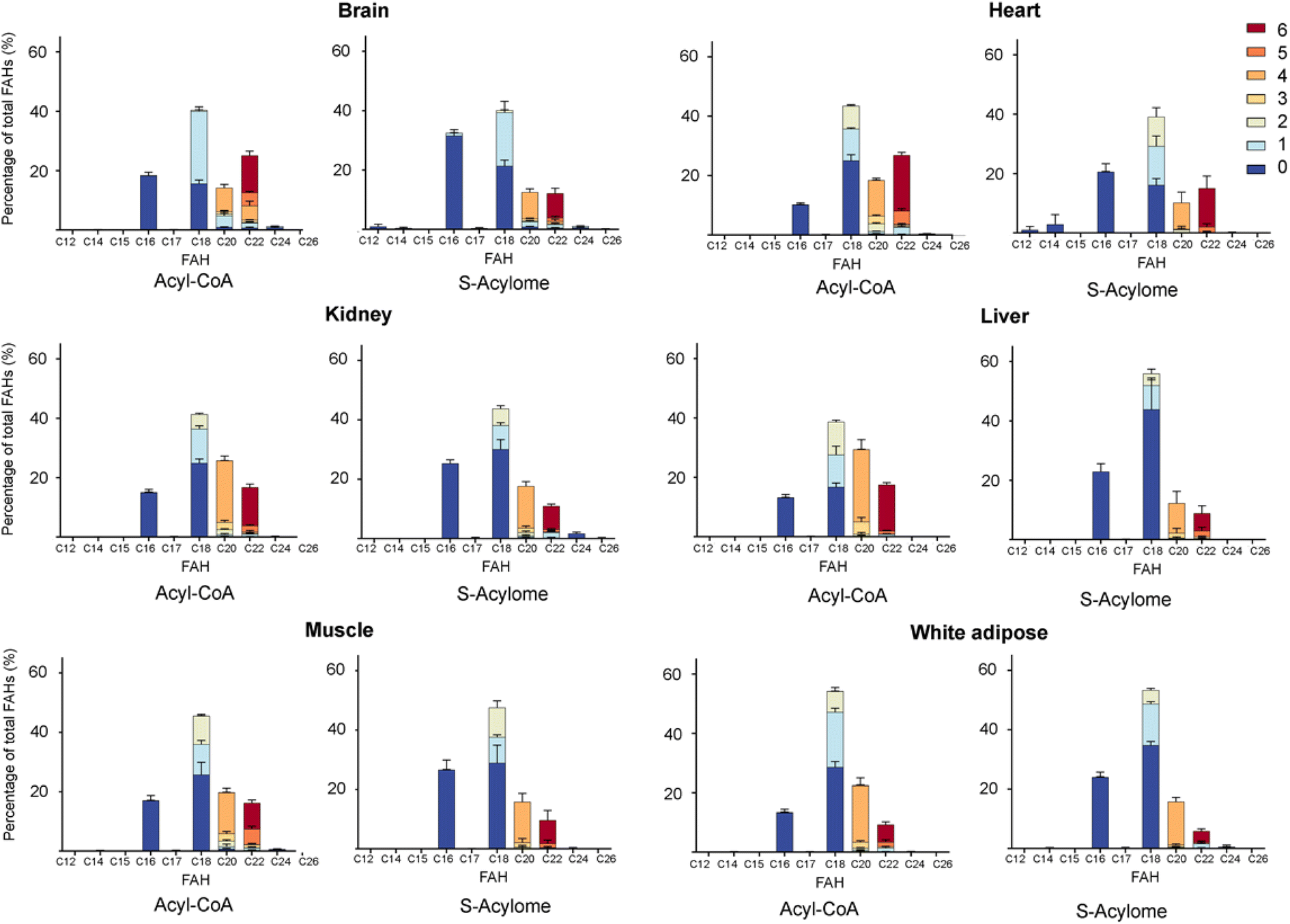 | ||
| Fig. 4 Acyl-CoA species and S-acylome of different tissues expressed as a percentage of total FAH detected. Colors indicate the number of unsaturations for a specific chain. Amounts are calculated to C19 FAH used as internal standard and for 0.6 mg of protein sample. Data are from 3 independent experiments (n = 3 replicates samples per experiment). All panels display mean with error bars representing standard deviation. Data for all the FAHs species that were quantified are displayed. Those that were not quantified showed insufficient signal intensity for the analyte. | ||
NH2OH can be applied to measure the acyl-CoA pools in tissue extracts
Acyl-CoAs are key metabolic intermediates of multiple physiologic and metabolic processes, such as fatty acid beta-oxidation, biosynthesis of lipids, and modification of proteins. As a result, great efforts have been spent on developing methods that allow for the detection and quantification of acyl-CoAs.28,29 However, acyl-CoAs are highly unstable in aqueous solution and are therefore susceptible to hydrolysis, and some of the reported methods require complex and time-consuming sample preparation approaches.30 Therefore, additional methods expanding the toolbox for acyl-CoA detection and quantification can contribute to overcoming these limitations and yield crucial insights into their role in different cellular processes. Moreover, apart from the substrate preferences displayed by each individual zDHHC enzyme, their choice of lipid donors can be also influenced by the tissue-dependent relative composition of the fatty acyl-CoA pools. Thus, it would be of great interest to compare the lipid pattern of the S-acylome in one specific tissue with its relative acyl-CoA composition.The first step in the previously reported characterization of the S-acylome includes simultaneous protein precipitation and sample delipidation. First, the addition of a chloroform/methanol/water mixture results in a three-phase system: a lower chloroform phase, an upper methanol–H2O (M/W) phase and a protein interphase. The upper phase is carefully removed and methanol is added again. The second centrifugation pellets the protein at the bottom, and the supernatant, containing methanol and chloroform (C/M) can be then removed. Thus, these two supernatants contain the cellular lipids including the acyl-CoA pool.
Structurally, acyl-CoA are thioesters formed between a fatty acid and a thiol, the coenzyme A, and are the major lipid-containing thioesters in the cells. Hence, treatment of the lipid fraction with NH2OH should result in the simple and rapid conversion of acyl-CoA to stable FAHs, thereby overcoming many of the described challenges in long acyl-CoA analysis. To initially address this possibility, the hydrolysis of the oleoyl-CoA upon treatment with a 2 M solution of neutral NH2OH was first monitored by HPLC. The complete disappearance of the thioester was already detected after 90 min (ESI Fig. S12†). Conversion of oleoyl-CoA to the corresponding FAH could be also observed after 2 h or an overnight treatment with NH2OH (ESI Fig. S13†).
Once proven the complete conversion of oleoyl-CoA into a FAH, this method was applied to the quantification of long-chain acyl CoA in different tissues. With this aim, the initial upper phase (M/W) and the second supernatant (C/M) were subjected to analysis. Briefly, both fractions were separately evaporated and treated with NH2OH. Lipids were extracted following the above-reported protocol and subsequently analyzed by LC-MS. Most of the FAHs were identified in the C/M fraction, whereas the M/W fraction only contained residual amounts of FAHs (<10%). Total extraction of the lipids was confirmed after a second washing step of the proteome with methanol, that revealed only traces of lipids (<1%). The most abundant acyl-CoA species detected were consistent with those identified in the literature and are displayed in Fig. 4.
Discussion
Heterogeneous lipidation of proteins is emerging as an additional regulator of protein function and localization. However, despite this increasing interest, there is still a limited number of methods enabling a straightforward identification and quantification of the lipids forming the S-acylome. Herein, we have established a method to profile the S-linked fatty acids by LC-MS upon conversion to fatty acid hydroxamates by treatment with NH2OH at neutral pH. Upon method optimization, we could prove that the resulting FAHs are stable, can be easily extracted using organic solvent mixtures with good recovery yields and analyzed by LC-MS with high sensitivity, resolution and coverage. Moreover, absolute quantification is allowed with the use of an internal standard that it is spiked into the sample before lipid extraction.The method was initially applied to a set of different cell types, four cancer and one non-cancer cell line. Cancer cells show an increased demand for fatty acids, that are obtained by biosynthesis upregulation, alterations in FA transport or storage, or through direct exogenous uptake. The altered lipid metabolism is employed as an energy source and causes also changes in membrane composition and dynamics that ultimately affect signaling pathways.31 Moreover, exposure to high-fat diets can also influence cancer progression and metastasis. Thus, higher levels of dietary C18 have been correlated with anti-cancer effects32 and a diet rich in palmitic acid (C16) promotes metastasis whereas diets rich in oleic (C18:1) or linoleic acid (C18:2) show a decreased frequency of developing metastatic tumors.33 However, whether these effects are mediated in part by S-acylation of proteins has not been fully elucidated. Interestingly, and in agreement with these previous observations, our results indicate that the non-cancer HEK293 cells displayed the highest levels of proteins modified with C18 and the lowest levels of proteins modified with C16, thereby suggesting that the observed effects of fatty acids can be in part due to protein modification events.
Cells in culture present an unnatural fatty acid profile, with low levels of PUFA and high levels of MUFA. This is caused due to their source of lipids, which is mainly fetal bovine serum (FBS), which only contains one-third of MUFA and 11% of PUFA compared to media made from human serum.24
Our results indicate that the composition of the S-acylome in cells or tissues is significantly different and in agreement with these previous observations. Thus, the S-acylome identified in different mice tissues displayed significantly higher levels of proteins modified with PUFA, which were in all the cases, except the brain tissue, higher than the fraction of proteins modified with MUFA (Fig. 5). On the contrary, cells in culture contained mainly proteins modified with saturated and monounsaturated FA (Fig. 2B).
 | ||
| Fig. 5 Relative percentage of saturated, monounsaturated (MUFA) and polyunsaturated (PUFA) fatty acids in the tested tissues. The horizontal line in this section represents mean values. | ||
So, caution should be taken when drawing conclusions from cells in culture, as they may not fully represent the whole S-acylome and their potential functional consequences. Moreover, the S-acylome seems to be able to respond to dietary changes, as treatment of HT29 cells with linoleic acid causes an increase of the proteins modified with C18:2, C20:3 and C20:4 fatty acids (Fig. 2C). Interestingly, a 24 h serum withdrawal did not substantially change the composition of the S-acylome or the S-acyl-CoA pool, but a ∼40% decrease in the total amount of acyl-CoA species could be observed (ESI Fig. S14†).
The overall factors influencing the S-acylation reaction are not fully elucidated. S-acylation may be regulated by substrate preferences of zDHHCs enzymes.6 Applying this methodology, we could confirm some of the reported substrate preferences. Thus, overexpression of zDHHC2 causes no relevant changes in the S-acylome, whereas overexpression of zDHHC3 and zDHHC7 results in a marked decrease of the proteins modified with the longer C18, C20 or C22 fatty acids, while there is an increase in the fraction of proteins modified with C14 or C16 fatty acids (Fig. 3).
Another important factor that could regulate S-acylation is the availability of acyl-CoA pools. We have demonstrated that the lipid profile in modified proteins responds to changes in dietary intake. However, it is still unknown whether the composition of the different pools of acyl-CoAs can also modulate the S-acylation reaction. Structurally, acyl-CoAs are formed by a fatty acid and a CoA linked by a thioester bond. Moreover, this currently established methodology enables the separation of the protein and the lipid fraction. Hence, since there are limited methods enabling the identification and quantification of the acyl-CoAs species among different tissues, mainly due to their reported low stability that complicates their analysis,34 we decided to explore if this approach could be also employed to identify the acyl-CoA pool. In addition, this simultaneous detection of the acyl-CoAs and the S-acylome in a single sample can be very useful for gaining novel insights into the existing links between metabolism and protein modification.
Interestingly, we have proved that acyl-CoAs can be converted to stable FAHs that can be then analyzed and quantified by LC-MS. Moreover, the method turned out to be suitable for the analysis of small quantities of tissues (∼25 mg/3 replicates) and provides a comprehensive profile of long and very long chain acyl-CoAs present in different tissues, such as heart, muscle, kidney, liver, brain and white adipose tissue. As observed in Fig. 4, the acyl-CoA profile shows significant differences along the tested tissues, that are mainly in agreement with previous reported results. Hence, brain tissue is significantly enriched in C16 and C18:1 species, heart tissue shows considerably high levels of C18:2 and C22:6, whereas kidney and liver display high levels of 20:4 acyl-CoAs. Moreover, when comparing the tissue-dependent acyl-CoA pools with the corresponding S-acylome, we can see in all the cases that the S-acylome does not completely resemble the acyl-CoA composition of the investigated tissues. On the contrary, the S-acylome is significantly enriched in saturated fatty acids, and to a minor extent in PUFA such as 20:4 and 22:6, while it displays lower levels of MUFA in all cases except for the brain, which contains considerable amounts of proteins modified with C16:1 and C18:1 (Fig. 5). These results suggest the existence of certain substrate specificities of the enzymes involved in their installation or removal and a potentially new mechanism of crosstalk between lipid composition and protein function.
Limitations of the study
Here, we measure the S-acylome in cells and tissues, as well as the acyl-CoAs present in different tissues by forming stable FAHs upon treatment of the corresponding proteins and lipid thioesters with NH2OH. While this method enables stable and sensitive detection of the FAHs formed during both protocols, some limitations should be discussed. Acyl-CoA species vary significantly in their polarity, and methods able to extract and analyze short-to-long species would be desirable. However, due to the complexity of the sample, there is only a limited number of methods able to make a comprehensive analysis of all these species in a single experiment or a single run.28,29,35Our described method is currently optimized for the detection of acyl-CoAs species that range from C12 to C26 carbons, and extraction methods and chromatographic separation will be optimized in the future for the identification of short and medium FAH species. In addition, as acyl-CoAs are the major lipid thioesters, most of the signals obtained from the lipid fraction should correspond to the acyl-CoA pool. However, some minor amounts of S-acyl glutathione species, derived from acyl-CoAs, may exist in equilibrium with acyl-CoAs or they can be formed post-extraction due the instability of acyl-CoA species.29 Currently, it is not possible to discern the ratio of FAH derived from S-acylated glutathione. Nevertheless, as NH2OH treatment of both thioester species will lead to the same FAHs, the method it is expected to prevent the loss of acyl-CoA signal due to this transformation.
Conclusions
Here we described the use of hydroxylamine to profile and quantify the fatty acids attached to proteins via thioester linkages, i.e. the S-acylome. This robust and simple methodology together with the reagent employed, commercially available and inexpensive, has enabled an unprecedented and extensive characterization of the S-acylome in different cell lines and tissues. Moreover, we could also apply this method to profile the lipid thioesters, i.e. acyl-CoAs, present in tissues. This strategy has enabled for the first time the correlation of the acyl-CoAs pool from a specific tissue with the lipidation profile of its proteome, enabling to explore the mechanistic relationship between acyl-CoA supply and one of the functional outputs of acyl-CoA metabolism, that is protein lipidation. We are convinced that this approach can strongly expand the toolbox of methods available to investigate S-acylation and contribute, as shown here for some zDHHC proteins, to deciphering the substrate specificities of the acylating and deacylating enzymes that tightly regulate it, which has been long considered an ongoing challenge.In addition, we are also convinced that this novel approach to detecting and quantifying acyl-CoA compounds can strongly contribute to the routine use of acyl-CoA as a biomarker in many physiological and pathological processes, such as fatty acid oxidation disorders.34 Moreover, the correlation of the acyl-CoA pool with the S-acylome can be very helpful in predicting the proteins and metabolic pathways involved in various disorders. All in all, these findings open up numerous questions for future investigations, such as what are the mechanisms governing substrate specificity, how the enrichment in saturated fatty acids takes place or the potential existence of tissue-specific effects.
Methods
Measurement of S-acylome in cells
Cells were seeded into T75 flasks and grown to a confluence of approximately 90%. After removal of the growing media by aspiration, cells were washed with 5 mL of phosphate-buffered saline (PBS) and collected using cell scrapers. The cell suspension was then centrifuged (400 g, 3 min, room temperature), the supernatant removed, and the cell pellet was washed with PBS (2 mL, 400 g, 3 min, room temperature) twice. Afterwards, the pellet was resuspended (1 × 107 cells per mL) with lysis buffer (50 mM Tris–HCl, 150 mM NaCl, pH 7.2) containing protease inhibitors, 1% NP-40 and 10 μM of palmostatin B. For the serum withdrawal experiments cells were grown to a confluence of approximately 75%. Then the media was aspirated, cells were washed with 5 mL of PBS and fresh media (DMEM or DMEM supplemented with 10% FBS) was added and the cells were left for 24 h at the incubator (37 °C; 5% CO2).Cell lysates were obtained by sonication at 0 °C (10 cycles, 5 seconds sonication followed by 5 seconds resting period, set at 50% power). After clarification (3000 g, 5 min, rt), the cell lysates were transferred into 15 mL plastic polypropylene tubes and protein precipitation was performed by adapting the protocol developed by Wessel and Flügge.20 Briefly, 4 volumes (V) of methanol, 1.5 V of chloroform and 3 V of H2Odd were added to the sample. A vortex mixer was used after each addition, to ensure a proper mixing. Centrifugation of the samples (6000 g, 20 min, rt) resulted in a two-phase mixture with the protein pellet in the interphase. The upper phase was carefully removed using a micropipette without touching the protein interphase. Next, 3 additional V of methanol were then added and the tubes were centrifuged again (6000 g, 10 min, rt). Finally, all the liquid was then removed and the resulting protein pellet was carefully dried under a stream of nitrogen.
A second washing step of the protein pellet with 0.3 mL of methanol was performed to evaluate potential lipid contamination. The organic phase was collected after centrifugation (6000 g, 10 min, rt), treated and analysed for FAHs. The precipitated proteome was resuspended in lysis buffer (50 mM Tris–HCl, 150 mM NaCl, pH 7.2, 1 × 107 cells per mL) and thoroughly mixed using a micropipette. A sample (20 μL of the diluted proteome and 180 μL of H2Odd) was then taken to determine the total protein concentration using the Bicinchoninic Acid Assay (BCA). After quantification, 600 μg portions of protein were distributed into 1.5 mL Eppendorf tubes. Then, an equal volume of either lysis buffer (for the blanks) or a 4 M solution of NH2OH in lysis buffer, previously adjusted to pH 7.4, was added to the samples, that were left overnight at room temperature with end-over-end rotation. The following day, 100 pmols of the internal standard (C19 FAH) were added to each sample and the solution was transferred into a glass vial. Lipid extraction was performed following the Bligh and Dyer protocol with some minor adjustments.23 To the sample, 3.75 V of a 1:2 (v/v) solution of chloroform and methanol, 1.25 V of chloroform and 1.25 V of H2Odd were sequentially added. A vortex mixer was used after each addition, to ensure a proper mixing. After 1–2 min, the solution separated into two phases. After removing the top aqueous phase, the organic phase (bottom) was taken with a micropipette and transferred into a conic UPLC glass vial, evaporated in a SpeedVac concentrator and stored at −20 °C until MS-based analysis. Before injection in the mass spectrometer, the dried samples were resuspended in 150 μL of UPLC-grade methanol. Samples were analyzed by liquid chromatography coupled with a high-resolution mass spectrometer (LC-HRMS) using an Acquity Ultra High-performance Liquid Chromatography (UHPLC) System (Waters, USA) connected to a time-of-flight (TOF; LCT Premier XE) as described above (Monitoring FAH formation from peptides by LC-MS assay). The concentration of FAHs was calculated in relation to the C19 FAH internal standard spiked into the sample (100 pmols).
zDHHC lipid specificity assays
HEK293T cells were cultured in Dulbecco's modified Eagle's medium (DMEM) media supplemented with 10% fetal bovine serum without penicillin–streptomycin.Cells were seeded in 10 cm dishes and incubated at 37 °C/5% CO2 24 hours before transfection. The following day, cells at 80% confluence were temporarily transfected using lipofectamine 2000 reagent, with a ratio of 2 μL lipofectamine per μg DNA used. 30 μg of zDHHC plasmid: pEF-zDHHC2-HA, pEF-zDHHC3-HA, pEF-zDHHC7-HA (or pEF-BOS-HA as a negative control) were used per each 10 cm dish. Cells were incubated for another 24 h, and then proteins were precipitated, treated with NH2OH and the lipids were released and analyzed as described above.
Measurement of S-acylome in tissues
The tissue was cut into smaller pieces while keeping it on ice. Then, 250 μL of lysis buffer (150 mM Tris–HCl, 100 mM NaCl, pH 7.2 containing protease inhibitors, 1% NP-40 and 5 mM palmostatin B) were added to 25 mg of tissue. The samples from soft tissues (liver, brain, kidney and white adipose tissue) were sonicated for 10 cycles (5 seconds sonication/5 seconds resting time), whereas samples from harder tissues (muscle and heart) were submitted to 2 rounds of 10 cycles of sonication. Samples were kept on ice during the whole process.To isolate the proteome, 4 volumes (V) of chloroform, 1.5 V of methanol and 3 V of H2Odd were added to the sample which was then centrifuged at 6.000 g for 20 min at 4 °C. The upper phase (H2O–methanol) was carefully removed using a micropipette without touching the protein interphase. Next, 3 additional V of methanol were added and the sample was centrifuged again at 6.000g for 10 min, at room temperature. Afterwards, the supernatant was removed and the protein pellet was gently dried at room temperature or under a stream of nitrogen. Then, the protein pellet was resuspended in 600–800 μL of lysis buffer (15 mM Tris–HCl, 100 mM NaCl containing protease inhibitors) and the total protein concentration was determined using the BCA assay. After quantification, portions containing 600 μg of protein were distributed into in 1.5 mL polypropylene tubes. Then, an equal volume of either lysis buffer (for the blanks) or a 4 M solution of NH2OH in lysis buffer, previously adjusted at pH 7.4, was added to the samples, that were left overnight at room temperature with end-over-end rotation. The following day, 100 pmols of the internal standard (C19:0 FAH) were added to each sample and the solution was transferred into a glass vial. Lipid extraction and analysis were performed as described above for cellular studies.
Measurement of S-acyl CoA pools in tissues
For the measurement of the S-acyl pools in tissues, the same protocol described for the measurement of the S-acylome was employed. However, in this case, the first upper phase (H2O–methanol) and the second supernatant obtained (CHCl3–MeOH) during the protein precipitation protocol were subjected to analysis. Hence, both fractions were separately evaporated in a SpeedVac concentrator and treated with lysis buffer or a 4 M solution of NH2OH in lysis buffer, previously adjusted to pH 7.4, overnight at room temperature with end-over-end rotation. Lipid were extracted following the above-reported protocol and analyzed by LC-MS.Data availability
The data supporting the findings of this study are available within the main text and in the ESI.†Author contributions
Conceptualization, G. T. and L. H. C.; chemical design and synthesis, C. B. H., D. C., A. T., J. B. B. C. and G. T.; assay development, sample processing and analysis, C. B. H., S. R., E. G. B., D. C., G. T and L. H. C.; original draft was written by G. T.; revision and edition of the manuscript were performed by all authors.Conflicts of interest
The authors declare no other competing interests.Acknowledgements
We would like to thank Josefina Casas, Eva Prats, Roser Chaler, Alexandre García, Christine Salaun and Kathrin Galistl for their invaluable assistance throughout this research. We would like to thank Gemma Fabriàs for the critical revision of the manuscript. CBH thanks MCIU for an FPU fellowship (FPU20/00203), EMBO for a Scientific Exchange Grant (10187) and the specialized group on Chemical Biology (GEQB) of the Spanish Royal Society of Chemistry (RSEQ) for a travel grant. This research was partially supported by the grant LCF/PR/HR20/52400006 funded by CaixaResearch, the grant PID2021-128902OB-I00I funded by CIN/AEI/10.13039/501100011033 and the grant 2021SGR00504 funded by AGAUR, Generalitat de Catalunya.References
- B. Chen, Y. Sun, J. Niu, G. K. Jarugumilli and X. Wu, Cell Chem. Biol., 2018, 25, 817–831 CrossRef CAS.
- L. H. Chamberlain and M. J. Shipston, Physiol. Rev., 2015, 95, 341–376 CrossRef CAS PubMed.
- C. Busquets-Hernández and G. Triola, Front. Mol. Biosci., 2021, 8, 659861 CrossRef.
- L. Muszbek, G. Haramura, J. E. Cluette-Brown, E. M. Van Cott and M. Laposata, Lipids, 1999, 34, S331–S337 CrossRef CAS PubMed.
- X. Q. Liang, Y. Lu, T. A. Neubert and M. D. Resh, J. Biol. Chem., 2002, 277, 33032–33040 CrossRef CAS.
- J. Greaves, K. R. Munro, S. C. Davidson, M. Riviere, J. Wojno, T. K. Smith, N. C. Tomkinson and L. H. Chamberlain, Proc. Natl. Acad. Sci. U. S. A., 2017, 114, E1365–E1374 CrossRef CAS.
- X. Q. Liang, A. Nazarian, H. Erdjument-Bromage, W. Bornmann, P. Tempst and M. D. Resh, J. Biol. Chem., 2001, 276, 30987–30994 CrossRef CAS PubMed.
- H. Nůsková, M. V. Serebryakova, A. Ferrer-Caelles, T. Sachsenheimer, C. Lüchtenborg, A. K. Miller, B. Brügger, L. V. Kordyukova and A. A. Teleman, Nat. Commun., 2021, 12, 1–15 CrossRef.
- L. V Kordyukova, M. V Serebryakova, L. A. Baratova and M. Veit, Virology, 2010, 398, 49–56 CrossRef.
- J. Schulte-Zweckel, M. Dwivedi, A. Brockmeyer, P. Janning, R. Winter and G. Triola, Chem. Commun., 2019, 55, 11183 CAS.
- G. Ji, R. Wu, L. Zhang, J. Yao, C. Zhang, X. Zhang, Z. Liu, Y. Liu, T. Wang, C. Fang and H. Lu, Anal. Chem., 2023, 14, 53 Search PubMed.
- L. M. Adams, C. J. Dehart, B. S. Drown, L. C. Anderson, W. Bocik, E. S. Boja, T. M. Hiltke, C. L. Hendrickson, H. Rodriguez, M. Caldwell, R. Vafabakhsh and N. L. Kelleher, J. Biol. Chem., 2023, 299, 102768 CrossRef CAS PubMed.
- L. Muszbek and M. Laposata, J. Biol. Chem., 1993, 268, 18243–18248 CrossRef CAS PubMed.
- F. Mohammadzadeh, V. Hosseini, A. Mehdizadeh, C. Dani and M. Darabi, IUBMB Life, 2018, 71, 340–346 CrossRef PubMed.
- A. Ech-Chahad, A. Minassi, L. Berton and G. Appendino, Tetrahedron Lett., 2005, 46, 5113–5115 CrossRef CAS.
- I. S. Rosenfeld, G. D'Agnolo and P. R. Vagelos, Anal. Biochem., 1975, 64, 221–228 CrossRef CAS PubMed.
- F. Vernon and J. H. Khorassani, Talanta, 1978, 25, 410–412 CrossRef CAS.
- M. G. Kokotou, C. Mantzourani, A. Bourboula, O. G. Mountanea and G. Kokotos, Molecules, 2020, 25, 3947 CAS.
- F. J. Dekker, O. Rocks, N. Vartak, S. Menninger, C. Hedberg, R. Balamurugan, S. Wetzel, S. Renner, M. Gerauer, B. Schölermann, M. Rusch, J. W. Kramer, D. Rauh, G. J. Coates, L. Brunsveld, P. I. H. Bastiaens and H. Waldmann, Nat. Chem. Biol., 2010, 6, 449–456 CrossRef CAS PubMed.
- D. Wessel and U. I. Flügge, Anal. Biochem., 1984, 138, 141–143 CrossRef CAS.
- J. Greaves and L. H. Chamberlain, Mol. Biol. Cell, 2006, 17, 4748–4759 CrossRef CAS PubMed.
- M. J. Edmonds, B. Geary, M. K. Doherty and A. Morgan, Sci. Rep., 2017, 7, 1–13 CrossRef CAS.
- E. G. Bligh and W. J. Dyer, Can. J. Biochem. Physiol., 1959, 37, 911–917 CrossRef CAS.
- P. L. Else, Prog. Lipid Res., 2020, 77, 101017 CrossRef CAS PubMed.
- B. J. Jenkins, K. Seyssel, S. Chiu, P.-H. Pan, S.-Y. Lin, E. Stanley, Z. Ament, J. A. West, K. Summerhill, J. L. Griffin, W. Vetter, K. J. Autio, K. Hiltunen, S. Hazebrouck, R. Stepankova, C.-J. Chen, M. Alligier, M. Laville, M. Moore, G. Kraft, A. Cherrington, S. King, R. M. Krauss, E. De Schryver, P. P. Van Veldhoven, M. Ronis and A. Koulman, Sci. Rep., 2017, 7, 44845 CrossRef CAS.
- B. C. Jennings and M. E. Linder, J. Biol. Chem., 2012, 287, 7236–7245 CrossRef CAS.
- M. S. Rana, P. Kumar, C. J. Lee, R. Verardi, K. R. Rajashankar and A. Banerjee, Science, 2018, 359, 6372 CrossRef PubMed.
- X. Liu, S. Sadhukhan, S. Sun, G. R. Wagner, M. D. Hirschey, L. Qi, H. Lin and J. W. Locasale, Mol. Cell. Proteomics, 2015, 14, 1489–1500 CrossRef CAS PubMed.
- A. M. James, A. A. I. Norman, J. W. Houghton, H. A. Prag, A. Logan, R. Antrobus, R. C. Hartley and M. P. Murphy, Cell Chem. Biol., 2022, 29, 1232–1244e5 CrossRef CAS.
- M. Singh, H. L. Elfrink, A. C. Harms and T. Hankemeier, Mol. Genet. Metab., 2023, 140, 107711 CrossRef CAS.
- L. A. Broadfield, A. A. Pane, A. Talebi, J. V. Swinnen and S. M. Fendt, Dev. Cell, 2021, 56, 1363–1393 CrossRef CAS PubMed.
- I. J. Tinsley, J. A. Schmitz and D. A. Pierce, Cancer Res., 1981, 41, 1460–1465 CAS.
- G. Pascual, D. Domínguez, M. Elosúa-Bayes, F. Beckedorff, C. Laudanna, C. Bigas, D. Douillet, C. Greco, A. Symeonidi, I. Hernández, S. R. Gil, N. Prats, C. Bescós, R. Shiekhattar, M. Amit, H. Heyn, A. Shilatifard and S. A. Benitah, Nature, 2021, 599, 485–490 CrossRef CAS.
- M. Singh, H. L. Elfrink, A. C. Harms and T. Hankemeier, Mol. Genet. Metab., 2023, 140, 107711 CrossRef CAS.
- M. Singh, L. A. Kiyuna, C. Odendaal, B. M. Bakker, A. C. Harms and T. Hankemeier, J. Chromatogr. A, 2024, 1714, 464524 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4sc02235a |
| ‡ Current address: Fundació Privada Institut de Recerca sobre Immunopatologies-Caixa IrsiCaixa, Campus Can Ruti, Badalona, Spain. |
| § Current address: Departament d'Infermeria Fonamental i Medicoquirúrgica, Escola d'Infermeria, Facultat de Medicina i Ciències de la Salut, Universitat de Barcelona. |
| This journal is © The Royal Society of Chemistry 2024 |